Automated flow injection system using extraction chromatography for the determination of plutonium in urine by inductively coupled plasma mass spectrometry
Dominic
Larivière
*a,
Thomas A.
Cumming
b,
Stephen
Kiser
a,
Chunsheng
Li
a and
R. Jack
Cornett
a
aRadiation Protection Bureau, Health Canada, 775 Brookfield Road, Address Locator 6302D1, Ottawa, ON, Canada K1A 1C1. E-mail: dominic_lariviere@hc-sc.gc.ca; Fax: +1 (613) 952-9071; Tel: +1 (613) 957-9039
bDepartment of Chemistry, Carleton University, 1125 Colonel By Drive, Ottawa, ON, Canada K1S 5B6
First published on 9th November 2007
Abstract
An automated system for on-line pre-concentration, separation and detection of plutonium in a urine sample was developed, based on the coupling of a multi-solvent delivery system, remotely-controlled switching modules, and an inductively coupled plasma mass spectrometer (ICP-MS). Effective separation between spectral and non-spectral interferences and Pu was performed viaTEVA selective extraction chromatography. Pu elution from the resin was performed using 0.01 M (NH4)2C2O4, directed to the ICP-MS through a switching module controlled by the multi-solvent delivery unit. The automated flow injection system (AFIS) enables the quantification of Pu isotopes for urinalysis at the sub-mBq L−1 range (DL: 0.21(239Pu), 0.19(240Pu) mBq L−1) in less than 15 min, with a chemical recovery exceeding 70%. The simplicity, speed, and automation of this approach make it attractive for radiological emergency response, especially considering its high daily sample throughput (>80). This throughput is the result of the faster flow rate used for the separation (up to 3 mL min−1) and the reusability of the extraction resin. If a calcium phosphate co-precipitation step is performed prior to loading the sample onto the TEVA resin, improvement in pre-concentration capacity is possible, making the AFIS usable for the assessment of occupational exposures.
Introduction
The development and use of nuclear technology in the 20th century has accentuated the need for biomonitoring of potentially exposed populations to various radionuclides.1 While routine biomonitoring for occupational health assessment of nuclear and military workers has been performed for years, unexpected events such as the nuclear accidents at Chernobyl, the 2000 Cerro Grande Fire, and the possibility of a radiological/nuclear terrorist threat have highlighted the need for large-scale public biomonitoring strategies.2–4 Biomonitoring of occupationally exposed populations requires analytical methods with a minimum detectable activity as low as sub-µBq L−1 and outstanding precision to reconstruct the dose estimate.5–8 Protocols designed for emergency response monitoring purposes have very different requirements from those used for routine monitoring. For example, the detection requirements would be much higher than for occupational exposure as it is used primarily to screen individuals.3 Pappas et al.9 have established the 239Pu action level in urine for emergency purpose as 4.8 mBq L−1, based on a single dose exposure of 500 mSv. This value is several orders of magnitude higher than the typical levels observed for occupational exposure.8 Emergency analysis would have to be performed promptly as results are required to provide a prognosis and determine treatment strategies for contaminated individuals.4,10,11 Higher sample throughputs are also desirable to cope with the large number of samples that will have to be processed following an event.11Amid the various anthropogenic radioisotopes potentially associated with a radiological/nuclear treat, plutonium is considered one of the most radiotoxic elements as a result of the relatively shorter half-lives (t½) of its most predominant radioisotopes (239Pu: 2.410 × 104 y; 240Pu: 6.537 × 103 y) when compared with other long-lived actinides such 238U (4.46 × 109 y), and the high energy α-emission associated with their decay. Many analytical methods and instrumentation have been applied to the detection of 239,240Pu, particularly in urine, a medium of predilection for biomonitoring. Among them, two distinctive approaches are discernable: radiation- and ion-counting. Radiation-counting measurements of Pu are generally performed either by fission track analysis (FTA)5–7 or alpha energy analysis (AEA).12–15 Unfortunately, these radiometric approaches have a significant time requirement for counting (up to a day per sample) and do not allow for the discrimination between 239Pu and 240Pu isotopes. Ion-counting instruments such as secondary ion mass spectrometry (SIMS),16resonance ionization mass spectrometry (RIMS),17 thermal ionization mass spectrometry (TIMS),8,14,18,19accelerator mass spectrometry (AMS),20,21 and inductively coupled plasma mass spectrometry (ICP-MS)9,22–31 have also been used to determine Pu isotopes in urine. Because ICP-MS can directly ionize analytes contained in an aqueous matrix, it is therefore understandable that this technique is principally used for the development of rapid analytical methods for Pu urinalysis. However, Pu determination by ICP-MS at the mBq L−1 range (1 mBq is equal to 0.435 and 0.119 pg for 239Pu and 240Pu, respectively) in urine is hampered by many issues, including isobaric and polyatomic interferences and non-spectral interferences.32,33
Nonetheless, the pre-concentration and separation of the analyte from the matrix constituents and interfering isotopes have been shown, to a large extent, to overcome the limitation of Pu urinalysis by ICP-MS.9,25–27,29–32 Most of the sample preparation protocols associated with Pu urinalysis have been based on a two-stage separation: bulk matrix constituent removal by co-precipitation and actinide pre-concentration and separation using extraction chromatographic resins.25–29 While these procedures are well suited for Pu urinalysis, they are not ideal for an emergency biomonitoring situation. First, the co-precipitation of Pu to reduce the volume of the urine sample (1 L or more) is a process that is time consuming and relatively labour intensive. Such a large volume of urine will most likely not be available in an emergency situation. Second, the time requirement to perform a two stage separation scheme is long, requiring as many as 12 hours per sample,25 an unacceptable delay in emergency response mode. For these reasons, many research groups have focused on developing analytical methods requiring decreased urine volume, thus avoiding the tedious co-precipitation step.9,23,24,31 They are all based on chromatographic separation of the bulk matrix and interfering analytes using either commercially available extraction resins9,24,31 such TEVA and TRU or ion-exchange resins such as MetPac™ CC-123 as the stationary phase. While the protocols developed by Hang et al.23,24 have significant application for the simultaneous detection of long-lived actinides in urine, they lack the selectivity to determine ultra-traces of Pu in urine as a result of peak overlapping with many other actinides, especially when undiluted urine samples were injected. The method presented by Pappas et al.9 is unique since it requires only a single mL of sample in order to be performed. Separation between 239Pu, 238U, and the bulk constituent of the urine matrix was done using the TEVA extraction chromatography resin. Using such resin and a desolvating introduction system, Pappas et al.9 were able to minimize the impact of 238U1H+ and other polyatomic interferences at m/z = 239 and achieve a limit of detection (LOD) of 3.2 mBq L−1, below 4.8 mBq L−1, the derived action level for 239Pu in urine. Epov et al.31 have also developed a similar protocol for 239Pu measurement in urine but designed around the TRU resin as extractant. Their approach involves the automation of the separation procedure by a flow injection unit. While such an approach has been used in the past for the pre-concentration and separation of 239Pu in various matrices such as soil and sediment,34–37 sea-water,38 nuclear related materials,39–41 vegetation42 and biological tissues,37 it was the first time that it had been applied to urine. The automation of the pre-concentration system presented by Epov et al.31 performs the pre-concentration, separation and detection of 239Pu in urine in less than 11 min with a LOD of 4.3 mBq L−1. Despite its high sample throughput, this system has some important limitations. Epov et al.31 demonstrated that the TRU resin reusability was limited to a single injection, unless sample digestion was performed prior to the loading phase. They also indicated that the separation between Pu and U was not complete, as the result of a co-extraction of U from the TRU resin during Pu elution. To minimize the detection of U-related interferences, they used CO2 gas inside a dynamic reaction cell (DRC) to minimize the 238U1HX+ interferences. However, the fact that this approach needs a particular type of instrumentation limits its widespread applicability.
In this study, the capability of TEVA resin for the automated pre-concentration and separation of Pu in urine was evaluated. In order to provide an analytical system that could be suitable for routine and emergency purposes, the pre-concentration, separation, and detection of the analyte were performed using an automated flow injection system (AFIS). The protocols developed allow the separation of the analyte from the bulk matrix and from interfering elements, as well as providing some level of pre-concentration within 15 min. The automation of the protocol also helps increase sample throughput, a critical factor in emergency response monitoring.
Experimental
Instrumentation
All measurements were performed on an Element2 (ThermoFinnigan, Bremen, Germany) sector-field inductively coupled plasma mass spectrometer (ICP-SFMS). A high sensitivity inlet system (Apex-HF, Elemental Scientific Inc., Omaha, NE) coupled with a Microflow perfluoroalkoxy (PFA) nebulizer was used as the sample introduction system. Instrument conditions for the Element2 ICP-SFMS and the Apex-HF were set to ensure high instrument sensitivity and good precision during measurements (Table 1).| Instrumental parameters | Element2 ICP-SFMS |
|---|---|
| Torch position | Optimized daily |
| Sample flow rate/mL min−1 | 1.0 |
| Gas flow/L min−1 | |
| Cooling | 16.0 |
| Auxiliary | 0.80 |
| Sample | 1.063 |
| RF power/W | 1225 |
| Guard electrode | On |
| Lenses/V | |
| Extraction | −2000 |
| Focus | −855 |
| X-deflection | 0.75 |
| Shape | 95 |
| Y-deflection | −3.56 |
| Detector voltage/V | 2230 |
| Sampling cone | 1.1 mm nickel |
| Skimmer cone | 0.8 mm nickel |
| m/z monitored | 239, 240, 242 |
| Number of passes | 1 |
| Number of replicates | 235 |
| Acquisition time/s | 213 |
| Dwell time/ms | 60 |
| Apex-Q system parameters | |
| Nebulizer | 1 mL min−1 PFA microflow |
| Spray chamber temperature/°C | 140 |
| Peltier-cooled multi-pass condenser temperature/°C | −5 |
Fig. 1 is a schematic diagram of the automated flow injection system (AFIS) developed, which is composed of three sections. The first section is a multi-solvent delivery system (600E unit, Waters, Milford, MA), which is used to deliver up to four different solvents in either an isocratic or gradient mode. The second section is composed of two biocompatible analytical-scale two-position, six-port switching modules (MX9900-000, Upchurch Scientific, Oak Harbor, WA). Biocompatible modules are used instead of stainless steel to minimize the risk of corrosion, due to the high acidity of the eluents, and possible sample contamination from metal leaching of the steel. Modules are controlled by the Empower 2 chromatography data software (Waters). The final section consists of the Apex-HF unit and the Element ICP-SFMS.
 | ||
| Fig. 1 Schematic of the automated flow injection system (AFIS) for the pre-concentration and separation of Pu in urine. | ||
Reagents and materials
Milli-Q water (High-purity, 18 MΩ cm) prepared by a Milli-Q® ultrapure water purification system (Millipore, Bedford, MA) was used for all reagent dilutions. Optima grade concentrated HNO3 (Fisher Scientific, Ottawa, ON) was used to prepare diluted solutions. ACS-certified ammonium oxalate monohydrate salt and concentrated optima grade HCl (both from Fisher Scientific) were used to prepare 0.1 M (NH4)2C2O4 and 5 M HCl eluent solutions, respectively. ACS-certified calcium nitrate tetrahydrate and ammonium hydrogen phosphate salts (Fisher Scientific) were used to prepare 1.25 M Ca(NO3)2 and 3.2 M (NH4)2HPO4 solutions, respectively, for Pu co-precipitation. Standard solutions of 239,240,242Pu (SRM-4330B, -4338A, and -4334G, respectively) and 243Am (SRM-4332D), used as spikes, were purchased from the National Institute of Standards and Technology (NIST) (Gaithersburg, MD). Uranium and thorium standard solutions were prepared from 1000 µg mL−1 Claritas PPT®-grade standard (Spex CertiPrep, Metuchen, NJ). Working standards were made by serial dilution of Pu standards into 3 M HNO3 medium. Urine was collected from healthy adult donors, homogenized, spiked with 239,240Pu, acidified to a final concentration of 1% (v/v) HCl and stored at 4 °C until measurement. In order to assess the efficiency of the automated protocol for use in case of a nuclear emergency, urine samples containing 239,240Pu, prepared by the bioassay section of the Radiation Surveillance and Health Assessment Division, Radiation Protection Bureau, Health Canada, were measured.Automated flow injection system
Detailed operation of the AFIS (with the exception of the loading/reloading step) is described in Table 2. Although the protocol proposed lasts only 870 s, the second sample can be injected only 60 s after the first injection, since the Empower 2 chromatography software requires 30 s to process the data and another 30 s to stabilize the eluent flow rate prior to injection.| Switching modules (SM) position | ||||||
|---|---|---|---|---|---|---|
| Step | Time/s | Medium | Flow rate/mL min−1 | SM-1 d | SM-2 d | SLP b |
| a The value in parentheses represents the time elapsed after the beginning of the step at which the switching process occurs. b Sample loop position: elute signifies that the medium is passing through the sample loop while load is the opposite. c Acquisition of data by the ICP-MS is triggered 100 s after the start of step 3. d SM-1 and SM-2 refer to switching modules shown in Fig. 1. | ||||||
| 1 | 360 | 3 M HNO3 | 3 | On | Off | Elute |
| 2 | 120 | 5 M HCl | 3 | On | Off | Load |
| 3c | 270 | 0.01 M (NH4)2C2O4 | 1 | On | On (100a) | Load |
| 4 | 30 | Milli-Q H2O | 4 | On | Off | Load |
| 5 | 90 | 3 M HNO3 | 3 | On | Off | Load |
| Total | 870 | |||||
| Step | Step description | |||||
| 1 | 3 M HNO3 is pumped through the sample loop to load the sample and rinse the residual elements from the resin | |||||
| 2 | 5 M HCl is pumped through TEVA to elute Th | |||||
| 3 | 0.01M (NH4)2C2O4 is pumped through TEVA to elute Pu | |||||
| 4 | Milli-Q water is used to rinse resin from any residual elements | |||||
| 5 | 3 M HNO3 is pumped through resin to pre-condition the resins for the next analysis | |||||
A stainless steel analytical grade column (Alltech, Columbia, MD), 4.6 mm id × 50 mm long, coated inside with polyether ether ketone (PEEK) was filled with TEVA resin (50–100 µm particle size, Eichrom Technologies Inc., Darien, IL). All transport and reagent lines used to design the FI unit were made of 0.762 mm id PEEK tubing (Alltech, Columbia, MD) with the exception of the transfer line between the second switching module (SM-2) and the PFA nebulizer, which was assembled using 1.1 mm id polytetrafluoroethylene (PTFE) tubing (Alltech, Columbia, IL). 10–32 PEEK high pressure fittings with PEEK ferrules (Upchurch Scientific) for coned ports were used to connect the switching modules, multi-solvent delivery system, and the analytical-grade column. A 10 mL sample loop (Rheodyne, Rohnert Park, CA) was used to ensure a constant volume during the sample loading phase.
System optimization, interference removal, and decontamination factor
In order to achieve the optimized pre-concentration and separation of Pu in urine, various eluents were tested. All optimization tests were performed with a human urine sample acidified to 3 M HNO3 and spiked with 265 mBq L−1 of 239Pu. Study of the minimum volume of eluent required to eliminate non- and weakly-retained matrix constituents was performed by directly monitoring the content of the rinse. Optimal conditions for the elution of Th were investigated by running 5 M HCl through the resin after the 3 M HNO3 rinse and monitoring its Th content. Finally, Milli-Q water, 0.01 and 0.1 M (NH4)2C2O4, 0.01 M HNO3, and 0.1 M HCl were evaluated to assess their efficiency of elution of Pu from the TEVA resin. The effect of the flow rate on the retention of Pu was studied subsequently by varying the uptake flow rate from 1 to 5 mL min−1 and comparing the signal measured during the elution to the results obtained at 0.5 mL min−1.The decontamination factor (DF) for various elements found in urine was calculated based on concentrations found in Pu fractions (step 3, Table 2). These fractions were collected and analyzed off-line using the same instrumentation and sample introduction as those specified in Table 1. Calculations were performed using eqn (1.1). [Analyte]Pu and [Analyte]blank represent the concentration of the analyte in the plutonium fraction collected after extraction for a spiked urine sample and a blank solution (3 M HNO3), respectively. [Analyte]urine and [Analyte]HNO3 represent the analyte concentration in the original urine sample, and in 3 M HNO3, respectively. PCF represents the pre-concentration factor of the volume collected. In this system, 10 mL of the sample was injected while 3 mL was collected as the final Pu fraction. Therefore, the PCF is equal to 3.33. To enhance the detection of the analytes tested (i.e., Li, Na, Rb, Ca, Be, Mg, Ca, Sr, Ba, Al, Fe, Pb, Bi, Th, U, and Am), the urine sample used in this part of the experiment was spiked to 100 µg L−1 of analyte using mono-elemental solutions or 1 Bq L−1 with 243Am. An internal standard, 115In, was added subsequently to each solution to correct for the possible variation of the ICP-MS signals among different solutions. UH+ interference was studied with urine spiked at levels of 10, 100, and 1000-fold higher than the original concentration found in the raw sample (approximately 10 ng L−1). 238U1H+/238U+ and 230,232Th12C+/230,232Th+ were measured by directly injecting a 10 µg L−1U and Th standard, respectively, in 0.01 M (NH4)2C2O4 into the ICP-SFMS at a flow rate of 1 mL min−1.
 | (1.1) |
In order to estimate the carry-over effect, a urine sample containing 3 Bq L−1 of 239Pu was measured, followed by three samples containing only 3 M HNO3. The 239Pu content of these samples was measured and used to assess the carry-over from each analysis.
Sample preparation
Sample preparation was performed as follows: 9.375 mL of concentrated nitric acid and 50 µL of a solution of 242Pu (13.37 ng g−1 in 3 M HNO3) were added to approximately 30 mL of human urine. Urine was then added to achieve a final sample volume of 50 mL. The 3 M HNO3 sample was shaken to homogenize and then analyzed. Prior to injection, the sample was filtered through a 0.45 µm nylon filter to remove any particulate that could clog the system. No further treatment was performed on the samples.In order to study the applicability of the AFIS for occupational health assessment, larger volumes of samples were subjected to calcium phosphate co-precipitation prior to the separation. The co-precipitation protocol used is a modification of the one proposed by Zoriy et al.27 One liter of urine was spiked with 50 µL of a 242Pu standard (see above) and a known volume of 239Pu and 240Pu and mixed using a stirring bar for 15 min. After mixing, 0.5 mL of 1.25 M Ca(NO3)2 and 0.2 mL of 3.2 M (NH4)2HPO4 were added. The sample was then heated to 40–50 °C. Finally, NH4OH was added to initiate the precipitation. The sample was then centrifuged, filtered, redissolved in 10 mL 3 M HNO3 and finally separated using the protocol presented in Table 2.
Results and discussion
Separation conditions and flow rates
In order to reduce the overall time requirement per sample, the minimal volume of eluent (HNO3) necessary to elute non- and weakly-retained analytes during the sample loading phase was investigated. As Horwitz et al.43 demonstrated, U is only partially retained on the TEVA (k′ = 15 at 3 M HNO3) and therefore, with a sufficient volume of eluent, it can be eluted from the resin. The complete elution of U occurs after 18 mL of 3 M HNO3. When the U signal, uncorrected by the internal standard (Tl) is used, it is possible to see that the major constituents of the bulk matrix are eluted before U. The change in internal standard signal occurs at 11 mL, which is consistent with the volume of sample injected (10 mL) after accounting for the dead volume (∼1 mL).When elevated concentrations of Th are present in the plasma in conjunction with carbon isotopes, an interfering signal is measurable at m/z = 242 (230Th12C+) and 244 (232Th12C+), the masses of two long-lived Pu isotopes commonly used for yield tracing. Using the current sample introduction system and 0.01 M (NH4)2C2O4 as solvent, a 232Th12C+/232Th+ ratio of (1.1 ± 0.1) × 10−6 was measured, which is equivalent to a signal of 15 ± 3 counts per second for a 10 µg L−1 232Th solution. In order to minimize Th-induced molecular interferences, an additional step aimed at removing Th from the sample was added. High molarities of HCl have been shown to elute Th from the TEVA resin effectively without affecting Pu retention.43 6 mL of 5 M HCl were determined experimentally to be necessary to completely elute Th. This step was added after the loading step.
Elution of Pu from TEVA was assessed using 5 different eluents: 0.01 M and 0.1 M (NH4)2C2O4, H2O, 0.1 M HCl, and 0.01 M HNO3. These eluents and molarities were chosen because TEVA shows a lower D for Pu in all of them.36,38,43,44Table 3 synthesizes the figures of merit of the 239Pu peak in each solvent. In addition to the eluting time at maximum peak and the integrated signal of the peak, the full width at half maximum signal (FWHM) and the full width at 5% of the maximum signal (FW5%M) were also determined. The last two parameters are indicators of the peak width and the level of peak tailing, respectively. Based on these results, it is obvious that H2O is not the most effective solvent for Pu elution, since a larger peak (FWHM = 30 s) was obtained at a much longer elution time (t = 179 s). 0.1 M (NH4)2C2O4 shows a very rapid elution (t = 156 s) and an especially narrow peak (FWHM = 22.9 s). However, the integrated signal obtained for this eluent is significantly lower than for the other tests, likely resulting from plasma overloading. In the case of 0.01 M HNO3, while the peak shape is excellent, the integrated signal was lower than that for 0.01 M (NH4)2C2O4 and 0.1 M HCl, probably due to either incomplete desorption from the resin or loss into the introduction system surface resulting from the low acidity of the solution. For 0.01 M (NH4)2C2O4 and 0.1 M HCl, the results of all the parameters evaluated were very similar. Therefore, both solvents could be used for Pu elution. In this study, 0.01 M (NH4)2C2O4 was chosen. An elution volume of 4.5 mL was found to be sufficient to completely elute Pu from TEVA resin.
| Eluent | Time/s | Integrated signal/cps | FWHM/s | FW5%M/s |
|---|---|---|---|---|
| Milli-Q water | 179 ± 11 | 139![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 378 ± 16688 378 ± 16688 |
30 ± 2 | 73 ± 5 |
| 0.01 M (NH4)2C2O4 | 164 ± 3 | 158219 ± 7519 |
25.0 ± 0.8 | 60 ± 1 |
| 0.1 M (NH4)2C2O4 | 156 ± 3 | 99908 ± 2688 |
22.9 ± 0.4 | 57.4 ± 0.8 |
| 0.1 M HCl | 169 ± 1 | 158191 ± 5810 |
24 ± 1 | 61.5 ± 0.5 |
| 0.01 M HNO3 | 169.6 ± 0.5 | 146153 ± 9994 |
25 ± 2 | 61 ± 2 |
In order to minimize the time requirement per sample, the sample loading flow rate was investigated at flow rates varying between 1 and 5 mL min−1. While the pressure increased exponentially with the increasing flow rate, the chemical recovery for Pu did not seem affected in a significant manner. This result is in agreement with those of other extraction-based experiments.39,45 Therefore, while theoretically a flow rate of 5 mL min−1 can be used as the sample uptake rate, experimentally a flow rate of 3 mL min−1 is more appropriate to reduce the risk of excessive pressure during the loading phase. In the case of the eluents injected into the plasma, the flow rate was voluntarily limited to 1 mL min−1 to prevent plasma overloading and to avoid exceeding the uptake flow rate recommended by the manufacturer for the micronebulizer.
Spectral and non-spectral interferences removal
It is well established that isobaric, polyatomic, and non-spectral interferences are among the most significant analytical challenges faced by mass spectrometrists, especially when dealing with ultra-trace analyses such as long-lived anthropogenic radionuclides.33,46 To evaluate the efficiency of the proposed protocol for the removal of spectral and non-spectral interferences, a urine sample was spiked with multiple mono-elemental solutions, subjected to the protocol, and the Pu fraction was collected for analysis. Based on eqn (1.1), the decontamination factors (DF) for many analytes were calculated (Table 4). Most of the major constituents of urine, such as alkalis and alkaline earths, are totally removed from the resin prior to Pu elution. Other elements such as Al, Fe, and Pb are significantly reduced, but not completely removed. In the case of other actinides that could interfere with Pu analysis, these were completely removed from the Pu fraction (better than 99.9%), demonstrating the specificity of this protocol. The experimental DF value obtained for U (7.1 × 103) is close to the DF estimated with TEVA distribution ratio measured by Horwitz et al.45 (3.8 × 103), confirming that the choice of this resin as extraction resin was judicious.Epov et al.31 have demonstrated that the concentration of uranium present in the sample has a critical impact on the detection of Pu, independent of the analytical system used. To evaluate the selectivity of the analytical protocol proposed, measurements of the background signal at m/z = 239 and 240 were carried out with increasing concentration of 238U. For the 238U concentration range tested (10–10000 ng L−1), the background signal observed did not vary, demonstrating the effectiveness of the protocol. Eqn (1.2) was used to estimate the maximum acceptable concentration of 238U ([U]max) in urine that could theoretically be measured, where [239Pu]QL is the 239Pu method quantification limit, UH+/U+ the hydride ratio, and DFU the uranium decontamination factor. A UH+/U+ ratio of 2.87 × 10−5 was measured using the system proposed. While this ratio is slightly higher than the one reported by others,32,47 even with a matching instrumental setup, this can be explained by three factors. First, the ratio was measured in 0.01 M (NH4)2C2O4 to reflect the actual ratio in the eluent used for Pu elution, instead of the 2% (v/v) HNO3 typically used. Second, no attempts were made to minimize the hydride formation into the plasma, the instrument was simply optimized for maximal sensitivity at m/z = 239. Finally, the Apex unit used by Boulyga and Heumann47 to measure uranium hydride ratio was equipped with a desolvating membrane, which has been know to produce drier plasma conditions,48 therefore reducing the possible formation of hydride. Based on this hydride formation ratio and the U decontamination factor (7.1 × 103), a 238U concentration higher than 76 µg L−1 is necessary to elevate the signal at m/z = 239 above the 239Pu method quantification limits (0.71 mBq L−1, which is equivalent to 0.309 pg L−1).
 | (1.2) |
Analytical performance
The most relevant analytical figures of merit for the AFIS for the pre-concentration and detection of Pu isotopes in urine were evaluated and are presented in Table 5. The instrument and method detection limits (based on 3σ for 12 replicates), measured using 3 M HNO3 and acidified blank urine, respectively, were found to be 0.12 and 0.21 mBq L−1 for 239Pu. The higher detection limit for the urine blank is the result of the higher background variability observable at m/z = 239 and the lower chemical recovery. The limits of detection in this work are better than those stated by Epov et al.31 (4.4 mBq L−1) and Pappas et al.9 (3.2 mBq L−1). However, note that in the case of the Epov et al.31 investigation, background was measured using 2% HNO3 and detection limits were based on a 10 mL volume of urine, whereas Pappas et al.9 used 3σ (both within and between run) on 20 runs of 1 mL of a base urine sample, making the absolute comparison between methods difficult. The proposed method also compares favorably to the Zoriy et al.27 method after accounting for the pre-concentration achieved by Ca3(PO4)2co-precipitation (2 mBq L−1).| Emergency protocol | Occupational exposure protocol | |||
|---|---|---|---|---|
| a N.A.: Not applicable as sample preparation is required. | ||||
| Sample volume/mL | 10 (8.125) | 1000 | ||
| Time of analysis/min | 14.5 | 14.5 | ||
| Sample frequency/sample d−1 | 99 | N.A.a | ||
| Precision for 10 mBq L−1 (% RSD, 12 replicates) | 4.6 | 3.8 | ||
| Chemical recovery (%, 24 samples) | 83 ± 3 | 72 ± 9 | ||
| 239Pu | 240Pu | 239Pu | 240Pu | |
| Absolute sensitivity/105 counts mBq−1 | 3.74 | 1.03 | 3.74 | 1.03 |
| Instrumental detection limits (3σ)/mBq L−1 | 0.12 | 0.14 | 0.0013 | 0.0015 |
| Method detection limits (3σ)/mBq L−1 | 0.21 | 0.19 | 0.0023 | 0.0021 |
| Method absolute detection limits/µBq | 1.7 | 1.5 | 0.019 | 0.017 |
| Method quantification limits (10σ)/mBq L−1 | 0.71 | 0.64 | 0.0078 | 0.0074 |
A sensitivity of 3.74 × 105 counts per mBq was calculated using the high sensitivity introduction system (Apex-Q) and the ICP-SFMS. This value is between the values reported by Zoriy et al.27 for the same instruments but with different sample introduction systems (0.9 and 6.02 × 105 counts mBq−1 for PFA-100 and DIHEN, respectively). Assuming that 100% of the sample is introduced into the plasma by the DIHEN, this indicates that the Apex-Q introduces approximately 62% of the sample into the plasma. This value is consistent with other nebulization yields33 for the same sample introduction system.
One of the concerns regarding the use of TEVA for Pu extraction is that it has a lower maximum distribution ratio than TRU.43 This should experimentally translate into a lower chemical recovery. In this study, recovery ranged from 77 to 88% with an average value of 83 ± 3%. These recoveries are lower than results reported by Epov et al.,31 who stated that using the TRU resin the recovery ranged from 70 to 100%, with most recoveries above 90%. Zoriy et al.27 reported a recovery average of 70% using TEVA, a result lower than the one observed in this study, probably resulting from loss in the co-precipitation step.
Although, the AFIS has a lower sample throughput than the protocol presented by Epov et al.31 (11 versus 14.5 minutes sample−1), the current methodology still has a significant theoretical daily throughput approaching 100 samples. Practically, the authors were able to perform 20 samples in a continuous 6-hour period, leading to a more realistic 80 samples per day. This sample frequency includes time required for column replacement and daily instrument optimization.
Memory effect and column reusability
In order to minimize the time associated with replacing and re-equilibrating the column between samples, the reusability of the TEVA extraction resin was investigated. Two indicators were used to assess the resin performance: (1) the carry-over percentage and (2) the 242Pu chemical recovery for multiple injections. The carry-over of one sample to another was evaluated by injecting a urine sample with high activity (239Pu = 3 Bq L−1), followed by 2 blank runs using 3 M HNO3. The results are presented in Fig. 2 and they indicate the presence of a small carry-over between samples. The carry-over was 1.3 ± 0.1% (n = 3) between samples. The percentages presented in Fig. 2 are similar to the ones reported with the same resin for the measurement of Pu in sea-water.38 Since the relative standard deviation of the measurements is approximately 4.6% (Table 5), the impact of carry-over is limited, especially for samples with a low level of activity. Based on this ratio and the detection limits of the analytical system, samples with an activity below 11.4 mBq L−1 will not produce a detectable signal from cross-contamination. A stronger complexing eluent (i.e., 0.1 M (NH4)2C2O4) and longer rinse time (step 4 in Table 2) were also tested but did not decrease the carry-over percentage.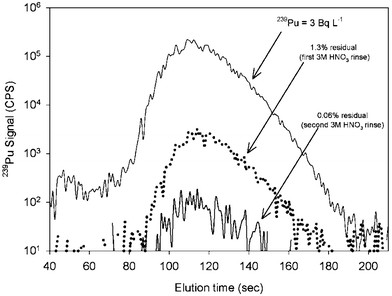 | ||
| Fig. 2 Memory effect observed in the subsequent rinses after a 3 Bq L−1 239Pu urine sample. Note that the y-axis is a log-scale. | ||
Resin reusability also was investigated by measuring the chemical recovery of 242Pu for 6 replicates performed on the same resin. The results show a 242Pu recovery ranging from 77.1 to 83.2%, with an average of 80.3 ± 2.1%. No statistical difference was observed between the first and the sixth urine sample measured using the same resin. This indicates that the resin can be reused at least 6 times without concern regarding the possible loss of extraction capacity for the TEVA resin. This result agrees with the results presented by Kim et al.,38 who did not observe any change in the TEVA resin performance after 10 injections.
Quantification of 239,240Pu in urine samples
The performance of the AFIS was evaluated using samples provided by the Radiation Protection Bureau as part of an emergency methods inter-comparison exercise that included ICP-MS, α-spectrometry, and TIMS.49 The measurements performed by this system proposed are in agreement with the expected activity for both Pu isotopes (Table 6). The activity range tested for the inter-comparison exercise also demonstrates the applicability of the method for a large range of Pu concentrations. When the results obtained by the newly developed system are plotted against the certified values, strong correlation between the results is observed (r2 = 0.9997 (239Pu) and 0.9996 (240Pu)). This indicates that the determination of 239Pu and 240Pu is not hampered by the presence of interference at m/z = 239 and 240, respectively, even at low activity, where interference would be most likely to occur. The performance of the system for determining the 240/239Pu mass ratio was also evaluated. The results are in agreement with all of the expected ratios, but show a higher variability for the low activity samples (HC–C and –D). The variability can be explained by the lower signal measured in these samples, which are more impacted by dark detector noise and fluctuations in the sample and blank signal.| Sample identification | This method/mBq L−1 | Expected activities/mBq L−1 | 240Pu/239Pu mass ratio measured | Expected 240Pu/239Pu mass ratio | ||
|---|---|---|---|---|---|---|
| 239Pu | 240Pu | 239Pu | 240Pu | |||
| a The expected activities and isotopic ratio are based on a round-robin inter-comparison for ICP-MS, α-spectrometry, and TIMS.49 b Samples spiked in Health Canada’s laboratory. | ||||||
| Emergency protocola | ||||||
| HC-A (12 replicates) | 289 ± 16 | 282 ± 19 | 297 ± 2 | 296 ± 2 | 0.274 ± 0.005 | 0.271 ± 0.003 |
| HC-B (12 replicates) | 994 ± 18 | 1176 ± 32 | 1002 ± 7 | 1246 ± 9 | 0.342 ± 0.006 | 0.339 ± 0.004 |
| HC-C (12 replicates) | 49 ± 2 | 76 ± 3 | 49.9 ± 0.3 | 75.1 ± 0.5 | 0.414 ± 0.011 | 0.410 ± 0.003 |
| HC-D (12 replicates) | 9.9 ± 0.4 | 5.2 ± 0.5 | 10.00 ± 0.07 | 4.90 ± 0.03 | 0.151 ± 0.016 | 0.133 ± 0.002 |
| HC-E (12 replicates) | 3099 ± 56 | 2201 ± 37 | 2994 ± 20 | 2250 ± 16 | 0.207 ± 0.001 | 0.205 ± 0.003 |
| Occupational protocol (Ca3(PO4)2co-precipitation prior to separation)b | ||||||
| S-1 | 0.97 ± 0.02 | 0.93 ± 0.03 | 0.95 ± 0.02 | 0.93 ± 0.02 | ||
| S-2 | 2.87 ± 0.05 | 1.86 ± 0.02 | 2.89 ± 0.09 | 1.86 ± 0.07 | ||
| S-3 | 3.69 ± 0.08 | 3.71 ± 0.16 | 3.79 ± 0.08 | 3.71 ± 0.11 | ||
| S-4 | 5.52 ± 0.37 | 1.90 ± 0.15 | 5.69 ± 0.22 | 1.86 ± 0.09 | ||
| S-5 | 7.50 ± 0.15 | 3.04 ± 0.11 | 7.58 ± 0.23 | 2.97 ± 0.08 | ||
The possibility of using the AFIS for occupational exposure assessment was also investigated. Prior to the pre-concentration and separation using the emergency methodology, a large volume of urine (approximately 1 L) was subjected to Ca3(PO4)2co-precipitation. This step achieves higher preconcentration factors (up to 100-fold if the final dilution is to 10 mL), leading to much lower detection limits (Table 5). Absolute detection limits of 19 and 17 nBq of 239Pu and 240Pu, respectively, were achieved when the automated process followed co-precipitation. This DL is sufficient to ensure the proper measurement of occupational exposure. While the analysis time remains the same (14.5 min), the time requirement for sample preparation becomes much longer (>1 d) due to the co-precipitation step. In addition, the chemical recovery is also impacted by the addition of the co-precipitation step (72 ± 9% versus 83 ± 3%). Assuming that the performances of the AFIS are not affected by the presence of large quantities of Ca3(PO4)2, the efficiency of the co-precipitation process for Pu would be approximately 90%. The chemical recovery observed (72 ± 9%) is similar to the one reported by Zoriy et al.27 (71%). Results obtained with the occupational exposure protocol (co-precipitation and automated separation) are also in excellent agreement with the expected activities based on the spiked levels (Table 6), confirming the capability of the methodology to measure minute quantities of 239Pu and 240Pu in urine samples.
Conclusion
The AFIS coupled with ICP-SFMS was successfully applied to the determination of Pu isotopes in urine. The AFIS demonstrates a high selectivity for Pu while minimizing the pre-concentration of possible interferences. While originally intended for urinalysis, the AFIS can be applied to the determination of Pu in any liquid matrix as the system eliminates the molecular interferences hampering Pu determination by mass spectrometry. The detection limits achieved (0.21 mBq L−1 for 239Pu) are more than 10-fold lower than the action level for Pu in urine during a nuclear emergency. The pre-treatment of the samples using co-precipitation prior to separation and detection using the AFIS allows the detection of Pu in the µBq L−1 range, the sensitivity required for occupational monitoring purposes. The high sample throughput of the AFIS is also a unique feature that is of the utmost importance for laboratories involved in radiobioassay measurements.Acknowledgements
Gerry Moodie and Renato Falcomer provided assistance during the sample preparation process. Support for this work was provided by the Canadian Chemical Biological Radiological and Nuclear (CBRN) Research and Technology Initiative (CRTI) and by Health Canada.References
- D. W. Moeller, Health Phys., 2005, 88, 676–696 CrossRef CAS.
- S. A. Hodgson, G. J. Ham, M. J. Youngman, G. Etherington and G. N. Stradling, J. Radiol. Prot., 2004, 24, 369–389 CrossRef CAS.
- D. L. Stricklin, A. Tjarnhage and U. Nygren, J. Radioanal. Nucl. Chem., 2002, 251, 69–74 CrossRef CAS.
- N. Green, Sci. Total Environ., 1993, 130, 207–218 CrossRef.
- L. C. Sun, A. R. Moorthy, E. Kaplan, J. W. Baum and C. B. Meinhold, Appl. Radiat. Isot., 1995, 46, 1259–1269 CrossRef CAS.
- M. E. Wrenn, N. P. Singh and Y. H. Xue, Radiat. Prot. Dosim., 1994, 53, 81–84 CAS.
- M. P. Krahenbuhl and D. M. Slaughter, J. Radioanal. Nucl. Chem., 1998, 230, 153–160 CAS.
- W. C. Inkret, D. W. Efurd, G. Miller, D. J. Rokop and T. M. Benjamin, Int. J. Mass Spectrom., 1998, 178, 113–120 CrossRef CAS.
- R. S. Pappas, B. G. Ting and D. C. Paschal, J. Anal. At. Spectrom., 2004, 19, 762–766 RSC.
- D. F. Flynn and R. E. Goans, Surg. Clin. North Am., 2006, 86, 601 Search PubMed.
- W. F. Blakely, C. A. Salter and P. G. S. Prasanna, Health Phys., 2005, 89, 494–504 CrossRef CAS.
- M. D. Erickson and N. A. Chieco, The Procedures Manual of the Environmental Measurements Laboratory HASL-300, US Department of Homeland Security, New York, 1997 Search PubMed.
- A. Alvarez and N. Navarro, Appl. Radiat. Isot., 1996, 47, 869–873 CrossRef CAS.
- S. E. Wagner, S. Boone, J. W. Chamberlin, C. J. Duffy, D. W. Efurd, K. M. Israel, N. L. Koski, D. L. Kottmann, D. Lewis, P. C. Lindahl, F. R. Roensch and R. E. Steiner, J. Radioanal. Nucl. Chem., 2001, 248, 423–429 CrossRef CAS.
- J. M. Diodati, N. Bonino and M. R. Huguet, J. Radioanal. Nucl. Chem., 1994, 182, 111–117 CAS.
- A. Amaral, P. Galle, C. Cossonnet, D. Franck, P. Pihet, M. Carrier and O. Stephan, J. Radioanal. Nucl. Chem., 1997, 226, 41–45 CAS.
- N. Erdmann, G. Herrmann, G. Huber, S. Kohler, J. V. Kratz, A. Mansel, M. Nunnemann, G. Passler, N. Trautmann, A. Turchin and A. Waldek, Fresenius’ J. Anal. Chem., 1997, 359, 378–381 CrossRef CAS.
- D. Lewis, G. Miller, C. J. Duffy, D. W. Efurd, W. C. Inkret and S. E. Wagner, J. Radioanal. Nucl. Chem., 2001, 249, 115–120 CrossRef CAS.
- N. L. Elliot, G. A. Bickel, S. H. Linauskas and L. M. Paterson, J. Radioanal. Nucl. Chem., 2006, 267, 637–650 CrossRef CAS.
- A. A. Marchetti, T. A. Brown, C. C. Cox, T. F. Hamilton and R. E. Martinelli, J. Radioanal. Nucl. Chem., 2005, 263, 483–487 CrossRef CAS.
- N. D. Priest, G. M. Pich, L. K. Fifield and R. G. Cresswell, Radiat. Res., 1999, 152, S16–S18 CAS.
- C. Bouvier-Capely, J. Ritt, N. Baglan and C. Cossonnet, Appl. Radiat. Isot., 2004, 60, 629–633 CrossRef CAS.
- W. Hang, C. Mahan, L. W. Zhu and E. Gonzales, J. Radioanal. Nucl. Chem., 2005, 263, 467–475 CrossRef CAS.
- W. Hang, L. W. Zhu, W. W. Zhong and C. Mahan, J. Anal. At. Spectrom., 2004, 19, 966–972 RSC.
- J. Kuwabara and H. Noguchi, J. Radioanal. Nucl. Chem., 2002, 252, 273–276 CrossRef CAS.
- N. Baglan, C. Cossonnet, P. Pitet, D. Cavadore, L. Exmelin and P. Berard, J. Radioanal. Nucl. Chem., 2000, 243, 397–401 CrossRef CAS.
- M. V. Zoriy, C. Pickhardt, P. Ostapczuk, R. Hille and J. S. Becker, Int. J. Mass Spectrom., 2004, 232, 217–224 CrossRef CAS.
- E. J. Wyse and D. R. Fisher, Radiat. Prot. Dosim., 1994, 55, 199–206 CAS.
- E. J. Wyse, J. A. MacLellan, C. W. Lindenmeier, J. P. Bramson and D. W. Koppenaal, J. Radioanal. Nucl. Chem., 1998, 234, 165–170 CAS.
- B. G. Ting, R. S. Pappas and D. C. Paschal, J. Anal. At. Spectrom., 2003, 18, 795–797 RSC.
- V. N. Epov, K. Benkhedda, R. J. Cornett and R. D. Evans, J. Anal. At. Spectrom., 2005, 20, 424–430 RSC.
- C.-S. Kim, C.-K. Kim, P. Martin and U. Sansone, J. Anal. At. Spectrom., 2007, 22, 827–841 RSC.
- D. Lariviere, V. F. Taylor, R. D. Evans and R. J. Cornett, Spectrochim. Acta, Part B, 2006, 61, 877–904 CrossRef.
- Y. Ohtsuka, Y. Takaku, K. Nishimura, J. Kimura, S. Hisamatsu and J. Inaba, Anal. Sci., 2006, 22, 309–311 CrossRef CAS.
- Y. Ohtsuka, Y. Takaku, J. Kimura, S. Hisamatsu and J. Inaba, Anal. Sci., 2005, 21, 205–208 CrossRef CAS.
- C. S. Kim, C. K. Kim, J. I. Lee and K. J. Lee, J. Anal. At. Spectrom., 2000, 15, 247–255 RSC.
- J. B. Truscott, P. Jones, B. E. Fairman and E. H. Evans, Anal. Chim. Acta, 2001, 433, 245–253 CrossRef CAS.
- C. S. Kim and C. K. Kim, Anal. Chem., 2002, 74, 3824–3832 CrossRef CAS.
- J. W. Grate and O. B. Egorov, Anal. Chem., 1998, 70, 3920–3929 CrossRef CAS.
- O. B. Egorov, M. J. O'Hara, O. T. Farmer and J. W. Grate, Analyst, 2001, 126, 1594–1601 RSC.
- L. Perna, M. Betti, J. M. B. Moreno and R. Fuoco, J. Anal. At. Spectrom., 2001, 16, 26–31 RSC.
- V. N. Epov, K. Benkhedda and R. D. Evans, J. Anal. At. Spectrom., 2005, 20, 990–992 RSC.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, S. L. Maxwell and M. R. Nelson, Anal. Chim. Acta, 1995, 310, 63–78 CrossRef CAS.
- M. V. Zoriy, P. Ostapczuk, L. Halicz, R. Hille and J. S. Becker, Int. J. Mass Spectrom., 2005, 242, 203–209 CrossRef CAS.
- E. P. Horwitz, M. L. Dietz, D. M. Nelson, J. J. Larosa and W. D. Fairman, Anal. Chim. Acta, 1990, 238, 263–271 CrossRef CAS.
- C.-S. Kim, C.-K. Kim, P. Martin and U. Sansone, J. Anal. At. Spectrom., 2007, 22, 827–841 RSC.
- S. F. Boulyga and K. G. Heumann, J. Environ. Radioact., 2006, 88, 1–10 CrossRef CAS.
- Elemental Scientific Inc, Overview Presentation of the ACM Module Peltier Cooled Nafion Membrane Desolvator, http://www.icpms.com/powerpoint/ACMmodule.pdf last accessed March 02, 2007.
- C. Li, D. Lariviere, S. Kiser, G. Moodie, R. Falcomer, M. Zamora, N. Elliot, V. N. Epov, R. D. Evans, K. Inn, H. Korasaki, R. S. Pappas, J. Smith and R. J. Cornett, Health Phys., 2007, to be submitted Search PubMed.
| This journal is © The Royal Society of Chemistry 2008 |