Synthesis, characterization, and photophysical properties of platinum(II) terpyridyl-based multi-component systems
Paul
Jarosz
a,
Jason
Thall
a,
Jacob
Schneider
a,
Duraisamy
Kumaresan
b,
Russell
Schmehl
*b and
Richard
Eisenberg
*a
aDepartment of Chemistry, University of Rochester, Rochester, New York 14627, USA
bDepartment of Chemistry, Tulane University, New Orleans, Louisiana 70118, USA
First published on 2nd September 2008
Abstract
Four new Pt(II) terpyridyl acetylide complexes are reported which possess either a viologen or nicotinamide unit attached as an electron acceptor via the benzylic carbon of the tolyl-terpyridine ligand. Specifically, the donor–chromophore–acceptor (D–C–A) triads prepared are [Pt(ttpy-MV)C≡C–C6H4–NH–CO–C6H2(OMe)3](PF6)3 [1], where ttpy-MV = 4′-([4-methyl-4,4′-bipyridin]-ylmethyl-phenyl)-[2,2′;6′,2″]terpyridine and C≡C–C6H4–NH–CO–C6H2(OMe)3 = N-(4-ethynylphenyl)-3,4,5-trimethoxybenzamide and [Pt(ttpy-Nd)C≡C–C6H4–NH–CO–C6H2(OMe)3](PF6)2 [2], where ttpy-Nd = 4′-([3-carboxamide-pyridin]-ylmethyl-phenyl)-[2,2′;6′,2″]terpyridine. The related chromophore–acceptor (C–A) dyads, [Pt(ttpy-MV)C≡CPh](PF6)3 [3], where C≡CPh = phenylacetylide and [Pt(ttpy-Nd)C≡CPh](PF6)2 [4], were also prepared. The syntheses of the above compounds are achieved by the reaction of a prepared cuprous phenylacetylide compound with the appropriate platinum terpyridyl chloride complex. This change from the CuI-catalyzed acetylide coordination, which requires basic conditions, results from the observed sensitivity of the pyridinium type acceptors to base. While the parent chromophore, [Pt(ttpy)C≡CPh]PF6 [5], where ttpy = 4′-p-tolyl-[2,2′;6′,2″]terpyridine, is brightly emissive in fluid solution, both of the aforementioned D–C–A triads 1 and 2 are nonemissive at room temperature. Of the related C–A dyads, 4 is emissive and 3 is very weakly emissive. Transient absorption studies reveal that the nicotinamide acceptor does not function as an oxidative quencher of the excited state. In contrast, the viologen acceptor appears to function as an oxidative quencher.
Introduction
Photoinduced charge separation is an essential process in light-to-chemical energy conversion. Towards this end, many efforts have been made to construct two and three component systems comprised of a chromophore, an electron donor, and an electron acceptor.1–41 Many such systems are based on octahedral Ru(II) bi- and terpyridyl chromophores.3,7,15,19,30,31 Our efforts in this regard have focused on analogous square planar bi- and terpyridyl complexes of Pt(II), which have the benefit of directionality in the charge transfer excitation from the metal to the heteroaromatic ligand. This directionality can be exploited in the design and placement of electron donating and accepting moieties.42–46 Once a system that can achieve long-lived charge separation has been synthesized, it can be coupled to catalysts for electron or hole collection to drive energy storing processes.1,3,8,24,30,32,33,35–39,47–49In this vein, the synthesis and characterization of a triad and two dyads, shown in Fig. 1 as complexes 6–8 and based on the platinum terpyridyl acetylide chromophore, have been described.45 For 6 and 8 a pyridinium acceptor moiety is attached to the chromophore via the ttpy benzylic C atom. While triad 6 was found to have no observable emission at room temperature in contrast to the bright 3MLCT luminescence of the chromophore, dyad 8 was seen to have an emission similar to that of the parent chromophore but at a significantly reduced intensity (19%). The latter result argued against rapid, efficient oxidative quenching of the 3MLCT excited state by the pyridinium acceptor. Consistent with that notion was the fact that the transient absorption (TA) spectrum of 8 mirrored that of the parent chromophore, thus revealing a lack of pyridyl radical formation.
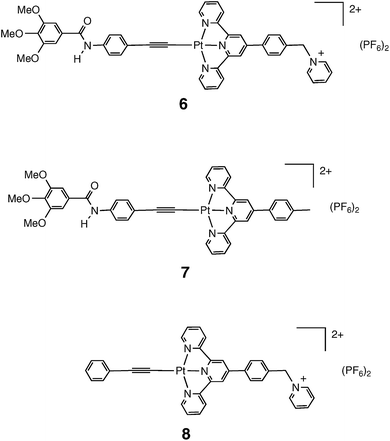 | ||
| Fig. 1 Triad and dyads prepared in previous studies. | ||
In order to examine whether the link between the chromophore and the acceptor was the problem in allowing facile electron transfer from the excited chromophore to the acceptor, other more readily reduced acceptor moieties attached to the chromophore in the same manner as 6 and 8 were employed. These acceptors include a benzyl nicotinamide cation and a mixed benzyl methyl viologen dication. The respective terpyridyl-acceptor ligands are shown in Fig. 2 and are denoted as ttpy-Nd and ttpy-MV.
 | ||
| Fig. 2 Ligands used to construct complexes 1–4. | ||
The present paper describes the new complexes produced with ttpy-Nd and ttpy-MV and the results of spectroscopic studies on these complexes regarding the effectiveness of charge transfer to the pyridinium-type acceptors. It should be noted that ttpy-MV has been employed previously by Sauvage and coworkers in the study of photoinduced charge separation in triad systems having a Ru(II) bis terpyridyl chromophore.3,7,15,19
Experimental
Materials
The chemicals 4′-p-tolyl-[2,2′;6′,2″]terpyridine (ttpy), N,N-dimethylformamide, dimethylsulfoxide, copper (I) iodide, ammonium hexafluorophosphate (NH4PF6), 3,4,5-trimethoxybenzoylchloride, 4-ethynylaniline (Aldrich), electrochemical grade tetrabutylammonium hexafluorophosphate (Fluka), biotech grade N,N-dimethylformamide and MeCN (Aldrich), and potassium tetrachloroplatinate (Strem Chemical) were used without further purification. Triethylamine was purified by distillation from KOH pellets. The syntheses of 4′-(p-bromomethylphenyl)-2,2′:6′2″-terpyridine (ttpy-Br), 1-methyl-4,4′ bipyridinium hexfluorphosphate, and Pt(DMSO)2Cl2 were carried out according to literature procedures.42,44,46,50 [Pt(ttpy)(C≡CPh]PF6 and [Pt(ttpy)(C≡C–C6H4–NH–CO–C6H2(OMe)3]PF6 [7] were used as previously prepared.45,46 Syntheses were performed under nitrogen with solvents purified by passing the degassed solvent through columns containing activated molecular sieves and activated alumina.51 All other reagents were of spectroscopic grade and used without further purification.Characterization
1H NMR spectra were recorded on a Bruker Avance-400 spectrometer (400 MHz). Mass determinations were accomplished by Electrospray Ionization Mass Spectrometry (ESIMS) and Atmospheric Pressure Chemical Ionization (APCI) using a Hewlett Packard Series 1100 mass spectrometer (Model A) equipped with a quadrupole mass filter. Cyclic voltammetry experiments were conducted on an EG&G PAR 263A potentiostat/galvanostat using a three electrode single compartment cell. A glassy carbon working electrode, Pt wire auxiliary electrode, and Ag wire reference electrode were used. For all measurements samples were degassed with nitrogen. 0.10 M Tetrabutylammonium hexafluorophosphate was used as the supporting electrolyte while ferrocene was employed as an internal redox reference. All redox potentials are reported relative to NHE following comparison to the observed ferrocenium/ferrocene (Fc+/Fc) couple (0.4 V vs. SCE for MeCN, 0.45 V vs. SCE for DMF),52 and all scans were done at 100 mV s−1. Absorption spectra were recorded using a Hitachi U2000 scanning spectrophotometer (200–1100 nm).Emission and excitation spectra were obtained using a Spex Fluoromax-P fluorimeter corrected for instrument response. Monochromators were positioned with a 2 nm band pass, and solution samples were degassed by at least three freeze–pump–thaw cycles prior to analysis. Frozen glass samples were prepared in butyronitrile in NMR tubes using a circular quartz-tipped immersion dewar filled with liquid nitrogen.
Transient absorption spectra were carried out using an Applied Photophysics LKS60 flash photolysis system with a Quantel Brilliant B Nd:YAG laser pumped OPOTEK OPO at various delays following excitation at various wavelengths between 420 nm and 500 nm. The spectra were assembled from decays measured at every 10 nm. Individual decays were analyzed as single or double exponentials using a Marquardt–Levenberg algorithm included with the LKS60 software. Kinetic analysis, where appropriate, was made by global analysis using the PRO-K software package (Applied Photophysics).
N-(4-Ethynyl-phenyl)-3,4,5-trimethoxybenzamide (3OMe-H)
This ligand was prepared by a modification of a previously reported procedure.45 A 100 mL round bottom flask was charged with 3,4,5-trimethoxybenzoylchloride (0.200 g, 0.83 mmol) and 4-ethynylaniline (0.100 g, 0.83 mmol) followed by 20 mL of chloroform and 0.4 mL of freshly distilled NEt3. The reaction was stirred at 40 °C for 12 h. The resulting solution was dried in vacuo to give a yellow oil. This was recrystallized from water and ethanol to give an off-white solid. Yield: 0.232 g (90%). 1H NMR (CDCl3): δ 8.39 (1H, br s), 7.76 (2H, d, J = 7 Hz), 7.52 (2H, d, J = 7), 7.20 (2H, s), 5.57 (1H, s), 3.98 (6H, s), 3.93 (3H, s). MS (APCI): m/z 312.1 [(M + 1)].4′-[p-(1-Methyl-4,4′-bipyridinium-1-yl)methyl-phenyl]-2,2′;6′,2″ terpyridine hexafluorophosphate [ttpy-MV](PF6)2
This ligand was prepared by a modification of a previously reported procedure.19 A 25 mL round bottom flask was charged with Br-ttpy (0.200 g, 0.50 mmol) and 1-methyl-4,4′ bipyridinium hexfluorphosphate (0.180 g, 0.57 mmol) followed by 30 mL of acetonitrile. The system was heated at reflux for 48 h, and the reaction was filtered to give a tan solid. The solid was treated with 50 mL distilled water and filtered again to remove any persisting Br-ttpy. To the filtrate, a saturated aqueous solution of NH4PF6 was added, and the resulting solution was stirred at room temperature for 3 h. The white solids were collected on a medium porosity glass frit, washed with distilled water, cold ethanol and diethyl ether. Yield: 0.174 g (43%). 1H NMR (DMSO-d6): δ 9.60 (2H, d, J = 7 Hz), 9.30 (2H, d, J = 7 Hz), 8.84 (2H, d, J = 7), 8.79 (2H, d, J = 6), 8.77 (2H, d, J = 6), 8.76 (2H, s), 8.72 (2H, d, J = 8), 8.11 (2H, d, J = 8), 8.09 (2H, t, J = 8), 7.88 (2H, d, J = 8), 7.58 (2H, t, J = 6), 6.09 (2H, s), 4.47 (3H, s). MS (positive-API-ES): m/z 636.0 [M − PF6]+.4′-(p-Nicotinamide-N-methylphenyl)-2,2′:6′,2″-terpyridine hexafluorophosphate (ttpy-Nd)PF6
This ligand was synthesized as previously reported.53 Yield: 0.961 g (96%). 1H NMR (DMSO-d6): δ 9.68 (1H, s), 9.35 (1H, d), 9.00 (1H, d), 8.80–8.78 (6H, m), 8.60 (1H, s), 8.32 (1H, dd), 8.20 (1H, s), 8.16 (2H, t), 8.08 (2H, d), 7.80 (2H, d), 7.63 (2H, td), 6.03 (2H, s). MS (positive API-ES): m/z 444.3 [M − PF6]+.[Pt(ttpy-MV)Cl](PF6)3
A 100 mL round-bottom flask was charged with [ttpy-MV](PF6)2 (0.090 g, 0.11 mmol), Pt(DMSO)2Cl2 (0.040 g; 0.10 mmol) and 10 mL MeCN. The mixture was stirred at reflux for 24 h. To this was added 10 mL of a saturated aqueous solution of ammonium hexafluorophospate. The resulting orange precipitate was collected by filtration and washed with water and ether before drying in vacuo. Yield: 0.076 g (65%). 1H NMR (DMSO-d6): δ 9.64 (2H, d, J = 7 Hz), 9.33 (2H, d, J = 7 Hz), 9.10 (2H, s), 9.07 (2H, d, J = 6), 8.92 (2H, d, J = 8), 8.88 (2H, d, J = 7), 8.78 (2H, d, J = 7), 8.63 (2H, t, J = 8), 8.37 (2H, d, J = 8), 8.06 (2H, t, J = 7), 7.98 (2H, d, J = 8), 6.10 (2H, s), 4.48 (3H, s). MS (positive API-ES): m/z 434.5 [(M − 2PF6)/2]+, 241.4 [(M − 2PF6)/3]+.[Pt(ttpy-Nd)Cl](PF6)2
This complex was prepared as previously reported.53 Yield: 0.580 g (60%) 1H NMR (CD3CN): δ 8.98 (1H, s), 8.78 (2H, s), 8.64 (1H, d, J = 6 Hz), 8.58 (1H, d, J = 8 Hz), 8.24 (2H, s), 8.14 (4H, m), 7.92 (1H, t, J = 7 Hz), 7.83 (2H, d, J = 8 Hz), 7.58 (2H, m), 7.45 (2H, d, J = 8 Hz), 6.97 (1H, br s), 6.42 (1H, br s), 5.64 (2H, s). MS (positive API-ES): m/z 337.7 [(M − 2PF6)/2]+.[Cu(C≡CPh)]n
This complex was prepared in a way similar to that previously reported.54–56 A 100 mL round bottom flask was charged with cuprous iodide (1.00 g, 5.2 mmol), hydroxylamine hydrochloride (0.34 g, 4.9 mmol). To this was added 30 mL of N2 sparged water and 10 mL of N2 sparged acetonitrile. The resulting solution was stirred at 0 °C under N2. To this was added 3 mL of 30% aqueous ammonium hydroxide solution dropwise, stopping when a faint blue color was observed. To this was added a solution of phenyl acetylene (0.500 g, 5.0 mmol) in 10 mL MeCN. This elicited a yellow precipitate. The suspension was allowed to stir for 30 min, at which time the MeCN was removed in vacuo. The remaining suspension was filtered, then washed with 1% aqueous ammonium hydroxide, water, and ether. The bright yellow solid was dried in vacuo and stored at 0 °C. Yield: 0.661 g (81%). Characterization of this compound was not possible by 1H NMR or MS. Further explanation is provided in the Discussion section.Cu(C≡C–C6H4–NH–CO–C6H2(OMe)3) (3OMe-Cu)
Attempts to prepare this complex in aqueous media frequently resulted in decomposition during drying. To avoid this problem, pure acetonitrile was used as the solvent. A 25 mL round bottom flask was charged with cuprous iodide (0.120 g, 0.63 mmol), 3OMe-H (0.200 g, 0.64 mmol), and MeCN (12 mL) then sealed with a septum and placed in an ice bath under N2. NEt3 (0.24 mL, 1.80 mmol) was delivered via syringe. The resulting yellow suspension was stirred at 0 °C for 30 min. The suspension was treated with diethyl ether (6 mL) and transferred to centrifuge tubes. After centrifugation, the supernatant was removed by decantation and the yellow solid washed with additional ether. After washing, the solid was dried with a stream of N2 and in vacuo to give an ochre solid. Yield: 0.260 g (74%). 1H NMR (DMSO-d6): δ 10.25 (1H, s), 7.78 (2H, d, J = 9 Hz), 7.48 (2H, d, J = 9), 7.26 (2H, s), 3.87 (6H, s), 3.73 (3H, s). MS analysis of this compound was not possible. Further explanation is provided in the Discussion.[Pt(ttpy-MV)(C≡CPh)](PF6)3 (3)
A 10 mL round bottom flask was charged with [Pt(ttpy-MV)Cl](PF6)3 (0.015 g, 0.013 mmol), Cu(C≡CPh) (0.005 g, 0.030 mmol) and 3 mL of DMF. The suspension was agitated with sonication for 15 min, eliciting a color change from yellow–orange to red. This suspension was stirred overnight under N2. To the resulting red solution was added 10 mL of diethyl ether, to give a dark red precipitate which was collected by filtration and washed with ether. Yield: 0.011 g (68%) 1H NMR (DMSO-d6): δ 9.63 (2H, d, J = 7 Hz), 9.33 (2H, d, J = 6), 9.30 (2H, d, J = 6), 9.12 (2H, s), 8.91 (2H, d, J = 8), 8.87 (2H, d, J = 6), 8.78 (2H, d, J = 6), 8.64 (2H, t, J = 8), 8.35 (2H, d, J = 8), 8.04 (2H, t, J = 7), 7.98 (2H, d, J = 8), 7.57 (2H, d, J = 8), 7.41 (2H, t, J = 7), 7.32 (1H, t, J = 7), 6.10 (2H, s), 4.48 (3H, s). MS (positive API-ES): m/z 467.7 [(M − 2PF6)/2]+, 263.1 [(M − 3PF6)/3]+. Anal. calcd for PtC41H32N5P3F18: C, 40.20; H, 2.61; N, 5.72. Found: C, 38.29; H, 2.17; N, 4.94.[Pt(ttpy-MV)(C≡C–C6H4–NH–CO–C6H2(OMe)3)](PF6)3 (1)
A 10 mL round bottom flask was charged with [Pt(ttpy-MV)Cl](PF6)3 (0.050 g 0.043 mmol), 3OMe-Cu (0.030 g 0.080 mmol) and 8 mL of DMF. The suspension was agitated with sonication for 15 min, eliciting a color change from yellow–orange to red. This suspension was stirred overnight under N2. To the resulting red solution was added 10 mL of diethyl ether, to give a dark red precipitate which was collected by filtration and washed with ether. Yield: 0.033 g (53%). 1H NMR (DMSO-d6): δ 10.24 (1H, s), 9.63 (2H, d, J = 6 Hz), 9.33 (2H, d, J = 4), 9.32 (2H, d, J = 5), 9.12 (2H, s), 8.91 (2H, d, J = 8), 8.87 (2H, d, J = 7), 8.77 (2H, t, J = 7), 8.64 (2H, t, J = 8), 8.35 (2H, d, J = 8), 8.06 (2H, t, J = 7), 7.98 (2H, d, J = 8), 7.80 (2H, d, J = 8), 7.58 (2H, d, J = 8), 7.33 (2H, s), 6.10 (2H, s), 4.48 (3H, s), 3.92 (6H, s), 3.78 (3H, s). MS (positive API-ES): m/z 571.7 [(M − 2PF6)/2]+, 333.0 [(M − 3PF6)/3]+. Anal. calcd for PtC51H43N6O4P3F18: C, 42.71; H, 3.00; N, 5.86. Found: C, 37.91; H, 2.60; N, 5.15.[Pt(ttpy-Nd)(C≡CPh)](PF6)2 (4)
A vial was charged with [Pt(ttpy-Nd)Cl](PF6)2 (0.050 g, 0.052 mmol), Cu(C≡CPh) (0.009 g, 0.050 mmol), and a 2.5 mL of DMF. The mixture was agitated with a sonicator and warmed with the heat gun until a clear, dark red solution was produced. Diethyl ether was added and a black precipitate formed. Precipitate was collected by decanting and vacuum dried. Yield: 0.045 g (87%). 1H NMR (DMSO-d6): δ 9.67 (1H, s), 9.38 (1H, d, J = 7 Hz), 9.20 (2H, d, J = 5 Hz), 9.06 (2H, s), 9.01 (1H, d, J = 8 Hz), 8.84 (2H, d, J = 8 Hz), 8.61 (1H, s), 8.57 (2H, t, J = 8 Hz), 8.36 (1H, t, J = 8 Hz), 8.25 (2H, d, J = 8 Hz), 8.22 (1H, s), 7.97 (2H, m), 7.89 (2H, d, J = 8 Hz), 7.50 (2H, d, J = 7 Hz), 7.34 (2H, t, J = 8 Hz), 7.26 (1H, t, J = 7 Hz), 6.05 (2H, s). MS (positive API-ES): m/z 370.1 [(M − 2PF6)/2]+. HRMS calcd. for PtC36H26N5O 738.17015. Found 738.17675. Anal. Calcd. for PtC36H27N5O4P2F12: C, 41.94; H, 2.62; N, 6.80. Found: C, 38.36; H, 2.29; N, 5.81.[Pt(ttpy-Nd)(C≡C–C6H4–NH–CO–C6H2(OMe)3)](PF6)2 (2)
A vial was charged with [Pt(ttpy-Nd)Cl](PF6)2 (0.050 g, 0.052 mmol), 3OMe-Cu (0.040 g, 0.104 mmol), and 2.5 mL DMF. The mixture was sonicated for 15 min, with occasional warming from heat gun. A clear, dark red solution was formed. Diethyl ether was added and a black precipitate formed. Precipitate was collected by decanting and vacuum dried. Yield: 54%. 1H NMR (DMSO-d6): δ 10.18 (1H, s), 9.66 (1H, s), 9.36 (1H, d, J = 6 Hz), 9.21 (2H, m), 9.05 (2H, s), 8.99 (1H, d, J = 8 Hz), 8.83 (2H, d, J = 8Hz), 8.60 (1H, s), 8.56 (2H, t, J = 8 Hz), 8.34 (1H, t, J = 7 Hz), 8.25 (2H, d, J = 8 Hz), 8.21 (1H, s), 7.94 (2H, m), 7.88 (2H, d, J = 8 Hz), 7.74 (2H, d, J = 8 Hz), 7.51 (2H, d, J = 8 Hz), 7.28 (2H, s), 6.04 (2H, s), 3.92 (6H, s), 3.77 (3H, s). MS (positive API-ES): m/z 474.8 [(M − 2PF6)/2]+. HRMS calcd. for PtC46H37N6O5 947.23896. Found 947.2395. Anal. calcd. for PtC46H38N6O5P2F12: C, 44.55; H, 3.07; N, 6.78. Found: C, 38.30; H, 2.94; N, 5.42.Results and discussion
Syntheses and characterization
The syntheses of the acceptor-derivatized terpyridyl ligands and their coordination chemistry to generate the corresponding Pt(II) chloride complexes proceed smoothly in accordance with previously established methodology (eqn (1)).45,46,53,57–61 However, when attempting to coordinate the arylacetylide ligand to either Pt(ttpy-MV)Cl(PF6)3 or [Pt(ttpy-Nd)Cl](PF6)2 using a CuI catalyst, product formation did not occur, and decomposition of the starting material was seen.45,53,62–66 The mechanism of decomposition was not immediately apparent, and an alternative route for coordination of the acetylide ligand was devised.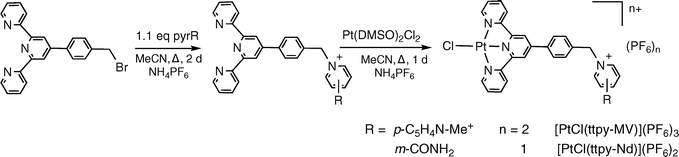 | (1) |
Copper acetylides have been prepared for a variety of synthetic applications and have also been shown to react with transition metal halides, including those of Pt(II).54–56,63,64,67 The conditions required for transmetalation reactions with metal halide complexes are generally very mild with short reaction times, and the reactions typically give the desired metal acetylide in reasonable to high yields. This method therefore was employed for the preparation of complexes 1–4 (eqn (2)) in place of the CuI-catalyzed procedure.
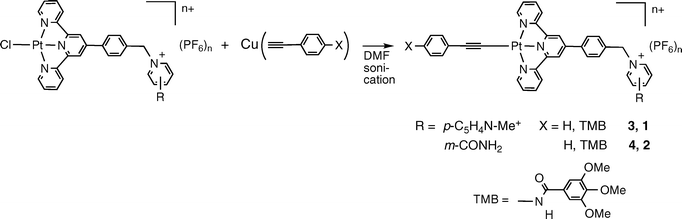 | (2) |
As pertains to characterization of the cuprous acetylides generated in this study, the solubility of Cu(C≡CPh) is too poor in conventional solvents to allow for either NMR or MS analysis, while Cu(C≡C–C6H4–NH–CO–C6H2(OMe)3) proved sufficiently soluble for characterization by 1H NMR spectroscopy. However, for the latter compound, MS analysis was unsuccessful in providing data attributable to the cuprous acetylide or fragmentation products thereof. Despite these difficulties in characterization, the cuprous acetylides were generally very active in the desired transmetallation reactions and their yellow color served as a reliable indicator of their formation and purity. Upon decomposition, these compounds are observed to become green, brown, and black, at which point they are no longer active.
After transmetallation, the 1H NMR spectra of the resulting Pt compounds may contain very broad resonances due to small amounts of Cu(II) in the sample. This impurity was successfully removed by dissolution of the crude material in acetonitrile followed by precipitation of the desired Pt complexes with a minimum amount of diethyl ether.
Sensitivity of the benzyl linkage to basic conditions
In an effort to remove the aforementioned Cu(II) species, several of the Pt complexes were washed with a dilute aqueous solution of ammonium hydroxide. After treating [Pt(ttpy-MV2+)Cl](PF6−)3 in such a fashion, a marked change was observed in the mass spectrum. Specifically, the mass values corresponding to [Pt(ttpy-MV2+)Cl](PF6−)3 were no longer observed, and a new peak was found at 842.0 amu with an isotope pattern typical of Pt containing compounds. The identity of the new complex was hypothesized as the alcohol derivative obtained by OH− substitution at the benzyl carbon atom for the viologen acceptor moiety. This is shown in eqn (3).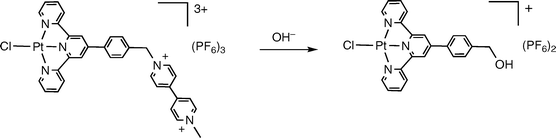 | (3) |
The substitutional reactivity suggested by eqn (3) was examined by NMR spectroscopy. Three samples were prepared each containing 1 mL of a 50 : 50 (v : v) mixture of CD3CN and D2O and 1 mg of [Pt(ttpy-MV)Cl](PF6)3. To one of the tubes 10 μL of triethylamine was added to give a modestly basic solution. To another was added 10 μL of trifluoroacetic acid to give a modestly acidic environment. No additional reagent was added to the remaining tube, which remained near neutral pH.
While the tubes containing neutral and acidic solutions showed no visible change in their proton resonances after one week of storage, the tube containing a basic solution had undergone a profound change immediately upon addition of the amine. While interpretation of the proton signals was complicated by the reduction in solubility upon reaction with amine, large changes in several resonances, particularly of the protons on the benzylic carbon atom to which the viologen moiety was bonded, were observed, indicative of reaction. The analogous Pt complexes bearing the nicotinamide and pyridinium acceptors were subsequently found to react with aqueous base in a similar fashion. Owing to the sensitivity of these compounds to hydrolysis, normal means of purification were not possible (i.e., chromatography), and attempts at recrystallization met with no success. The viologen species were found to be particularly unstable, and studies of their photophysical properties had to be performed on freshly prepared solutions.
A key conclusion from these observations is that while the benzylic attachment of the pyridinium type acceptors is synthetically easy and feasible for studies of photoinduced charge separation in non-aqueous media, this linkage is too unstable to be employed for the development of robust, multi-component systems for eventual H2 generation and artificial photosynthesis.
Absorption and emission spectra
Absorption spectra of all complexes were taken at 1.0 × 10−5 M concentration in degassed MeCN at room-temperature (Fig. 3 and 4). For analogous compounds, higher energy absorptions (λ < 350 nm) have previously been assigned as intraligand transitions originating within the acetylide and terpyridyl ligands, and the same assignments are applicable here.45,46,62,68–70 Regarding the central chromophore 5, the less intense absorption centered at λ ∼ 420 nm has been assigned previously as a 3MLCT transition corresponding to dπ(Pt) → π*(tpy).45,46,62,69–74 The absorptions observed for compounds 1–4 between 400–500 nm are likewise assigned as this 3MLCT transition typical of the chromophore. As was also observed for related multi-component compounds, the λmax of this transition is slightly red shifted for triads 1 and 2 relative to the dyads 3 and 4.45,46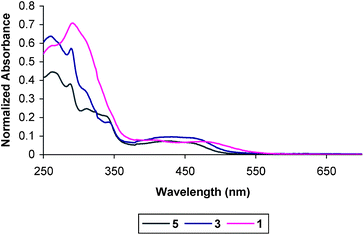 | ||
| Fig. 3 Room temperature absorption spectra of the chromophore (5), C–A dyad (3) and D–C–A triad (1) in MeCN. | ||
 | ||
| Fig. 4 Room temperature absorption spectra of the chromophore (5), C–A dyad (4) and D–C–A triad (2) in MeCN. | ||
Emission spectra were taken at 1.0 × 10−5 M in degassed MeCN at room-temperature. Previous reports have shown that 5 is strongly emissive, and that the donor–chromophore (D–C) dyad, 7, with the trimethoxybenzamide donor is nonemissive.45 This suggests that the trimethoxy donor efficiently quenches the excited state of the chromophore by electron transfer. The nonemissive behavior of triads 1 and 2 reaffirms this observation. From Fig. 5, it can be seen that the emission intensity from a solution of the C–A dyad 3 is also nearly completely quenched at the same concentration as 5. The quantum yield for 3 was determined to be ∼0.002 (using Ru(bpy)32+ as a standard with ϕrelem = 0.062). Solutions of 3 become more strongly emissive upon standing, which is consistent with the observed tendency to cleave the ttpy-acceptor connection by substitution at the benzylic C atom (eqn 3). The spectrum shown is of a freshly prepared solution taken immediately after degassing with an N2 sparge.
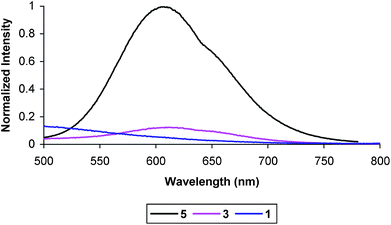 | ||
| Fig. 5 Room temperature emission spectrum of 5, 3 (2×), and 1 (2×) at 1 × 10−5 M in degassed MeCN. Excitation wavelength 420 nm. | ||
A solution of the nicotinamide C–A dyad, 4, exhibited luminescence with intensity lower than that observed for 5 and comparable to that for the previously reported C–A dyad based on the pyridinium acceptor.45 The quantum yield of 4 was found to be 0.008, which is similar to that previously reported for the closely related dyad 8 at 0.011.45
Low temperature emission studies were conducted in butyronitrile solution in NMR tubes, using liquid nitrogen in a circular quartz-tipped dewar. Triads 1 and 2 were found to be brightly emissive at 77 K using this method (Fig. 6). 1 also clearly exhibited a vibronic progression, which has been observed in related platinum terpyridyl arylacetylide complexes.45,46,62,68 That the triads were emissive in a frozen glass but were non-emissive in a room temperature solution is consistent with a charge transfer quenching process having a significant solvent reorganizational barrier.
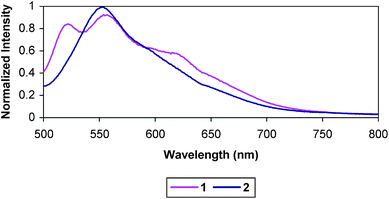 | ||
| Fig. 6 Emission of 1 and 2 at 77 K in butryonitrile. | ||
Electrochemistry
Cyclic voltammetry studies were performed on all of the complexes in either MeCN or DMF with 0.1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte. All of the redox potentials are reported relative to NHE based on the Fc/Fc+ couple used as an internal redox standard and using values of 0.4 V vs. SCE taken for the Fc/Fc+ couple in MeCN and 0.45 V vs. SCE in DMF . The electrochemistry of the parent chromophore 5 has been reported previously.46 The dyads and triads in the present study show the expected number of oxidation and reduction waves, and the results are listed in Table 1. Chromophore complex 5 exhibits two reduction potentials at −0.56 and −1.06 V. Both dyad 3 and triad 1 undergo an additional irreversible reduction at ca. −0.1 V (Table 1), while dyad 4 and triad 2 exhibit broad, overlapping reductions with an onset of −0.46 V. All of the complexes show an irreversible oxidation at 1.5–1.6 V while the complexes having the trimethoxybenzene donor (1, 2) exhibit another, less anodic oxidation at 1.05–1.2 V.| Compound | Oxidation E1/2/V | Reduction E1/2/V |
|---|---|---|
| a Irreversible under experimental conditions. b Broad, overlapping couples (potential of reduction onset). Potential in V vs. NHE (see the Experimental section). | ||
| 5 | 1.49a | −0.56, −1.06a |
| 3 | 1.62a | −0.12, −0.42b, −1.12a |
| 1 | 1.62a, 1.20a | −0.07, −0.46b, −1.11a |
| 4 | 1.56a | −0.46b, −1.16a |
| 2 | 1.56a, 1.05a | −0.46b, −1.11a |
| ttpy-MV | −0.11, −0.76, −1.12a | |
| ttpy-Nd | −0.81b |
From the electrochemical behavior of free ligand ttpy-MV and complexes 1 and 3, the reduction at −0.07 to −0.12 V is assigned as the first viologen reduction. For ligand ttpy-MV, the clearly defined reduction at −0.76 V is assigned as the second viologen reduction. In the case of complexes 1 and 3, this reduction overlaps with the first terpyridine reduction to give a broad signal with an onset of ca. −0.46 V. For dyad 4 and triad 2, there is also a very broad reduction with an onset of −0.46 V, but it is not clear if this broadness is caused by overlap of the terpyridine reduction with that of nicotinamide, since the reduction of nicotinamide seems to occur nearer to −0.8 V. This reduction is not clearly defined in the free ligand ttpy-Nd, but is observed in the model compound, 1-benzyl nicotinamide, and is consistent with literature values for analogous species.75,76
For the parent chromophore and the dyad and triad systems, the irreversible oxidations at 1.5–1.6 V correspond to the Pt(II) → Pt(III) oxidation, whereas the irreversible oxidations for triads 1 and 2 at 1.20 and 1.05 V, respectively, are likely the trimethoxybenzamide oxidation. Similar observations have been reported previously for analogous platinum terpyridine systems.45,46,62,77–80
Transient absorption studies
To probe the nature of charge separation in the complexes, nanosecond transient absorption studies were performed on 1–4 at 298 K in MeCN. The spectra were assembled from decays measured at every 10 nm. The results are shown in Fig. 7–11. The spectra were collected immediately after excitation at 470 nm (pulse < 5 ns); the large negative absorptions between 450–490 nm are due to scattered light from the laser.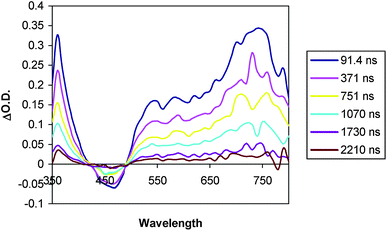 | ||
| Fig. 7 Transient absorption spectrum of C–A dyad 4 (0.08 mM in MeCN) at room temperature in MeCN (λexc = 440 nm). | ||
 | ||
| Fig. 8 Transient absorption spectrum of D–C–A triad 2 (0.08 mM in MeCN) at room temperature in MeCN (λexc = 460 nm). | ||
 | ||
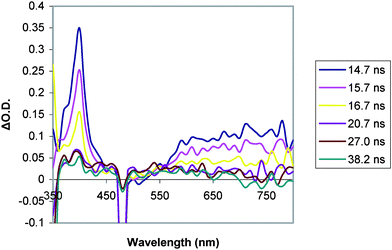
| Fig. 9 Transient absorption spectrum of C–A dyad 3 (0.08 mM in MeCN) at room temperature in MeCN (λexc = 440 nm). | ||
 | ||
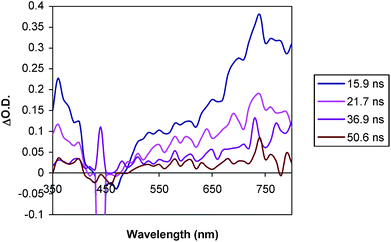
| Fig. 10 Transient absorption spectrum of D–C–A triad 1 (0.08 mM in MeCN) at room temperature in MeCN (λexc = 460 nm). | ||
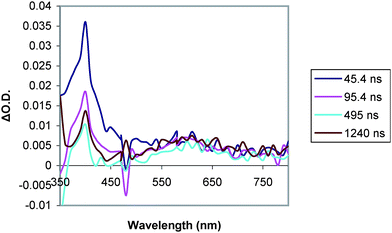 | ||
| Fig. 11 Transient absorption spectrum of D–C–A triad 1 (0.08 mM in MeCN) at room temperature in MeCN (λexc = 460 nm) taken on a longer timescale. | ||
The transient absorption spectrum of the C–A dyad 4 (Fig. 7) is indistinguishable from previous data reported for the parent chromophore 5 collected under the same conditions.45,46 This indicates that the TA spectrum of 4 is actually arising from the chromophore and that charge transfer to the nicotinamide acceptor is not occuring in this system. Both 4 and 5 give a positive absorption at 360 nm and a broad absorption between 520–800 nm with a maximum at 740 nm and a bleach at 420–460 nm which is still observed when the excitation wavelength is varied. All the features decay with a rate constant of 1.2 × 106 s−1 in the case of 4, which is similar to that previously reported for related C–A dyad 8 (1.6 × 106 s−1) as well as parent chromophore 5 (1.4 × 106 s−1).45,46
The partial quenching of emission observed in the C–A dyad 4 (Fig. 12) suggests that there are non-radiative pathways available which are not present in the parent chromophore. As there is no evidence that electron transfer quenching is occurring, the reduction in luminescence intensity may be caused by some other non-radiative mechanism as yet undetermined. This is consistent with previous observations in studies of the pyridinium acceptor dyad, 8.45 The luminescence lifetime of 4 was determined to be 250 ns in MeCN, as compared with 620 ns for the parent chromophore 5 collected under the same conditions. This is shorter than the lifetime observed in the transient absorption experiments (830 ns), which is also the case for 5.
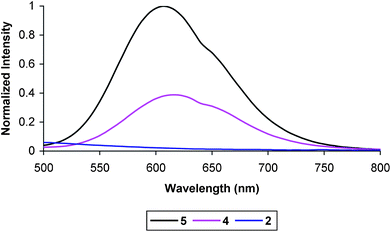 | ||
| Fig. 12 Room temperature emission spectrum of 5, 4 (2×), and 2 (2×) at 1 × 10−5 M in degassed MeCN. Excitation wavelength 420 nm. | ||
The transient absorption features of D–C–A triad 2 (Fig. 8) are also experimentally identical to those previously reported for the related D–C dyad 7.45 These both exhibit an absorption at 400 nm and a broad absorption pattern between 550–800 nm which decays in less than 20 ns. These spectral features are consistent with trimethoxybenzene+• which has a strong absorption at 400 nm and provide no clear evidence for the generation of any other species. The electronic spectrum of the nicotinamide radical has not been previously reported, but is known to react rapidly with protons or to dimerize to give species with absorptions centered near 340 nm in both cases.76,81,82
In contrast, the viologen-bearing C–A dyad 3 gives transient absorption signals (Fig. 9) which are similar to that of parent chromophore 5, but these features decay at a much higher rate (k ∼ 108 s−1). In addition, there is a sharp feature at 400 nm which is not seen in the transient absorption spectra of 5, the spectrum of which is nearly identical to that of 4 shown in Fig. 7. This absorption clearly decays with a different rate than other features, although quantitative analysis of the kinetics of this decay was not possible due to overlap with other features.
It may be that the transient absorption at 400 nm results from an impurity resulting from the decomposition of the complex via the hydrolysis process described above. However, given that the solutions were freshly prepared and immediately characterized, we expect the degree of decomposition to be minimal. Also, the product of this decomposition is a singly alkylated bipyridine, which is not expected to function as an electron acceptor. Therefore, it is more probable that the absorption is that of a reduced viologen species resulting from intramolecular charge transfer.
None of the transient data for dyad 3 could be fit with a single exponential, indicating that two or more decay pathways are operative. Additionally, in determining the luminescence lifetime, two components were observed, one with a lifetime of ∼10 ns, and another, less intense component with a lifetime of 480 ns, which is likely the result of a small amount of the chromophore product from hydrolysis of the dyad. This chromophore would be expected to have a lifetime similar to that of the parent chromophore, 5.
Absorptions from a reduced viologen+• would be expected with λmax near to 397 and 610 nm, with the former being fairly sharp and the latter broad.15,19 These absorptions would overlap strongly with the spectral features of the excited state of the chromophore, except for the sharp peak near 400 nm, which is clearly observed in the transient spectrum (Fig. 9). This may serve to explain the complicated kinetics observed in the decays of the transient features. Thus, it seems that the attached viologen species is acting as an oxidative quencher of the excited state of the chromophore. This analysis is also consistent with the nearly complete quenching of luminescence intensity relative to 5 in solution (Fig. 5).
The broad envelope observed between 550–800 nm in the transient absorption spectra of dyads 3 and 4 and triads 1 and 2 has been previously assigned as absorption of the ttpy−• radical anion generated in the 3MLCT excited state of the chromophore.19,25,45,46 However, a recent report by Castellano et al. suggests that this assignment is incomplete.83 In this work, transient absorption studies of the chromophore in the presence of a reductive quencher demonstrate that the intensity of the spectral features of the excited state attributed to the terpyridyl radical anion diminish upon reduction of the complex.
The D–C–A triad 1 gives transient signals (Fig. 10) that are different than triad 2 and D–C dyad, 7. The profile of the absorption between 550 and 800 nm is different, and the transient absorption does not return to the baseline on the timescale of the measurement as was the case for triad 2 and dyad 3. Spectra obtained on a longer timescale reveal the presence of an extremely weak (ΔO.D. < 0.02), but long-lived absorption at 400 nm and a similarly weak broad absorption between 520 and 700 nm (Fig. 11). No such signals were detected in measurements of triad 2 under the same conditions.
As with C–A dyad 3, the decay of the transient absorption features of triad 1 could not be fit to a single exponential, consistent with a sequence involving initial excited state electron transfer followed by recombination. At 400 nm there appears to be a fast decay component (k > 108 s−1) as well as a slower one. Since the absorption of trimethoxybenzene+• overlaps with that of a reduced viologen radical, particularly at 400 nm, the analysis is complicated further. The fast component observed at 400 nm is most likely due to back electron transfer from the reduced chromophore to the oxidized trimethoxybenzene, similar to that observed in dyad 7. The weak, long-lived absorptions observed in 1 are not consistent with the excited state of the chromophore since the strong absorption features between 700 and 800 nm are missing. The longer lived component of the spectrum in Fig. 9 strongly resembles that of the reduced viologen and could be interpreted as the combined contributions of a trimethoxybenzene+• and viologen+• at low concentration. If this is the case, it suggests that the fully charge separated state, D+–C–A− is capable of forming, but with low efficiency. However, once formed, it appears to have a lifetime in excess of a microsecond. The possible pathways which could lead to this state are illustrated in Fig. 13. Based on the transient absorption data of the related dyads 3 and 7 and triad 1, it can be inferred that k1 ≥ k3, k6 (D–C+–A− to D–C–A) > k5, and that k2 > k4.
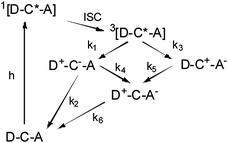 | ||
| Fig. 13 A schematic representation of the charge transfer events following excitation of D–C–A triad 1. | ||
Estimates of the free energies for the electron transfer processes can be obtained by application of the Rehm Weller equation (eqn (4)). For calculations using eqn (4), the oxidation of the chromophore, ED°, is estimated as 1.6 V vs. NHE from electrochemical data. The excited state energy, ΔE* is taken as the λmax of the frozen emission of the complex, or 550 nm (2.2 eV), and the work term is estimated as 0 for an intramolecular charge transfer.
| ΔG = |ED° − EA°| − ΔE* + w | (4) |
Based on the electrochemical behavior of triad 2 and dyad 4, the ligand ttpy-Nd, and previous studies for related compounds, the benzyl nicotinamide reduction potential can be estimated as −0.8 V vs. NHE.76 Using this value, oxidative quenching by the nicotinamide acceptor is predicted to be unfavorable at 0.2 eV, consistent with the lack of evidence for electron transfer quenching for C–A dyad 4. In contrast, oxidative quenching by the viologen acceptor would be predicted to be exergonic. With the first viologen reduction occurring at ca. −0.1 V vs. NHE, the driving force for electron transfer is calculated to be favorable by more than 0.5 eV. Independently, it has also been established that methyl viologen is a very efficient oxidative quencher of 5 with rate constants approaching the diffusion limit.84 The observation of what appears to be rapid excited state decay for C–A dyad 3 (Fig. 9) but no long-lived electron transfer products, is consistent with back electron transfer of C+–A− being fast.
The possibility that the benzylic –CH2– prevents efficient charge transfer in dyad 3 was proposed previously for the case of the pyridinium acceptor, but this hypothesis could not be fully supported,44 and several other systems have been reported with charge transfer across benzylic bridges at high rates.15,19,30,43,45,46,53 The current observations are consistent with electron transfer across the benzylic linkage occurring at a moderate rate (k < 108 s−1). The design of future triads and dyads will employ synthetic strategies which connect the chromophore and acceptor via different linkages to avoid the instability found in the present study with the benzylic connection when nucleophiles or base are present. If in future studies the same acceptor species are used, they will provide a way to probe the electronic effect of the bridging group directly.
Conclusions
The D–C–A triads [Pt(ttp-MV2+)(p-C≡C–C6H4–NH–CO–C6H2(OMe)3)](PF6)3 (1) and [Pt(ttpy-Nd+)(p-C≡C–C6H4–NH–CO–C6H2(OMe)3](PF6)2 (2) have been prepared along with the corresponding C–A dyads 3 and 4. All of these complexes exhibit characterization data consistent with their assigned structures and similar to previously reported analogous compounds. The syntheses of these complexes employ cuprous acetylides, as opposed to the standard copper iodide-catalyzed method, in which excess base is required to deprotonate the acetylene ligand. This change was made to accommodate the observed sensitivity of the benzylic linkage to basic conditions. This observation is of particular interest, as pyridinium based acceptors have been fairly ubiquitous in the design of molecular dyads and triads. As anticipated, the D–C–A triads 1 and 2 are nonemissive, whereas the parent chromophore 5 is brightly emissive in solution. The viologen-bearing C–A dyad 3 is also very weakly emissive, suggesting that a charge transfer quenching process is operative. In contrast, the nicotinamide-bearing C–A dyad 4 is only slightly less emissive than the parent chromophore and virtually identical to a previously reported C–A dyad 8 with a pyridinium acceptor. Transient absorption shows no evidence for charge separation into the nicotinamide acceptor, which parallels previous results with dyad 8 and is consistent with a thermodynamic analysis of the charge transfer process. In contrast, the transient absorption features of the viologen-bearing complexes shows evidence that the viologen acts as an oxidative quencher, but with a modest rate constant (k ∼ 108 s−1). These observations lend support to the previously proposed hypothesis that the benzylic linkage slows charge transfer in these systems.45 This may also explain why triad 1 does not form the fully charge-separated state with high efficiency. Nevertheless, when this state is formed, it appears to have a long lifetime, in excess of a microsecond.Acknowledgements
We wish to thank the Division of Basic Sciences, U.S. Department of Energy for financial support of this research (Grant DE-FG02-90ER14125). We also want to thank Dr Paul Merkle for his help with the lifetime measurements for complexes 3, 4, and 5.References
- D. Gust, T. A. Moore, L. R. Makings, P. A. Liddell, G. A. Nemeth and A. L. Moore, J. Am. Chem. Soc., 1986, 108, 8028–8031 CrossRef CAS.
- P. A. Liddell, D. Barrett, L. R. Makings, P. J. Pessiki, D. Gust and T. A. Moore, J. Am. Chem. Soc., 1986, 108, 5350–5352 CrossRef CAS.
- J. S. Krueger, J. E. Mayer and T. E. Mallouk, J. Am. Chem. Soc., 1988, 110, 8232–8234 CrossRef CAS.
- P. Seta, E. Bievenue, A. L. Moore, P. Mathis, R. V. Bensasson, P. Liddell, P. J. Pessiki, A. Joy, T. A. Moore and D. Gust, Nature, 1985, 316, 653 CrossRef CAS.
- L. Flamigni, N. Armaroli, F. Barigelletti, V. Balzani, J. P. Collin, J. O. Dalbavie, V. Heitz and J. P. Sauvage, J. Phys. Chem., 1997, 101, 5936–5943 Search PubMed.
- M. D. Hossain, M. Haga, H. Monjushiro, B. Gholamkhass, K. Nozaki and T. Ohno, Chem. Lett., 1997, 573 CrossRef CAS.
- B. J. Coe, D. A. Friesen, D. W. Thompson and T. J. Meyer, Inorg. Chem., 1996, 35, 4575–4584 CrossRef CAS.
- S. C. Hung, A. N. Macpherson, S. Lin, P. A. Liddell, G. R. Seely, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1995, 117, 1657–1658 CrossRef CAS.
- D. Kuciauskas, P. A. Liddell, S. C. Hung, S. Lin, S. Stone, G. R. Seely, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. B, 1997, 101, 429–440 CrossRef CAS.
- A. N. Macpherson, P. A. Liddell, S. Lin, L. Noss, G. R. Seely, J. M. Degraziano, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1995, 117, 7202–7212 CrossRef CAS.
- D. G. McCafferty, D. A. Friesen, E. Danielson, C. G. Wall, M. J. Saderholm, B. W. Erickson and T. J. Meyer, Proc. Natl. Acad. Sci. USA, 1996, 93, 8200–8204 CrossRef CAS.
- A. L. Moore, T. A. Moore, D. Gust, J. J. Silber, L. Sereno, F. Fungo, L. Otero, G. SteinbergYfrach, P. A. Liddell, S. C. Hung, H. Imahori, S. Cardoso, D. Tatman and A. N. Macpherson, Pure Appl. Chem., 1997, 69, 2111–2116 CAS.
- G. Steinberg-Yfrach, P. A. Liddell, S. C. Hung, A. L. Moore, D. Gust and T. A. Moore, Nature, 1997, 385, 239–241 CrossRef CAS.
- J. P. Sumida, P. A. Liddell, S. Lin, A. N. Macpherson, G. R. Seely, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. A, 1998, 102, 5512–5519 CrossRef CAS.
- J. A. Treadway, P. Y. Chen, T. J. Rutherford, F. R. Keene and T. J. Meyer, J. Phys. Chem. A, 1997, 101, 6824–6826 CrossRef CAS.
- L. Flamigni, I. M. Dixon, J. P. Collin and J. P. Sauvage, Chem. Commun., 2000, 2479–2780 RSC.
- I. M. Dixon, J. P. Collin, J. P. Sauvage, F. Barigelletti and L. Flamigni, Angew. Chem., Int. Ed., 2000, 39, 1292 CrossRef CAS.
- H. Imahori, K. Tamaki, D. M. Guldi, C. P. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS.
- J.-P. Collin, S. Guillerez, J.-P. Sauvage, F. Barigelletti, L. De Cola, L. Flamigni and V. Balzani, Inorg. Chem., 1991, 30, 4230–4238 CrossRef CAS.
- M. Hissler, A. Harriman, A. Khatyr and R. Ziessel, Chem.–Eur. J., 1999, 5, 3366–3381 CrossRef CAS.
- A. Harriman, M. Hissler and R. Ziessel, Phys. Chem. Chem. Phys., 1999, 1, 4203–4211 RSC.
- A. Harriman, M. Hissler, A. Khatyr and R. Ziessel, Chem. Commun., 1999, 735–736 RSC.
- J. M. DeGraziano, A. N. Macpherson, P. A. Liddell, L. Noss, J. P. Sumida, G. R. Seely, J. E. Lewis, A. L. Moore, T. A. Moore and D. Gust, New J. Chem., 1996, 20, 839–851 Search PubMed.
- D. Gust, A. L. Moore and T. A. Moore, Acc. Chem. Res., 1993, 26, 198–205 CrossRef CAS.
- P. Laine, F. Bedioui, E. Amouyal, V. Albin and F. Berruyer-Penaud, Chem.–Eur. J., 2002, 8, 3162–3176 CrossRef CAS.
- A. Harriman, F. Odobel and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 5481–2 CrossRef CAS.
- R. Ballardini, V. Balzani, M. Clemente-Leon, A. Credi, M. T. Gandolfi, E. Ishow, J. Perkins, J. F. Stoddart, H.-R. Tseng and S. Wenger, J. Am. Chem. Soc., 2002, 124, 12786–12795 CrossRef.
- L. A. Lucia, D. G. Whitten and K. S. Schanze, J. Am. Chem. Soc., 1996, 118, 3057–8 CrossRef CAS.
- R. D. Fossum, M. A. Fox, A. M. Gelormini and A. J. Pearson, J. Phys. Chem. B, 1997, 101, 2526–2532 CrossRef CAS.
- E. Danielson, C. M. Elliott, J. W. Merkert and T. J. Meyer, J. Am. Chem. Soc., 1987, 109, 2519–20 CrossRef CAS.
- K. A. Maxwell, M. Sykora, J. M. DeSimone and T. J. Meyer, Inorg. Chem., 2000, 39, 71–75 CrossRef CAS.
- D. M. Kaschak and T. E. Mallouk, Abstr. Pap. Am. Chem. Soc., 1997, 214, 358.
- D. M. Kaschak, S. A. Johnson, C. C. Waraksa, J. Pogue and T. E. Mallouk, Coord. Chem. Rev., 1999, 185–186, 403–416 CrossRef CAS.
- D. M. Kaschak, J. T. Leau, C. C. Waraksa, G. B. Saupe, H. Usami and T. E. Mallouk, J. Am. Chem. Soc., 1999, 121, 3435–3445 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS.
- D. Gust, Nature, 1997, 386, 21–22 CrossRef CAS.
- D. Gust and T. A. Moore, J. Photochem., 1985, 29, 173–184 CrossRef CAS.
- D. Gust, T. A. Moore, A. L. Moore, S.-J. Lee, E. Bittersmann, D. K. Luttrull, A. A. Rehms, J. M. DeGraziano, X. C. Ma, F. Gao, R. E. Belford and T. T. Trier, Science, 1990, 248, 199–201 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- S. R. Greenfield, W. A. Svec, D. Gosztola and M. R. Wasielewski, J. Am. Chem. Soc., 1996, 118, 6767–6777 CrossRef CAS.
- S. L. larson, C. M. Elliot and D. F. Kelley, J. Phys. Chem., 1995, 99, 6530–6539 CrossRef CAS.
- J. E. McGarrah and R. Eisenberg, Inorg. Chem., 2003, 42, 4355–4365 CrossRef CAS.
- J. E. McGarrah, Y.-J. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510–4511 CrossRef CAS.
- T. Wadas, S. Chakraborty, Q. M. Wang, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2005, 44, 2628–2638 CrossRef CAS.
- S. Chakraborty, T. J. Wadas, H. Hester, C. Flaschenreim, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6284–6293 CrossRef CAS.
- S. Chakraborty, T. J. Wadas, H. Hester, R. Schmehl and R. Eisenberg, Inorg. Chem., 2005, 44, 6865–6878 CrossRef CAS.
- C. A. Bignozzi, J. R. Schoonover and F. Scandola, Prog. Inorg. Chem., 1997, 44, 1–95 CAS.
- F. D. Lewis, R. L. Letsinger and M. R. Wasielewski, in Proceedings of the Twenty-Second DOE Solar Photochemistry Research Conference, U.S. Department of Energy, Westfields International Conference Center, Chantilly, Virginia, 1998, pp. 10–11 Search PubMed.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- E. H. Yonemoto, R. L. Riley, Y. I. Kim, S. J. Atherton, R. H. Schmehl and T. E. Mallouk, J. Am. Chem. Soc., 1992, 114, 8081–8087 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- T. J. Wadas, Q.-M. Wang, Y.-j. Kim, C. Flaschenreim, N., B.T. and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef CAS.
- C. E. G. Castro, E.J. and D. C. Owsley, J. Org. Chem., 1966, 31, 4071–4078 CrossRef CAS.
- R. D. C. Stephens and C.E., J. Org. Chem., 1963, 28, 3313–3315 CrossRef CAS.
- C. E. H. Castro, R., V. K. Honwad, A. M. Malter and S. W. Moje, J. Am. Chem. Soc., 1969, 91, 6464–6470 CrossRef CAS.
- C.-W. Chan, C.-M. Che, M.-C. Cheng and Y. Wang, Inorg. Chem., 1992, 31, 4874–4878 CrossRef CAS.
- C.-W. Chan, L.-K. Cheng and C.-M. Che, Coord. Chem. Rev., 1994, 132, 87–97 CrossRef CAS.
- M. Howe-Grant and S. J. Lippard, in Inorganic Syntheses, ed. D. H. Busch, John Wiley and Sons, New York, 1980, vol. XX Search PubMed.
- R. Romeo and L. M. Scolaro, Inorg. Synth., 1998, 32, 153–158 CAS.
- G. Arena, G. Calogero, S. Campagna, L. M. Scolaro, V. Ricevuto and R. Romeo, Inorg. Chem., 1998, 37, 2763–2769 CrossRef CAS.
- Q.-Z. Yang, L.-Z. Wu, Z.-X. Wu, L.-P. Zhang and C.-H. Tung, Inorg. Chem., 2002, 41, 5653–5655 CrossRef CAS.
- K. Sonogashira, Y. Fujikura, T. Yatake, N. Toyoshima, S. Takahashi and N. Hagihara, J. Organomet. Chem., 1978, 145, 101–108 CrossRef CAS.
- K. Sonogashira, T. Yatake, Y. Tohda, S. Takahashi and N. Hagihara, J. Chem. Soc., Chem. Commun., 1977, 291 RSC.
- K. Sonogashira, S. Takahashi and N. Hagihara, Macromol. Chem., 1997, 10, 879.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627–630 CrossRef CAS.
- I. B. Ara, J.R., E. Eguizábal, J. Forniés, J. Gómez and E. Lalinde, Organomet. Chem., 2003, 670, 221–234 CrossRef CAS.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476–4482 CrossRef CAS.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong, C.-C. Ko and K.-K. Cheung, Inorg. Chem., 2001, 40, 571–574 CrossRef CAS.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506–6507 CrossRef CAS.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong, X.-X. Lu, K.-K. Cheung and N. Zhu, Chem.–Eur. J., 2002, 8, 4066–4076 CrossRef CAS.
- V. W.-W. Yam and V. W.-M. Lee, J. Chem. Soc., Dalton Trans., 1997, 3005–3010 RSC.
- V. W.-W. Yam, K. M.-C. Wong and N. Zhu, Angew. Chem., Int. Ed., 2003, 42, 1400–1403 CrossRef.
- C.-K. Hui, B. W.-K. Chu, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2002, 41, 6178–6180 CrossRef CAS.
- A. J. Bard, Encyclopedia of Electrochemistry of the Elements. Organic Section, Marcel Dekker, New York, 1986, vol. XV Search PubMed.
- K. S. V. E. Santhanam and P.J., J. Am. Chem. Soc., 1973, 95, 5482–5490 CrossRef CAS.
- V. W. W. Yam, R. P. L. Tang, K. M. C. Wong and K. K. Cheung, Organometallics, 2001, 20, 4476–4482 CrossRef CAS.
- K. M.-C. Wong, W.-S. Tang, B. W.-K. Chu, N. Zhu and V. W. W. Yam, Organometallics, 2004, 23, 3459–3465 CrossRef CAS.
- A. E. Brodsky, L. L. Gordienko and Degtiarev, Electrochim. Acta, 1968, 13, 1095–1100 CrossRef.
- C. R. Bock, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1975, 97, 2902–2911 CrossRef.
- P. O'Neill, S. Steenken and D. Schulte-Frohlinde, J. Phys. Chem., 1975, 79, 2773–2779 CrossRef CAS.
- N. Ichinose, T. Tanaka, S. Kawanishi, T. Suzuki and K. Endo, J. Phys. Chem. A, 1999, 103, 7923–7926 CrossRef CAS.
- E. Shikhova, E. O. Danilov, S. Kinayyigit, I. E. Pomestchenko, A. D. Tregubov, F. Camerel, P. Retailleau, R. Ziessel and F. N. Castellano, Inorg. Chem., 2007, 46, 3038–3048 CrossRef CAS.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |