Engineering materials and biology to boost performance of microbial fuel cells: a critical review
Antonio
Rinaldi†
a,
Barbara
Mecheri†
a,
Virgilio
Garavaglia
b,
Silvia
Licoccia
a,
Paolo
Di Nardo
c and
Enrico
Traversa
*a
aNAST Center & Department of Chemical Science and Technology, University of Rome “Tor Vergata”, Via della Ricerca Scientifica, 00133, Rome, Italy. E-mail: traversa@uniroma2.it; Fax: +39-06-72594328; Tel: +39-06-72594492
bAlintec Scarl, Via Garofalo 39, 20133, Milan, Italy
cNAST Center & Department of Internal Medicine, University of Rome “Tor Vergata”, Via della Ricerca Scientifica, 00133, Rome, Italy
First published on 4th August 2008
Abstract
In less than a decade the levels of performance of microbial fuel cells (MFCs) in terms of current output, voltage, and power density have grown tremendously according to steady exponential trends. Achievements occurred over the past 2–3 years have been particularly impressive. This is due partly to a better understanding of the biological aspects of this multidisciplinary technology, but also to systematic work undertaken by several research groups worldwide aimed at improving and optimizing aspects related to materials and system configuration. Aim of this review is to outline the current perspective about MFCs by focusing on the recent major advances in the areas of materials and engineering. MFCs are promising devices to address sustainability concerns both in terrestrial and space applications.

| From top left clockwise. Enrico Traversa is a Full Professor of Materials Science and Technology, Silvia Licoccia is a Full Professor of Chemistry, Antonio Rinaldi and Barbara Mecheri are Postdoctoral Researchers at the Department of Chemical Science and Technology of the University of Rome “Tor Vergata”. Their main research activities are in the development of nanostructured materials for energy, environmental and biomedical applications, focusing on polymeric and solid oxide fuel cells and gas sensors for environmental monitoring. Paolo Di Nardo is a Researcher at the Department of Internal Medicine of the University of Rome “Tor Vergata”, studying cardiovascular physiopathology, cardiac tissue engineering, and space biomedicine. Virgilio Garavaglia (absent in the photo) is a microbiologist with a joint appointment at Alintec Scarl, LTD and the University of Milan at the Department of Plant Science. |
1. Opening remarks: historical highlights, current trends, and sustainability
Luigi Galvani observed the bioelectric phenomenon first in 1790,1 but it was not until the beginning of the 20th century that microbial fuel cells (MFCs) were discovered. Generally speaking, a MFC is a bioreactor that converts chemical energy stored in the chemical bonds of organic compounds to electrical energy through catalytic reactions of microorganisms. The earliest work on MFC dates back to 1912 and is due to Potter. He described the production of electrical energy from living cultures of Escherichia coli and Saccharomyces.2 His work was not to receive any considerable attention until 1931, when Cohen was able to produce a voltage larger than 35 V from MFCs connected in a series.3 MFCs became popular in the 1960s, when the National Aeronautics and Space Administration (NASA) in USA carried out further research to assess their application in space missions.4 However, relatively little was understood about how these MFCs functioned and about fuel oxidation. New insight came from the studies by Allen and Bennetto in the 1980s,5,6 who discovered that current density and power output could be greatly enhanced by using electron mediators to accelerate the electron transfer rate from microbes to the anode substrate. Unfortunately, toxicity and instability of synthetic mediators are major impediments to their use for practical MFC applications. More recently, though, scientists found out that some microbes can use “safe” natural compounds as mediators, such as their own microbial metabolites (endogenous mediators). The next significant advance occurred when some microbes were found to transfer electrons directly to the anode,7 rendering MFCs a viable technology to generate electric power from biomass. The first laboratory-scale MFC prototype was developed by Bruce Logan,8,9 who demonstrated that electricity is generated when microbial foodstuffs, such as glucose and acetate, or even organic compounds in wastewater are fed to the bacteria.All such time serried contributions awakened the general interest in MFCs and triggered a spiral of research achievements that have steadily raised the performance levels by several orders of magnitude over a decade. For example, Fig. 1 displays some published data10–18 representative of maximum power densities (PD) achieved in the whole time span for two types of MFCs, i.e. the ones with aqueous and membraneless air–cathodes, marked with “*”, and those using pure fuels (e.g.glucose, acetate), marked with “o”. The data indicate a 5 orders of magnitude increase in PD in merely 10 years. The increasing trend of PD in time (t) may be roughly approximated for each category by an exponential model PD ∝ exp(k⋅t) corresponding to the regression lines in the log-linear plot, with k = 1.4 (R2 = 87%) for “*” and k = 0.7 (R2 = 50%) for “o”. The two MFC types displayed are not only well suited to highlight clear trends, but represent MFC types that once were far apart in performance. The initial gap has progressively thinned with time and nowadays the PD levels are of the same order, as hinted by the crossover between the trending lines. However, such a convergence is to a large extent a consequence of the greater effort poured into the development of MFCs running on low cost fuel (e.g. wastewaters) in the wake of sustainability concerns, which determined an almost-double growth rate of this class of MFCs. Note worthily, the highest PD output reported to date appears to be 4300 mW m−2 achieved back in 200419 through the use of ferricyanide as a catholyte, but sustainability considerations10,13 drew the attention of the research community elsewhere.
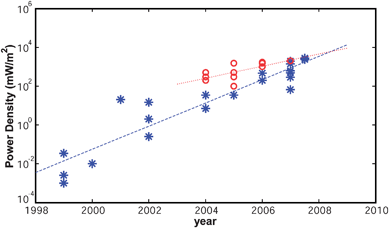 | ||
| Fig. 1 (A) Power density trends based on published results for aqueous and membraneless air–cathode MFCs (*) and for pure fuel MFCs (o); the slopes of the fitted lines shown are k = 1.4 (R2 = 87%) and k = 0.7 (R2 = 50%) respectively. | ||
The increase in PD, although initiated by crucial advances on the biological side, lately have been driven largely by substantial improvements on the engineering side, e.g. materials, cell architecture, buffer solution. As an emblematic example, the PD value of 2400 mW m−2 in Fig. 1 (adapted from13) was achieved by Logan and coworkers14 through a continuous evolution of a basic MFC design capable of 292 mW m−2 in 2004. Without substantial changes in the working bacteria, the power passed orderly to 494 mW m−2 by removing the proton exchange membrane, to 1210 mW m−2 by reducing electrodes spacing, to 1330 mW m−2 and then to 1640 mW m−2 by increasing the solution conductivity, to 1970 mW m−2 by adopting improved anode substrate, and finally to 2400 mW m−2 by optimizing the cell layout and by deploying brush anodes. The membrane removal in the late configurations rendered a hybrid design with an air–cathode wet by aqueous solution on one side, which induced us to pool membraneless air–cathode data together with aqueous cathode in Fig. 1.
The current PD trends are encouraging but PD levels—both in terms of W m−2 and W m−3 as the MFC power is customarily normalized either to the electrode surface or to the liquid volume, respectively—should increase substantially to render MFC technology feasible for commercial applications. For example, according to estimates by Rabaey and Verstraete,20 volumetric PD around 500–1000 W m−3 would allow MFC deployment for treatment of large wastewater flows, but maximum values are still of the order of 200–250 W m−3.21,22 Nonetheless, recent figures are significant and, when comparing the MFC power output with other alternative energy production systems in Fig. 2, the “credibility gap” with respect to conventional fuel cells (FCs) postulated by Bullen, and coworkers not long ago is already shrinking.23 But, even when such a gap should narrow and disappear, the difference between the power range of chemical FCs and MFCs would certainly remain large, resulting in a minimum overlapping application-wise between the two technologies. As depicted in Fig. 2, the gap would indeed soon disappear based on the forecast by Torres and coworkers, who achieved in 2008 a current density of 10 A m−2 and envision power densities of 5 W m−2 and 1000 W m−3 for a typical operational voltage of 0.5 V.24 Of course, the actual power yield will strictly depend upon advances in scalability of lab-prototypes, which is a major technical challenge. The MFC upscaling is currently bounded by the cost of materials and available cell designs. For example, researchers all over the world are striving to substitute costly Pt-catalysed cathodes (priced at US$98 per Watt) with inexpensive equipotent materials, such as granular graphite cathodes (priced US$79 per Watt).25 Technical feasibility and cost arguments alone cannot justify the enormous interest surrounding a technology that evidently is not likely to compete with conventional FCs (at least in the near future) in terms of efficiency and power output (Fig. 2).
 | ||
| Fig. 2 Schematics of current positioning of MFC technologies based on power output in comparison with other sustainable energy production technology adapted from ref. 23. | ||
The competitive advantages of MFCs lay elsewhere, namely in the sustainability challenges faced by modern society. Although nanotechnology and biotechnology are main enabling factors that “pushed” the technological development, sustainability has provided a strong “pull” action. In a broad sense “sustainability” has to do with a regime of limited and constrained resources, and is a characteristic of a process or a system to be maintained in a certain state at a certain level of performance indefinitely. After a silent period, MFC research has revamped at a time where a global effort is being made by the industrial world to move away from fossil fuels towards a sustainable economy based on a pool of renewable energy resources with low environmental impact. In this context, MFC technology looks very attractive since it can potentially address two sustainability problems at once: energy supply and wastewater management. In the near future, in fact, MFCs might allow energy production from “poor” waste (e.g. sludge or wastewater) while providing inexpensive waste treatment alternatives.
Besides terrestrial applications, MFCs retain also a potential for space applications. The achievement of the International Space Station, which is the largest system ever accomplished and functioning away from Earth, has paved the way to more ambitious goals of human explorations of space. Thus, long-term space missions and human settlements on the Moon and on Mars are currently undergoing serious evaluation by space agencies worldwide. Human missions of longer duration (e.g. years) and higher complexity pose significant problems of sustainability related to severely limited equipment and supplies in an environment hostile to human survival. For example, managing organic waste from human metabolism becomes an issue of crucial importance. Storage and dismissal of wastewater cannot be handled as in current missions, instead new methods are needed for safe and efficient waste disposal. One requirement is the development of technologies enabling a “closed loop of waters” onboard. Securing a power supply throughout the mission is another essential problem. As the mission time increases, sufficient amounts of energy cannot be stored beforehand on Earth but rather are produced “in situ”, either by exploiting energy sources in space or by producing it in the spacecraft/station. MFC technology can aid the solving of both challenges simultaneously. It is not incidental that research in this area was recently resumed at NASA even though traditional FCs have been preferred in NASA space programs since the 1960s,4 when the bioelectrochemical complexity of MFCs undermined reliability and operational stability of early devices. Many space applications present less scalability problems since they do not require large liquid flows. Payload limitations may also encourage exploitation of miniature-MFCs. Such devices are currently under investigation and eventually might be used in the biomedical compartment to power small biomedical devices, e.g. biosensors and implants.22 After these opening remarks and considerations, the review will focus on engineering aspects related to individual components as well as to the overall MFC architecture. Many excellent reviews are already available discussing at length both fundamental and specific aspects of MFCs, e.g.20,26–29 Several of them concern biological aspects10,23,30–35 while fewer are concentrated on materials and engineering aspects. In an effort to keep the overlap at a minimum, after a brief discourse about the current perspective of MFC biology, the present review will emphasize the developments of anodes, cathodes, electrolytes, and cell architectures occurring in the last few years.
2. MFC basic principles
A basic double-chambered MFC consists of anode and cathode compartments separated by an ion exchange membrane (or alternatively by a salt bridge) and connected by an external electric circuit (Fig. 3). | ||
| Fig. 3 Schematics of a double-chamber MFC with a PEM membrane and oxygen reduction at the cathode. | ||
In the anode compartment a biofilm, of bacteria (the catalysts), is laid upon an anode substrate to form a bioanode immersed in a solution of organic matter (fuel) fed to the compartment in either continuous or batch mode. Some of the bacteria involved in catalysis might also reside in the electrolyte solution. The bacteria, first oxidize the fuel (the electron donor) through their metabolism, freeing electrons, protons and/or other cations, and then transfer these electrons to the anode substrate through a number of mechanisms, e.g. direct contact, nanowires or mobile electron shuttles (mediators). The electrons from the bioanode pass to the cathode compartment through the external electric circuit, while selected ions move across the electrolyte membrane to close the circuit.
A reduction reaction takes place in the cathode compartment, mostly in the presence of oxygen. For example, when a proton exchange membrane is used, migrated protons and electrons combine with oxygen to form water at the cathode. A biofilm may optionally be employed also here to catalyse the reduction reaction. Although, anodophile and cathodophile bacteria belong to different species, the former ones being typically anaerobic and the latter ones aerobic. It is also possible to expose the cathode directly to air and eliminate the need of a cathode chamber altogether, yielding single chamber MFCs. This solution offers simpler design and cost savings.
A FC basically works as a voltage source with an internal resistance. As soon as the current is drawn, the voltage drops due to various losses, bringing the actual cell potential to be always lower than the attainable theoretical voltage.
The cell voltage is described by the following equation:
| Ecell = [Ecathode − |ηact, c + ηconc.c| ] − [Eanode − |ηact, a + ηconc.a|] − ηohm | (1) |
In MFCs, the activation polarization is due to the activation energy needed by the fuel and the oxidant to undergo reactions at each electrode. Such effect depends upon (i) the current density flowing through the anode, (ii) the electrochemical properties of the electrodes, (iii) the presence of electrochemical mediators, and (iv) the operation temperature. Hence, the decrease of these type of losses can be achieved in several ways, such as by increasing the electrode surface area, by improving the electrode catalysis, by increasing the operation temperature, and/or by deploying catalytic biofilms at the electrodes enriched with electro-mediating compounds.
Concentration polarization determines energy losses associated with mass transport and is due to the accumulation of reaction products and the depletion of reactants in the electrolyte near the two electrodes. Stirring and/or bubbling as well as a special MFC reactor configuration helps to reduce the concentration gradient.
Ohmic losses are due to the electronic flow through the electrodes, the external resistance, the current collectors, and the contacts, as well as the ionic flow through the electrolyte. According to the Ohm's law, ηohm can be expressed as “i*R”, where “i” is the electrical current through the cell and “R” is the total cell internal resistance obtained from the sum of the above mentioned contributions. Ohmic losses can be reduced by minimizing the electrode spacing, by using membranes with lower resistance, and by increasing the solution conductivity compatibly with bacteria survival.
The performance of a MFC in terms of power generation, current output, and electric efficiency ultimately depends upon a complex array of parameters. As we shall discuss, the type and the efficiency of the active mechanisms enabling bioelectrochemical energy conversion in a MFC (e.g. the complex respiratory chain underlying fuel degradation) are of paramount importance in defining the electric performance, but materials, system layout, and operating conditions are equally key factors.
3. MFC microbiological aspects: an overview
The attention paid to biocatalysts and bioelectrochemical aspects is documented in the series of referenced review papers. They illustrate the extensive search for optimal ways to transfer electrons from the microorganisms to the fuel cell anode and discuss a number of possible electron transfer processes enabling—to different extents—the production of electricity in MFCs.The biological basis of any MFC rests in the degradative metabolism responsible for the release of energy and the breakdown of complex materials substrates (e.g. sugar, proteins, or lipids) within the organism, also known as cell catabolism. Metabolism may happen either by respiration (i.e. any of several energy-yielding oxidative reactions occurring in living matter that decouple electrons and protons to establish an electrochemical gradient) or by fermentation (i.e. a dismutation consisting of an enzymatically controlled transformation of a single organic compounds into two products without decoupling electrons and protons). All of them may be active, separately or simultaneously, in a MFC. Respiration takes place in the presence of heterotrophic microorganisms (requiring complex organic compounds of nitrogen and carbon for the metabolism) and the amount of attainable energy is expressed by the free energy (ΔG0) corresponding to the oxidation of the organic compounds:
| ΔG0 = nF [E0donor − E0acceptor] | (2) |
| C6H12O6 + 6O2 → 6CO2 + 6H2O; ΔG0 = − 2895 KJ mol−1 | (3) |
Under anoxic conditions, instead, some bacteria carry out anaerobic respiration by using nitrates, sulfates, carbon dioxide, metal ions, or fumarate as terminal electron acceptors, yielding a lower energy gain than in aerobic conditions due to the less positive redox potential of these oxidants compared to oxygen. Alternatively, in the absence of oxygen, many microorganisms perform fermentation. The microorganisms may convert the chemical energy of a substrate into electricity, but they must retain a certain amount of free energy (ΔG0biol.) required for cell anabolism, i.e. the constructive part of metabolism concerned particularly with the synthesis of macromolecules needed for cell survival and reproduction. Therefore, the free energy actually available for the generation of electricity (ΔG0elec.) is lower than the total theoretical value ΔG0:
| ΔG0elec. = ΔG0 − ΔG0biol. | (4) |
The energy amount for bacterial vitality (ΔG0biol.) is evidently necessary for long-term MFC operation and often represents the driving force directing the organisms to follow certain electron transfer mechanisms. However, should the biological energy become too high, the current output would lower too much in favor of stronger cell growth and unwanted biomass formation. As a consequence of these conflicting tendencies, an optimal balance between suitable metabolic and electron transfer paths has to be determined to allow, at the same time, continued MFC operations and maximum electric energy output.
In the recent past, considerable research has been devoted to the identification of microorganisms capable of completing anaerobic respiration onto an extra-cellular electron acceptor in the anode compartment. From such investigations, a few iron-reducing microbial species emerged as being able to transfer electrons from their metabolism directly to the anode, e.g. Aeromonas hydrophila, Geobacter metallireducens, Geobacter sulfurreducens, Rhodoferax ferrireducens, Pseudomionas aeruginosa, Clostridium butyricum, Enterococcus gallinarum, Shewanella oneidensis, and Shewanella putrefaciens. It is not true, however, that “all” iron-reducing bacteria can transfer electrons to the anode.36 During biological evolution, many electron-transfer mechanisms were developed by different microorganisms to complete respiration through an extracellular pathway by either donating or harvesting electrons for their own metabolism.37 There are two main mechanisms, as depicted schematically in Fig. 4 and Fig. 5. The first mechanism (Fig. 4) features a “direct electron transfer”, implying a physical/electrical contact between the bacterial cell membrane and the anode substrate (Fig. 4a). The mechanism requires the microorganisms to possess outer membrane redox macromolecules (i.e.cytochrome) that enable electrons to transfer to the anode. Recently, it has been demonstrated that some microbial strains can develop electronically conducting molecular pili (nanowires) to establish physical electrical connections with an electrode not directly in contact with the cell (Fig. 4b). Such pili are connected to cytochromes, which permit electron transfer to the outside of the cell. The second mechanism (Fig. 5) features a “mediated electron transfer”, where the electron passage happens by means of soluble redox mediators: either exogenous ones, which could be natural or synthetic, or endogenous electron “shuttles”. Any electron mediator must (i) be able to physically contact the electrode surface, (ii) be electrochemically active, and (iii) have a standard potential as close as possible to the redox potential of the substrate or significantly negative compared to the oxidant. However, synthetic exogenous mediators have to be continuously supplied to the MFC and are toxic, which are disadvantages that endanger their environmental and technological sustainability. Exogenous mediators, though, are not necessary when the mediation process is carried out by shuttles produced by the bacteria itself as secondary metabolites, i.e. endogenous redox mediators.38 More importantly, in the case of shuttles, the bacteria can better control the bioelectrochemical activity in response to a range of working conditions, being able to regulate both the electrons throughput and the synthesis of its own transporters. In the other case, in fact, the adaptivity of the bacteria can rely just on pacing electrons production on the availability of exogenous carriers added in the solution.
 | ||
| Fig. 4 Schematics of Direct Electron Transfer mechanism via (a) membrane bound cytochromes and (b) via electronically conducting nanowires (pili). | ||
 | ||
| Fig. 5 Schematics of Mediated Electron Transfer mechanism via added (exogenous) or secreted (endogenous) mediators. | ||
It is not surprising that most “electrical” bacteria belong in anaerobic environments, since “lack of oxygen” represents the prime evolutionary cause for the development of extracellular respiration, which is the basis of all electron transfer mechanisms. Nonetheless, an exception is made for the direct electron transfervia conductive nanowires, all other modes are exploitable by both anodophilic (anaerobic) or cathodophilic (aerobic) bacteria.
Nowadays, two key ideas stand out as generally agreed upon and contribute fundamentally to the current perspective about MFC biocatalysts, namely:
1. Despite the extreme plasticity exhibited by some bacteria, no single bacterial strain should be trusted when a complex substrate (fuel) is used, because a consortium of microorganisms yield better and more reliable performances;
2. Both types of electron transfer mechanisms to the electrode (i.e. the direct and mediated ones, in all the variants in Fig. 4 and Fig. 5) may coexist in an MFC.
Mixed-culture MFCs usually render better electric performance compared to the pure-culture counterparts.39 This scenario may be regarded as a tight collaboration amongst the interdependent and competing bacterial species contributing to the consortium aimed at the full fuel substrate degradation. The pictorial representation in Fig. 6 shows a first group of fermentation bacteria, breaking complex molecules into energy-rich reduced metabolites suitable for the anaerobic respiration of a second bacterial group. Finally, some bacteria in the latter group are able to carry out an extra-cellular respiration when provided with a proper anode substrate, while the remaining ones take advantage of co-existing “adjuvant” bacterial strains.
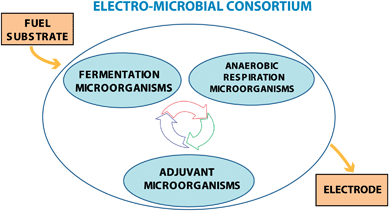 | ||
| Fig. 6 Schematic representation of microbial consortium including three types of interdependent microorganisms cooperating in fuel degradation and electron transfer to the electrode. | ||
The importance of adjuvant microorganisms for a correct ecology of the consortium and, hence, for greater energy production was highlighted recently.36,40,41 As a matter of fact, the proper choice of microorganisms must be regarded as one of the key parameters in MFC engineering.
4. MFC materials and architectures
The assessment of different materials and MFC architectures is a cumbersome task since, in the beginning, findings were obtained and reported in an erratic fashion. Currently, there is still a lack of standards related to MFC construction, characterization, and operating conditions, which does not permit either an organic classification of existing devices or a direct comparison. The need to establish a general framework within the MFC research community is rather urgent. However, the situation has progressively improved, partly due to some previous reviews and to the first book dedicated to MFCs,26,42 which are notable attempts to organize the results in a systematic manner and provide a sort of guideline to researchers. In this respect, the “MFC web platform” (http://www.microbialfuelcell.org/) deserves a mention, it is an initiative contributed to by research groups active in the area and is aimed at promoting knowledge and best practices within the research community. Nevertheless, the problem persists and systematic quantitative comparisons are not always possible. Furthermore, MFCs are complex systems governed by several factors mutually interconnected. For example, it may be hard to pinpoint whether an improvement in performance is due to better materials or to a novel architecture, unless a systematic product development and/or statistical techniques for “design of experiments” is carried out to separate and assess both the main effects and the interactions of the control parameters. Such approaches, although desirable, are still rarely encountered and, admittedly, do require large and time consuming experimental campaigns.4.1. Anode substrate
The bioanode is perhaps the defining element of MFC technology and is often the limiting factor for a high power output and performance. Having discussed biofilms, we now turn to the anode substrate, which lately has been the subject of a considerable amount of research. The anode material and its structure can directly affect bacteria attachment, electron transfer, and substrate oxidation. Various materials, including non-corrosive stainless steel,43 plain graphite,44carbon paper,45carbon cloth, felt, or foam,7 reticulated vitreous carbon,46,47graphite granules,41,48 and graphite fiber brushes13 have been used as anodes, due to their stability in a microbial inoculum mixture, high conductivity, and high specific surface area. Notably, the graphite brushes already mentioned above and used in a cubic air–cathode MFC yielded a maximum PD of 2400 mW m−2 (with an internal resistance Rint = 8 Ω), which is considerably larger than the 1640 mW m−2 obtained with a plain carbon cloth (Rint = 31 Ω). In addition, these were commercial brushes mounted onto a non corrosive metal core, which are much cheaper than carbon cloth and are advantageous for scaling up the technology. Brushes leverage on the surface area but have low electrocatalytic activity for the anodic biofilm. Such an example demonstrates the importance of proper engineering of the anode substrate.Different strategies have been pursued to enhance the anode–microbes electrical interfacing. For example, Park and Zeikus showed that the maximum power output of E. coli-based MFCs increased up to 152 mW m−2—a value three orders of magnitude larger than the power achieved with an unmodified graphite felt electrode—by embedding into the anode substrate Mn(IV) and Fe(III) covalently linked to neutral red (NR) to mediate electron transfer from microbes to the anode material.49 Alternatively, a large power density was achieved by ammonia gas treatment of carbon cloth at elevated temperatures.14 Besides, other types of electrocatalytic materials such as polyaniline(PANI)–Pt composites, have been used to improve current generation, owing to their straightforward processability, electrical conductivity, and the environmental stability of polyaniline.50–52
Even though such approaches have contributed to some extent to the improvement of anode performance, it is well known that the use of Pt on the electrodes is the most effective strategy to enhance electrocatalysis. In fact, Pt is the most widely used electrocatalyst in the fuel cell field, behaving also as a very efficient catalyst towards the electrochemical oxidation of organic molecules. However, the use of costly Pt as an electrocatalyst has been already denounced as a major drawback for MFC mass production. Major efforts have been focused on overcoming the problem by either minimizing the Pt loading amount or finding Pt substitute materials. Several routes have been explored to these ends. To minimize the Pt loading, some authors deposited a Pt nanolayer on a carbon paper anode and cathode via an e-beam evaporator, obtaining effective and low-cost Pt electrodes.53Tungsten carbide anodes have reached performance levels comparable to Pt in MFC applications.54,55
Anode performance can be boosted also by increasing the surface area and the biocompatibilty of the substrates. Scott et al.56 prepared graphite felt anodes modified with (i) C/PANI composites, (ii) carbon nanofibers, or (iii) nitric acid carbon activation. They demonstrated that the modified anode materials have superior performance in terms of power density when compared to the unmodified graphite felt. This improvement was a likely result of the increase in surface area, which multiplies the number of sites for microbial colonization, as well as in anode biocompatibility, the latter being linked to surface functionalization by the quinoid groups of PANI or by the formation of quinone groups on the carbon surface by the carbon activation treatment. The maximum power densities obtained with the modified anodes, compared to plain graphite felt (9.5 mW m−2), increased in order for carbon nanofibers (26.1 mW m−2), for C/PANI composite (26.5 mW m−2), and for activated carbon anodes (28.4 mW m−2), which further proves the effect of anode-pretreatments on the final composition/quality of the microbial communities colonizing the anode.57
Other authors58 sought additional improvements by adding carbon nanotubes (CNTs) to PANI to simultaneously enhance both specific surface area and charge transfer capability. Purposely fabricated “CNT-PANI” nanocomposites delivered indeed greater electrochemical activity for the MFC anode reaction. Besides high conductivity and extended active surface area, CNTs also seem to have a beneficial impact on the biocompatibility of the electrodes towards the working bacteria, which is an equally important factor in MFCs. A biocompatibility test related to the carbon structures was performed in a MFC incorporating Escherichia coli (as bacteria) and methylene blue (as electron mediator), to verify that there is a greater biocompatibility with nanotubes than with regular carbon fibres or other carbon forms.59
Finally, another interesting option was proposed by Zhang et al.,60,61 who investigated graphite–polytetrafluoroethylene composite films as anodes for E. coli-based MFCs and observed that the polytetrafluoroethylene (PTFE) content in the anode film affected the catalytic activity of the electrochemically activated E. coli. They obtained a power density of 760 mW m−2 with a composite anode containing 30% PTFE in absence of exogenous mediators.
Besides materials, there are other aspects as relevant to the anode performance. The anode potential, for example, is an important factor controlling the interaction between bacteria and the electrode substrate, thus defining the power generation of a MFC. From an electrochemical viewpoint, the anode potential should be as low as possible and the cathode potential as high as possible to achieve the highest electrical energy output. Moreover, from a biolectrochemical point of view, it was found that a more positive anode potential imparts a greater stimulus for the bacterial colonization (i.e. adhesion and growth), resulting in a higher biocatalyst density,62 in a faster start-up of the electricity generation, and often in larger current generation. However, for set current and cathode potential, the anode potential should be as low as possible to maximize the electrical energy output of a MFC, leading to a trade-off between the anode potential and the thickness of the biofilm. The anode potential needs to be carefully tuned to maximise the electric current and the power output. Finkelstein and coworkers suggested the existence of an optimal range for the anode potential, determined by the tendency of microorganisms to adapt their electron transferring system to a level just below the minimum anode potential.62 Yet, this aspect was further investigated by Aelterman et al.,21 who operated three identical reactors fed with acetate (theoretical anode potential of −496 mV vs. Ag–AgCl) at “poised” anode potentials of 0, −200, and −400 mV vs. Ag–AgCl, and found that −200 mV was the best anode potential to sustain enhanced current and maximum power generation (31.1 mW, 199 W m−3). The increased current was a consequence of higher biomass densities causing stronger bacterial competition for the available electrode surface and substrate.
Diffusion limited regimes of mass transport are another critical issue governing the maximum protonic transport at the anode. Biofilm thickness is certainly crucial to this end, together with a buffer solution that is known to reduce ohmic resistance in an MFC by both increasing water conductivity and enhancing proton transfer between anode and cathode chambers. Torres et al.24 determined that protons were mainly transported out of the biofilm by protonating the conjugate base of the buffer solution at high current densities, which is directly linked to the buffer diffusion transport in and out of the biofilm. With non-limiting acetate concentrations, in fact, the current density grew by increasing buffer concentration from 2.21 to 9.3 A m−2 at 12.5 mM and 100 mM phosphate buffer medium at a constant anode potential of E = −0.35 V vs. Ag–AgCl. Apparently ion migration was not as important as the phosphate diffusion inside the biofilm, which depends on its thickness. Besides diffusion, the pH of the buffer medium is also affected by the film thickness and in turn influences sensibly the current density. Reportedly, current densities varied from 10 A m−2 at pH = 8 to half this value at pH = 6.5. If the buffer concentration gradient through the thickness is not too large, then the pH can be maintained in a range compatible with bacterial growth and metabolism, else the pH drops causing bacterial inhibition and lowering current, which therein occurred at pH ≈ 6. In other experiments by Fan et al.63 a 38.6 % increase in PD occurred by shifting the pH from 7 to 9 in a bicarbonate buffer solution. Their set-up consisted of a single-chamber air–cathode MFC (with a carbon cloth–Pt–PTFE cathode) featuring a characteristic CEA (cloth electrode assembly) anode, which delivered a maximum PD of 2770 mW m−2 at pH = 9. That, seemingly is the highest value ever reported for this cell architecture (better than brush anodes) and approaches MFCs based on ferricyanidic catholyte (3000–4300 mW m−2). However, this achievement was attributed to the optimization of the bicarbonate buffer solution, since the best PD performance lowered to 1800 mW m−2 when phosphate buffer was used. This result provides a sound proof that the buffer is a primary tunable parameter to maximize anode performance, and seems even more relevant when considering that bicarbonate is cheaper than phosphate.
Collectively taken, the work by Torres et al.24 and Fan et al.63 point out that electron transport may not be limiting the current density of an MFC as much as the proton transport. Anode substrates should be designed to increase the contact between the biofilm and the bulk liquid. A high specific surface area of an electrode substrate would provide a significant benefit only when it translates into an increased biofilm–liquid interfacial area, where H+ transport occurs.
In conclusion, the “inefficiencies” of the anodic processes have not been either fully identified or solved yet, and represent an area for major improvement. As a final remark, it was recently highlighted that there is a possibility that organic fuel may get stored inside anodophilic microbes under certain conditions (e.g. initial high fuel conditions) to be consumed during starvation. Freguia et al. assumed the existence of such storage mechanism64 by estimating that about 57% of the current generated in one experiment occurred after depletion of the external carbon source.
4.2. Cathode
The cathode is responsible for transferring the electrons to the terminal electron acceptor, i.e.oxygen in most cases, and is currently the major bottle-neck preventing the application of MFCs for electricity generation. Like conventional FCs, the high overpotential of the oxygen reduction reaction renders non-catalysed cathodes rather inefficient. Catalysts and/or artificial electron mediators are generally required.65 Analogously to the anode, Pt mounted on carbon black is widely used also as an electrocatalyst for oxygen reduction, both in traditional FCs and MFCs.66–68 Due to its high cost, reducing the Pt amount and/or finding substitutes to catalyse oxygen reduction is perhaps even more urgent and challenging than for the anodes. Cheng at al.11 examined the effect of Pt loading (0.1–2 mg cm−2) on the electrochemical performance of the cathodes in a bacteria-free electrochemical cell using chronopotentiometry. Unlike what can be expected from conventional FCs where the performance is greatly affected by Pt loading,69 they found that lowering the Pt loading by a factor of 5 caused only a small reduction in the overall MFC performance, which may be acceptable considering the cost of the catalyst and the large surface areas needed for MFC operation. Of course, this result should be assessed in light of the substantial differences between FCs and MFCs, since ambient temperature, ionic strength, and mostly neutral pH may pose major thermodynamic and kinetic constraints on MFCs cathode performance.69 The Pt cathode also endures a performance loss resulting from the lack of tolerance to fuel components (poisoning) and/or to pH increase during MFC operations.70,71 Two main approaches are being pursued to replace Pt at the cathode. The first one uses non-Pt catalysts that nonetheless contain non-abundant precious metals, typically based on Pd72 or Ru.73 The other approach seeks Pt replacement with plentiful non-precious materials, such as Fe and Co. Such materials act as suitable electron mediators between the cathode and oxygen, owing to the high commuting rate between their redox states. Park and Zeikus49 developed a Fe3+–graphite cathode, thus developing a less expensive MFC system with high levels of power generation. By using E. coli as the biocatalyst coupled with a Fe3+–graphite cathode and a Mn4+–graphite anode, they obtained 325 mA m−2 of current density and 91 mW m−2 of PD. Lead dioxide was also considered as an alternative cathode catalyst in a double-chamber MFC utilizing glucose.74 Even though this kind of catalyst allows for higher power generation and lower production costs, its stabilization on the cathode should be improved to prevent dissolution.Catholytes such as ferricyanide68,75 or permanganate76 were also investigated for optimizing cathodic reactions. Such redox mediators serve as a terminal electron acceptor instead of oxygen, allowing for a power output as large as 258 W m−3.41 As mentioned earlier, despite the fact that ferricyanide-based catholytes have achieved greater power production than oxygen systems, these liquid-state electron acceptors are generally deemed unsustainable for practical uses due to the regeneration requirement of the chemicals and environment-related issues.77
The non-precious cathode catalysts that have attracted most attention over the years are based on pyrolized metal porphyrins and phthalocyanine, with cobalt and iron porphyrins viewed as the most promising precursors, providing inexpensive and efficient alternatives to Pt-based catalysts for conventional FCs.78 Some authors have investigated the use of transition metal macrocycles as MFC cathodes. Zhao et al. performed a fundamental study to investigate the influence of the composition of the cathodic electrolyte solution on the electrocatalytic oxygen reduction reactions using noble-metal free catalysts compared to Pt.71,79 They used iron phthalocyanine (FePc) and cobalt tetramethoxyphenylporphyrin (CoTMPP) as the oxygen reduction catalyst for a biohydrogenE.coli-based MFC, studying the effect of pH and concentration of the electrolyte on the cell performance. The Co-based material performed better than the iron compounds, which may be due to a stronger back binding between oxygen and cobalt and/or to the catalyst preparation procedure. Noteworthily, Zhao et al.71 also warned about the performance of the cathode being controlled mainly by anode limitations. Therefore, the absence of power loss upon reduction of Pt-loading reported in69 could just be a result of the cathode not being the limiting factor in that experimental setting.
Further studies11,12 compared the performance of the Pt-based cathodes to that of several transition metal macrocycles (i.e.CoTMPP, FePC, CoPc, CuPc, MnPc), demonstrating that CoTMPP and FePC are suitable candidates for Pt replacement at the MFC cathode.
Nanostructured materials can be exploited too, to facilitate the oxygen reduction reaction at the cathode surface. Freguia et al.25 proposed a strategy to limit the oxygen reduction overpotential which focused on surface area rather than on various catalysts. Thus, instead of lowering the activation energy through a catalyst, they used a non-catalysed material with a high specific surface area (i.e. highly porous granular graphite). Power outputs as high as 21 W m−3 or 50 W m−3 (over cathode total volume and cathode liquid volume, respectively) were achieved in a MFC fed continuously with acetate.
4.3. Biocathode
The growth of microorganisms in the cathode chamber with the consequent formation of biofilms on the cathode is hard to avoid in MFCs, especially when the electrolyte membrane is absent.45,80,81 Rather than preventing microbes from depositing on the cathode, bacteria could be used as biocatalysts to accept electrons from the cathode substrate underneath.29 Biocathodes offer a different path to avoid the use of noble or non-noble catalysts for oxygen reduction, which increases substantially the viability and sustainability of MFCs (even though further research is needed to minimize the start-up period for these cathodes). He et al.82 extensively reviewed the development and experimental progresses of biocathodes in MFCs, elucidating several possible biological cathodic processes, which included oxygen reduction.Among recent studies on biological cathodes, Gregory et al.83 demonstrated that bacteria can take up electrons from a graphite electrode without hydrogen as an intermediate electron shuttle for nitrate and fumarate reduction. Recently, it has been shown that a bioanode oxidizing acetate could be combined with a biocathode, reducing nitrate to nitrogen gas.84 Seawater biofilms growing on a stainless-steel cathode were found to be able to catalyse the oxygen reduction, using the electrons delivered by the cathode.85 Dumas et al.43 checked the effectiveness of a stainless steel cathode covered with a seawater biofilm formed during MFC operations, revealing its promising aptness for sediment MFCs.
Leptothrix discophora SP-6, i.e. a type of manganese (Mn) oxidizing bacteria, has been known to accumulate Mn oxides from the aqueous environment and biomineralize Mn oxides, suggesting its potential use as a cathodic reactant in a new generation of MFCs featuring a biocathode. Aerobic biocathodes have been tested in freshwater with manganese as an electron shuttle between a graphite electrode and L. discophora.86 Also, an acetate oxidizing tubular MFC was built by combining the anode with an open air biocathode in freshwater conditions.87 The results from the latter group indicated that cathode-driven microbial growth is possible at high cathodic potentials (above 0 mV), which excludes the occurrence of cathodic hydrogen evolution and subsequent bacterial hydrogen consumption. As a follow up on this theme, Rabaey et. al.88 investigated open-air carbon cathodes colonized by bacterial strains able to reduce oxygen, i.e. the final acceptor of the electrons provided by the solid-phase cathode. Although best results were achieved with a mixed population mainly of Proteobacteria and Bacteroidetes, the use of selected pure microbial cultures yielded a threefold increase in PD compared to the same non-inoculated MFC used as a control. The marked decrease in activation losses proved that bacteria acted as true catalysts—perhaps the most sustainable long-term catalysts—for the oxygen reduction reaction. More work is needed to optimize this result and overcome the technical difficulties, e.g. pH control and surface modification of cathode substrates, but these studies suggest interesting possibilities for sustainable and low cost MFCs.
4.4. Electrolyte
Generally speaking, FCs are classified by their electrolyte material. Among them, polymer electrolyte membrane (PEM) FCs have drawn significant interest, since they deliver high power density while operating at relatively low temperatures. They also offer advantages in terms of low weight and volume, compared to other FCs.89 The proton exchange membrane is the most critical component in the PEM-FC configuration. It provides a separation between the fuel and the oxidant agent, but at the same time allows for transport of positive charges to compensate the electron transport. Nafion® has set the industry standard for PEM and is used in almost all current PEM-FCs. Its properties have been extensively reviewed.90Nafion consists of a hydrophobic fluorocarbon backbone to which hydrophilic sulfonic acid groups are linked. Because of its PTFE backbone, Nafion is chemically inert in both oxidizing and reducing atmospheres, whereas the high concentration of the sulfonic acid groups is responsible for its high proton conductivity. Due to its good characteristics, Nafion has also been widely investigated for MFC applications7,50,67 whilst only a few reports deal with different electrolyte materials.91–96 On the other hand, large-scale commercialization of Nafion-based devices is limited by its high cost, and even more by its oxygen permeability (lit. 9.3 · 10−12 mol cm s−197), which is a major problem for MFC applications. Min et al.93 measured the amount of oxygen that diffuses through the Nafion membrane by evaluating the oxygen accumulation in the anode chamber under abiotic conditions, and put forward evidence to show how such oxygen diffusion to the anode reduces Coulombic efficiency.Currently, polyether ether ketone (PEEK) is a promising polymer actively being researched by the FC community to overcome the drawbacks of Nafion.98,99 PEEK is a low-cost polymer with good thermal stability and mechanical properties. Proton conductivity for this polymer can be achieved by sulfonation (sulfonated PEEK, i.e. SPEEK), and the degree of sulfonation (DS) can be readily tuned by controlling the parameters of the sulfonation reaction. Both for Nafion and SPEEK, ionic clusters coexist with hydrophobic domains according to models proposed in the past.100 However, the nanoseparation of hydrophobic and hydrophilic entities is greatly reduced for SPEEK with respect to Nafion due to, the lower acidity of the sulfonic acid groups, to the absence of fluorinated groups, and to the higher mechanical stiffness of the polymer backbone induced by the presence of aromatic groups. All these features impact upon the proton transport properties, as well as the permeability towards reactants. In fact, SPEEK-based electrolytes exhibit oxygen permeability one order of magnitude lower than Nafion-like polymers,101 as would be expected from the narrower separation between hydrophobic and hydrophilic domains. The permeability towards a reactant can be further reduced by decreasing the DS and/or by doping the polymeric matrix with inorganic components.102–104 Such types of electrolytes may have a relevant impact in the MFC field, but there is no evidence yet of their use (to the best of our knowledge).
The use of Nafion in MFCs has been related to further practical drawbacks caused by the side effects of other cation transport onto protons.105 In fact, Gil et al.106 observed a decrease in the pH of the anode chamber and an increasing pH in the cathode chamber because the proton transport through Nafion seemed to be slower than both the proton production rate in the anode chamber and the proton consumption rate in the cathode chamber. Liu and Logan67 operated a single-chamber MFC in the presence and absence of a Nafion membrane and found a reduced power output when Nafion was present. In a traditional PEM-FC, protons are the only cation species present in the system, whereas in a MFC, operating at neutral pH conditions, the concentration of other cation species has to be taken into account. The electrolyte conductivity is controlled by both the ion mobility and the interaction of ions with water and micro-structural features of the membrane. Typically, moving ions carry water molecules along, thus limiting the cation rate transport across the hydrophilic channels of the membrane. However, the H+ conduction in the membrane is larger than for other cations because it relies heavily on the distinctive Grotthus mechanism. According to the latter, protons can “jump” between neighbouring water molecules via a bonding-debonding process that enables protons to be highly mobile.107,108,109 Nevertheless, despite the lower mobility of other cations (i.e,Na+, K+, NH4+, Ca2+, and Mg2+), their concentration in a MFC environment is several orders of magnitude larger than proton concentration.
In this context, Rozendal et al.110 quantified the membrane cation transport in an operating MFC and evaluated the consequences of this transport for MFC application with wastewaters. They report that cation species other than protons are primarily responsible for the transport of positive charges across Nafion membranes. This effect causes an accumulation of these cations at the cathode and a consumption of protons in the cathode reaction, leading to an increased pH in the cathode chamber and to decreased MFC performance. Zhao et al.71 also performed experiments where porphyrin-modified cathodes were used with Nafion membranes, suggesting that pH gradients and enhanced cation transport at neutral pH across Nafion membranes have a crucial role in reducing MFC efficiency.
These drawbacks pushed research to find new electrolytes which prevent the above described effects of Nafion-based MFC. Anion-exchange membranes may represent an effective option, involving hydroxide ions diffusing from the cathode through the membrane and forming water at the anode. For the first time, a bipolar membrane, consisting of cation and anion exchange membranes joined together in a series, was applied to a MFC.111 In the bipolar membrane water dissociates viaelectrodialysis, supplying protons that migrate through the cation exchange membrane towards the cathodes, while hydroxide ions migrate through the anion exchange membrane towards the anode. This successfully led to a pH decrease in the cathodic compartment. Moreover, on the basis that phosphate anions are often present at high concentrations in MFCs, Kim et al.94 examined the possibility of using an anion exchange membrane to allow proton transferviaphosphate anions. The performance of an anion exchange membrane was tested and compared to Nafion in terms of power densities, Coulombic efficiencies, and permeability to oxygen and acetate as the substrate. The anion exchange membrane produced power densities up to 610 mW m−2 and Coulombic efficiency (72%), both larger than those achieved with Nafion.
Alternatively, membranes can be omitted from the MFC configuration, fabricating membraneless single-chamber devices, as discussed later. Nevertheless, none of the available solutions seem to be optimal and innovative electrolytes or membraneless MFCs based on new concepts and architectures are crucial for the development of future MFCs.
4.5. MFC design and architecture
The electrical performance of MFC systems are largely affected by the mutual interplay amongst the individual components listed above. Substantial improvements descend from cell design, architecture and assembly. Such advances usually occurred as a “gradual” evolution from the two chambers scheme in Fig. 3 taken here as the basic design. Air–cathode MFCs, for example, were developed step-wise from the basic scheme to optimize the cell design when oxygen is used as an oxidant. The basic rationale consists of exposing the cathode directly to air at least on one side (Fig. 7a) to eliminate limitations in oxygen supply to the electrode due to mass transport issues. Further refinements, mostly due to Logan and co-workers,12–15 resulted in a single-chamber design (Fig. 7b), where the cathode is bonded onto the electrolyte on one side, yielding a cell with much lower internal resistance than the basic, and a power output strongly dependent on the fuel substrate. From a historical perspective, such a technical solution is certainly not entirely innovative and neither was it invented with MFCs, since it has long been used in conventional FCs. | ||
Fig. 7 Evolutionary representation of air–cathode MFC from a two-chamber configuration in Fig. 3: (A) exposition of cathode to air at least on one side; (B) single chamber design with cathode mounted onto the electrolyte; (C) membrane-less design, (D) pump-less and chamber-less designs (A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anode; C:cathode; M:membrane). :anode; C:cathode; M:membrane). | ||
The next step saw the removal of the electrolyte leading to the membraneless air–cathode design in Fig. 7c, with high performance and much lower costs.50,67,112,113 In the absence of the membrane, though, a significant oxygen crossover to the anode chamber takes place and the cathode may be contaminated by microbes, which is accompanied with the reduction of Coulombic efficiency due to the consumption of a diffusing oxygen quota for bacterial growth on the cathode at the expense of substrate oxidation in the anode chamber. The bacterial colonization of the cathode may also undermine long-term operation. Biffinger et al.95 suggested a pump-less and chamber-less design (Fig. 7d), where nanoporous polymer membranes based on nylon, cellulose, or polycarbonate may inhibit this type of cathode fouling without sensible loss of power compared to Nafion. By using nanoporous membranes rather than Nafion they fabricated MFCs that could be deployed with anaerobic bacteria in aerobic environments, similar to membraneless MFCs where there is no Nafion and significant oxygen crossover to the anode occurs.
More recently, research efforts have been focusing on optimization of mass transport and fluid circulation. Some cell designs have been elaborated to leverage their importance, such as the ones in Fig. 8. Fig. 8a shows a scheme of “flux through the anode” MFC, derived from the membraneless air–cathode cell in Fig. 7c operated in continuous flow mode,11,114 where the fuel is forced across a porous anode towards the cathode. This configuration proved to increase the maximum PD of air–cathode systems both using glucose and wastewater as fuel. Nevertheless, serious issues, related to anode clogging (especially for unfiltered wastewater) and/or excessive biofilm growth, persist and have not been addressed yet.
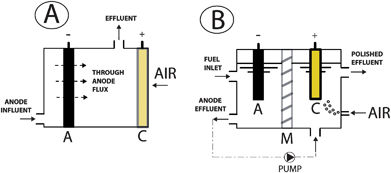 | ||
| Fig. 8 Sample stratagems related to mass transport: (A) fluid-flow through the anode; (B) anode effluent fed to cathode in a dual chamber MFC with sequential flow. | ||
The second example in Fig. 8b (unrelated to the air–cathode class) features a dual chamber MFC with sequential flow, where the anode effluent is fed to the aerated biocatalysed cathode for better oxidation (i.e. greater fuel consumption and quality of final effluent) and high electric efficiency.115 For example, an initial content of CH3COOH was almost totally consumed (>99%), passing from a concentration of 500 mg L−1 (pH 7.1) at the anode influent to 10–200 mg L−1 (pH 5.8–6.5) at cathode influent, and finally to <5 mg L−1 (pH 7.0–7.5) at the end outlet. From a microbial standpoint, the quasi-complete oxidation is a result of the cooperation between anaerobic bacteria at the anode and aerobic bacteria at the cathode (both of cathodophilic and heterophilic types). The large quota of fuel depleted in the cathode chamber does not contribute to the electrical power of the cell but is a boost in waste elimination performance, rendering sequential flow MFCs as promising candidates for waste treatment purposes. Coulombic efficiencies were also high (65–95%), due in part to the extra proton transfer mechanism provided by the mass transport from anode to cathode via the liquid stream, and in part to the stability of the catholyte pH unlike typical liquid separated cathodes. Notably, the cathodic biofilm outperformed non-catalysed graphite cathode in terms of current output, suggesting a sustainable alternative to Pt and other chemical catalysts. Nevertheless, several drawbacks exist, e.g. the strict control of loading rate necessary to avoid turning the cathode into a simple aerobic heterotrophic reactor.
We made implicit reference to planar electrodes to illustrate general ideas, but other types of architectures do exist. For example, the cited tubular MFCs, although less studied so far, offer several potential advantages over the planar counterparts, e.g. better scalability in continuous flow operations and greater air–cathode surface.116,117 Sedimentary MFCs are another important class—both in terms of fuel and overall layout—and are reviewed elsewhere.29,118
Independently of the selected architecture, a parametric optimization of the geometry can lead to major improvements and drive costs down. Researchers25,116,119 have looked into optimizing the relative surface areas of the anode–membrane–cathode assembly. Oh et al. found, for example, that power generation may depend on the surface areas of PEM membrane and cathode relative to that of the anode. More precisely, while the anode is rarely a problem, the PEM area became a limiting factor for power output when its value was smaller than that of the electrodes because the internal resistance increased too much. Some attention was also devoted to electrode spacing, which can be reduced to increase PD,11,46,116 but the effect of the relative spacing of the electrodes and the PEM has never been fully investigated yet. It is probably fair to state that parametric optimization of geometry would benefit from a more systematic research approach, say, based on statistical design of experiments. Publications in this area appear mostly like self-contained “proof-of-concept” studies, limited in scope and hard to link together. In prospective, better understanding of geometrical factors may heavily impact the upscaling and downscaling (miniaturization) of MFC technology.
Finally, in this brief outlook (by no means exhaustive), it is noteworthy of mentioning that MFC stacking represents a valid strategy to overcome limitations in voltage and power of single units. Oh and Logan120 demonstrated that cell stacking is a non trivial route and may result in voltage reversal. By analyzing a two-cell stack, they demonstrated that substrate concentration affects voltage generation and observed that voltage reversal may occur in a variety of situations (e.g. by temporary fuel starvation of one of the MFC units) but not when operating the stack in continuous mode and/or at lower current densities. However, problems related to stacking and voltage reversal are not associated just to MFCs but are well known in traditional FCs.
Conclusions
Even though MFC technology is not yet mature to deliver marketable products, the exponential trends in electrical performances, along with continued costs reduction and bioelectrical efficiency, should not be underestimated, especially in a context of growing environmental concerns. In the short term, MFC research seems geared towards systems running on sludge or wastewater to tackle two major problems of sustainable development at once, i.e. it delivers a low-cost waste treatment technology that simultaneously allows for energy production from alternative fuels. Space exploration may also stimulate MFC development due to a distinctive set of engineering requirements, more shifted towards reliability, durability, and usage of certain fuel substrates rather than electrical performance.As discussed here, advances in materials and engineering are greatly responsible for such developments. New materials are necessary to yield simplified electrodes which are able to replace the current (relatively) expensive ones. However, to render MFCs as an economical and reliable technology, more systematic and multidisciplinary research must be undertaken, e.g. by setting standards and protocols, by conducting statistically designed experiments, and (most of all) by increasing the degree of transversal interaction amongst diverse disciplines, such as microbiology, electrochemistry, chemical engineering, electrical engineering, materials science, and nanotechnology. The current lack of standards ranks high in the list of priorities, together with technical problems about scalability and costs of the technology. Finally, education may represent another exploitable front to effectively raise the general awareness of MFCs and attract researchers.
References
- L. Galvani, Ex Typographia Instituti Scientarium, Bologna, 1971 Search PubMed.
- M. C. Potter, Proc. R. Soc. London, Ser. B, 1912, 84, 260 CAS.
- B. Cohen, J. Bacteriol., 1931, 21, 18 CAS.
- J. H. Canfield, B. H. Goldner and R. Lutwack, NASA Technical Report, Magna Corporation, Anaheim, CA, 1963, 63 Search PubMed.
- C. F. Thurston, H. P. Bennetto, G. M. Delaney, J. R. Mason, S. D. Roller and J. L. Stirling, J. Gen. Microbiol., 1985, 131, 1393 CAS.
- R. M. Allen and H. P. Bennetto, Appl. Biochem. Biotechnol., 1993, 39/40, 27 Search PubMed.
- S. K. Chaudhuri and D. R. Lovley, Nat. Biotechnol., 2003, 21, 1229 CrossRef.
- B. E. Logan, Environ. Sci. Technol., 2004, 38, 160A CrossRef CAS.
- B. Min, J. R. Kim, S. E. Oh, J. M. Regan and B. E. Logan, Water Res., 2005, 39, 4961 CrossRef CAS.
- B. E. Logan and J. M. Regan, Trends Microbiol., 2006, 14, 512 CrossRef CAS.
- S. Cheng, H. Liu and B. E. Logan, Environ. Sci. Technol., 2006, 40, 2462 CrossRef.
- E. H. Yu, S. Cheng, K. Scott and B. Logan, J. Power Sources, 2007, 171, 275 CrossRef.
- B. Logan, S. Cheng, V. Watson and G. Estadt, Environ. Sci. Technol., 2007, 41, 3341 CrossRef CAS.
- S. Cheng and B. E. Logan, Electrochem. Commun., 2007, 9, 492 CrossRef.
- J. R. Kim, S. H. Jung, J. M. Regan and B. E. Logan, Bioresour. Technol., 2007, 98, 2568 CrossRef CAS.
- S. Venkata Mohan, S. Veer Raghavulu, S. Srikanth and P. N. Sarma, Curr. Sci., 2007, 92, 1720.
- J. Menicucci, H. Beyenal, E. Marsili, R. A. Demir and Z. Lewandowski, Environ. Sci. Technol., 2006, 40, 1062 CrossRef CAS.
- Y. Fan, H. Hu and H. Liu, Environ. Sci. Technol., 2007, 41, 8154 CrossRef CAS.
- K. Rabaey, N. Boon, S. D. Siciliano, M. Verhaege and W. Verstraete, Appl. Environ. Microbiol., 2004, 70, 5373 CrossRef CAS.
- K. Rabaey and W. Verstraete, Trends Biotechnol., 2005, 23, 291 CrossRef CAS.
- P. Aelterman, S. Freguia, J. Keller, W. Verstraete and K. Rabaey, Appl. Microbiol. Biotechnol., 2008, 78, 409 CrossRef CAS.
- J. C. Biffinger, J. Pietron, R. Ray, B. Little, R. Bradley and A. Ringeisen, Biosens. Bioelectron., 2007, 22, 1672 CrossRef CAS.
- R. A. Bullen, T. C. Arnot, J. B. Lakerman and F. C. Walsh, Biosens. Bioelectron., 2006, 21(11), 2015 CAS.
- César I. Torres, A. Kato Marcus and B. E. Rittmann, Biotechnol. Bioeng., 2008, 100, 872 CrossRef.
- S. Freguia, K. Rabaey, Z. Yuan and J. Keller, Electrochim. Acta, 2007, 53, 598 CrossRef CAS.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Vestraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181 CrossRef CAS.
- B. E. Logan and J. M. Regan, Environ. Sci. Technol., 2006, 1, 5172 CrossRef.
- Z. Du, H. Li and T. Gu, Biotechnol. Adv., 2007, 25, 464 CrossRef CAS.
- H. Rismani-Yazdi, S. M. Carver, A. D. Christy and O. H. Tuovinen, J. Power Sources, 2008, 180, 683 CrossRef CAS.
- D. R. Lovley, Curr. Opin. Biotechnol., 2006, 17, 327 CrossRef CAS.
- K. Rabaey, J. Rodríguez, L. L. Blackall, J. Keller, P. Gross, D. Batstone, W. Verstraete and K. H. Nealson, ISME J., 2007, 1, 9 CrossRef CAS.
- U. Schröder, Phys. Chem. Chem. Phys., 2007, 9, 2619 RSC.
- A. J. M. Stams, F. A. M. de Bok, C. M. Plugge, M. H. A. van Eekert, J. Dolfing and G. Schraa, Environ. Microbiol., 2006, 8, 371 CrossRef CAS.
- T. Narihiro and Y. Sekiguchi, Curr. Opin. Biotechnol., 2007, 18, 273 CrossRef CAS.
- L. T. Angenent, K. Karim, M. H. Al-Dahhan, B. A. Wrenn and R. Domíguez-Espinosa, Trends Biotechnol., 2004, 22, 477 CrossRef CAS.
- R. Hanno, L. Martin, K. P. Nevin and D. R. Lovley, Appl. Environ. Microbiol., 2007, 73(16), 5347 CrossRef.
- J. A. Gralnick and D. K. Newman, Mol. Microbiol., 2007, 65, 1 CrossRef.
- M. E. Hernandez and D. K. Newman, Cell. Mol. Life Sci., 2001, 58, 1562 CrossRef CAS.
- S. Jung and J. M. Regan, Appl. Microbiol. Biotechnol., 2007, 77, 393 CrossRef CAS.
- T. H. Pham, N. Boon, P. Aelterman, Cluwaert, L. De Schamphelaire, L. Vanhaecke, K. De Maeyer, M. Höfte, W. Verstraete and K. Rabaey, Appl. Microbiol. Biotechnol., 2008, 77, 1119 CrossRef CAS.
- P. Aelterman, K. Rabaey, HT. H. Pham, N. Boon and W. Verstraete, Environ. Sci. Technol., 2006, 40, 3388 CrossRef CAS.
- B. E. Logan, Microbial Fuel Cells, John Wiley & Sons, New York, NY, 2008 Search PubMed.
- C. Dumas, A. Mollica, D. Féron, R. Basséguy, L. Etcheverry and A. Bergel, Electrochim. Acta, 2007, 53, 468 CrossRef CAS.
- K. Rabaey, G. Lissens, S. D. Siciliano and W. Verstraete, Biotechnol. Lett., 2003, 25, 1531 CrossRef CAS.
- H. Liu, S. Cheng and B. E. Logan, Environ. Sci. Technol., 2005, 39, 658 CrossRef CAS.
- K. Rabaey, P. Clauwaert, P. Aelterman and W. Verstraete, Environ. Sci. Technol., 2005, 39, 8077 CrossRef CAS.
- Z. He, S. D. Minteer and L. T. Angenent, Environ. Sci. Technol., 2005, 39, 5262 CrossRef CAS.
- Z. He, N. Wagner, S. D. Minteer and L. T. Angenent, Environ. Sci. Technol., 2006, 40, 5212 CrossRef CAS.
- D. H. Park and J. G. Zeikus, Biotechnol. Bioeng., 2003, 81, 348 CrossRef CAS.
- U. Schröder, J. Niessen and F. Scholz, Angew. Chem., Int. Ed., 2003, 42, 2880 CrossRef.
- J. Niessen, U. Schröder, M. Rosenbaum and F. Scholz, Electrochem. Commun., 2004, 6, 571 CrossRef CAS.
- D. A. Lowy, L. M. Tender, J. G. Zeikus, D. H. Park and D. R. Lovley, Biosens. Bioelectron., 2006, 21, 2058 CrossRef CAS.
- H. I. Park, U. Mushtaq, D. Perello, I. Lee, S. K. Cho, A. Star and M. Yun, Energy Fuels, 2007, 21, 2984 CrossRef CAS.
- M. Rosenbaum, F. Zhao, U. Schröder and F. Scholz, Angew. Chem., Int. Ed., 2006, 45, 6658 CrossRef CAS.
- M. Rosenbaum, F. Zhao, M. Quaas, H. Wulff, U. Schröder and F. Scholz, Appl. Catal., B, 2007, 74, 262.
- K. Scott, G. A. Rimbu, K. P. Katuri, K. K. Prasad and I. M. Head, Trans IChemE, Part B, Process Saf. Environ. Prot., 2007, 85, 481 Search PubMed.
- J. L. Liu, D. A. Lowy, R. G. Baumann and L. M. Tender, J. Appl. Microbiol., 2007, 102, 177 CrossRef CAS.
- Y. Qiao, C. M. Li, S.-J. Bao and Q.-L. Bao, J. Power Sources, 2007, 170, 79 CrossRef CAS.
- A. Morozan, I. Stamatin, L. Stamatin, A. Dumitru and K. Scott, J. Optoelectron. Adv. Mater., 2007, 9, 221 Search PubMed.
- T. Zhang, C. Z. Cui, S. L. Chen, X. P. Ai, H. X. Yang, P. Shen and Z. R. Peng, Chem. Commun., 2006, 21, 2257 RSC.
- T. Zhang, Y. Zeng, S. Chen, X. Ai and H. Yang, Electrochem. Commun., 2007, 9, 349 CrossRef CAS.
- D. A. Finkelstein, L. M. Tender and J. G. Ziekus, Environ. Sci. Technol., 2006, 40, 6990 CrossRef CAS.
- Y. Fan, H. Q. Hu and H. Liu, J. Power Sources, 2007, 171, 348 CrossRef CAS.
- S. Freguia, K. Rabaey, Z. Yuan and J. Keller, Environ. Sci. Technol., 2007, 53, 598 CAS.
- A. K. Shukla, P. Suresh, S. Berchmans and A. Rajendran, Curr. Sci., 2004, 41, 2915.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220 CrossRef CAS.
- H. Liu and B. E. Logan, Environ. Sci. Technol., 2004, 38, 4040 CrossRef CAS.
- S. E. Oh, B. Min and B. E. Logan, Environ. Sci. Technol., 2004, 38, 4900 CrossRef CAS.
- Z. Qi and A. Kaufman, J. Power Sources, 2003, 113, 37 CrossRef CAS.
- S. L. J. Gojkovic, T. R. Vidakovic and D. R. Durovic, Electrochim. Acta, 2003, 48, 3607 CrossRef CAS.
- F. Zhao, F. Hamisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Hermann, Environ. Sci. Technol., 2006, 40, 5193 CrossRef CAS.
- J. L. Fernandez, V. Raghuveer, A. Manthiram and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 13100 CrossRef CAS.
- N. A. Alonso-Vante and H. Tributsch, Nature, 1986, 323, 431 CAS.
- J. M. Morris, S. Jin, J. Wang, C. Zhu and M. A. Urynowicz, Electrochem. Commun., 2007, 9, 1730 CrossRef CAS.
- S. Venkata Mohan, R. Saravanan, S. Veer Raghavulu, G. Mohanakrishna and P. N. Sarma, Bioresour. Technol., 2008, 99, 596 CrossRef CAS.
- S. J. You, Q. L. Zhao, J. N. Zhang, J. Q. Jiang and S. Q. Zhao, J. Power Sources, 2006, 162, 1409 CrossRef CAS.
- B. E. Logan and J. M. Regan, Environ. Sci. Technol., 2006, 40, 5172 CAS.
- M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2000, 104, 11238 CrossRef CAS.
- F. Zhao, F. Harnisch, U. Schroder, F. Scholz, P. Bogdanoff and I. Herrmann, Electrochem. Commun., 2005, 7, 1405 CrossRef CAS.
- O. Hasvold, H. Henriksen, E. Melvaer, G. Citi, B. O. Johansen, T. Kjonigsen and R. Galetti, J. Power Sources, 1997, 65, 253 CrossRef CAS.
- D. E. Holmes, D. R. Bond, R. A. OLNeill, C. E. Reimers, L. R. Tender and D. R. Lovley, Microb. Ecol., 2004, 48, 178 CrossRef CAS.
- Z. He and L. T. Angenent, Electroanalysis, 2006, 18, 2009 CrossRef CAS.
- K. B. Gregory, D. R. Bond and D. R. Lovley, Environ. Microbiol., 2004, 6, 596 CrossRef CAS.
- P. Clauwaert, K. Rabaey, P. Aelterman, L. DeSchamphelaire, T. H. Pham, P. Boeckx, N. Boon and W. Verstraete, Environ. Sci. Technol., 2007, 41, 3354 CrossRef CAS.
- A. Bergel, D. Feron and A. Mollica, Electrochem. Commun., 2005, 7, 900 CrossRef CAS.
- A. Rhoads, H. Beyenal and Z. Lewandowski, Environ. Sci. Technol., 2005, 39, 4666 CrossRef CAS.
- P. Clauwaert, D. Van der Ha, N. Boom, K. Verbeken, M. Verhaege, K. Rabaey and W. Verstraete, Environ. Sci. Technol., 2007, 41, 7564 CrossRef CAS.
- K. Rabaey, S. T. Read, P. Clauwaert, S. Freguia, P. L. Bond, L. L. Blackall and J. Keller, ISME J., 2008, 1.
- B. D. McNicol, D. A. J. Rand and K. R. Williams, J. Power Sources, 2001, 100, 47 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef CAS.
- H. J. Kim, H. S. Park, M. S. Hyun, I. S. Chang, M. Kim and B. H. Kim, Enzyme Microb. Technol., 2002, 30, 145 CrossRef CAS.
- B. H. Kim, H. J. Kim, M. S. Hyun and D. H. Park, J. Microbiol. Biotechnol., 1999, 9, 127 CAS.
- B. Min, S. Cheng and B. E. Logan, Water Res., 2005, 39, 1675 CrossRef CAS.
- J. R. Kim, S.-E. S.ChengOh and B. E. Logan, Environ. Sci. Technol., 2007, 41, 1004 CrossRef CAS.
- J. C. Biffinger, R. Ray, B. Little and B. R. Ringeisen, Environ. Sci. Technol., 2007, 41, 1444 CrossRef CAS.
- M. Grzebyk and G. Pozniak, Sep. Purif. Technol., 2005, 41, 312.
- V. I. Basura, P. D. Beattie and S. Holdcroft, J. Electroanal. Chem., 1998, 458, 1 CrossRef CAS.
- J. Rozière and D. J. Jones, Annu. Rev. Mater. Res., 2003, 33, 503 CrossRef CAS.
- B. Mecheri, A. D'Epifanio, M. L. Di Vona, E. Traversa, S. Licoccia and M. Miyayama, J. Electrochem. Soc., 2006, 153, A463 CrossRef CAS.
- T. D. Gierke, G. E. Munn and F. C. J. Wilson, J. Polym. Sci., Polym. Phys. Ed., 1981, 19, 1687 CrossRef.
- V. S. Silva, B. Ruffmann, S. Vetter, M. Boaventura, A. M. Mendes, L. M. Madeira and S. P. Nunes, Electrochim. Acta, 2006, 51, 3699 CrossRef CAS.
- V. S. Silva, B. Ruffmann, H. Silva, Y. A. Gallego, A. Mendes, L. M. Madeira and S. P. Nunes, J. Power Sources, 2005, 140, 34 CrossRef CAS.
- S. Licoccia, M. L. Di Vona, A. D'Epifanio, Z. Ahmed, S. Bellitto, D. Marani, B. Mecheri, C. de Bonis, M. Trombetta and E. Traversa, J. Power Sources, 2007, 167, 79 CrossRef CAS.
- B. Mecheri, A. D'Epifanio, E. Traversa and S. Licoccia, J. Power Sources, 2008, 178, 554 CrossRef CAS.
- Z. Samec, A. Trojanek, J. Langmaier and E. Samcova, J. Electrochem. Soc., 1997, 144, 4236 CrossRef CAS.
- C. G. Gil, I. S. Chang, B. H. Kim, M. Kim, J. K. Jang and H. S. Park, Biosens. Bioelectron., 2003, 18, 327 CrossRef CAS.
- T. Okada, G. Xie, O. Gorseth, S. Kjelstrup, N. Nakamura and T. Arimura, Electrochim. Acta, 1998, 43, 3741 CrossRef CAS.
- K.-D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637 CrossRef CAS.
- E. Traversa, Sens. Actuators, B, 1995, 23, 135–156 CrossRef.
- R. A. Rozendal, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5206 CrossRef CAS.
- A. ter Heijne, H. V. M. Hamelers, V. de Wilde, R. A. Rozendal and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5200 CrossRef CAS.
- C. E. Reimers, L. M. Tender, S. Ferig and W. Wang, Environ. Sci. Technol., 2001, 2001(35), 192 CrossRef.
- D. R. Bond, D. E. Holmes, L. M. Tender and D. R. Lovley, Science, 2002, 295, 48.
- D. Sell, P. Kramer and G. Kreysa, Appl. Microbiol. Biotechnol., 1989, 31, 211 CAS.
- S. Freguia, K. Rabaey, Z. Yuan and J. Keller, Water Res., 2008, 42, 1387 CrossRef CAS.
- Y. Zuo, S. Cheng, D. Call and B. E. Logan, Environ. Sci. Technol., 2007, 41, 3347 CrossRef CAS.
- K. Rabaey, P. Clauwaert, P. Aelterman and W. Vestraete, Environ. Sci. Technol., 2005, 39, 8077 CrossRef CAS.
- F. Rezaei, T. L. Richard, R. A. Brennan and B. E. Logan, Environ. Sci. Technol., 2007, 41, 4053 CrossRef CAS.
- S.-E. Oh and B. E. Logan, Appl. Microbiol. Biotechnol., 2006, 70, 162 CrossRef CAS.
- S.-E. Oh and B. E. Logan, J. Power Sources, 2007, 167, 11 CrossRef CAS.
Footnote |
| † These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2008 |