DOI:
10.1039/B813501K
(Paper)
Dalton Trans., 2008, 6953-6965
Impact of the linker groups in bis(7-azaindol-1-yl) chelate ligands on structures and stability of Pt(N,N-L)R2 complexes†
Received
4th August 2008
, Accepted 24th September 2008
First published on 3rd November 2008
Abstract
New organoplatinum(II) complexes with the general formula of Pt(N,N-L)R2, where L is a bis(7-azaindol-1-yl) chelate ligand, R is a methyl or a phenyl, have been synthesized and investigated with the aim to understand the impact of the linker group in the L ligand on the structure and the stability of the Pt(II) complexes. The L ligands used in our investigation belong to two classes: class I with an aliphatic linker between two 7-azaindolyl groups such as -CH2- (BAM), -(CH2)3- (1,3-BAPr), and -(CH2)4- (1,4-BABu), class II with an aromatic linker such as 1,2-phenyl (1,2-BAB) 1,3-phenyl (1,3-BAB) and 1,4-dihydronaphthalene-1,4-epoxide (BAHE). The structures of these new mononuclear Pt(II) complexes have been determined by single-crystal X-ray diffraction analyses. For the 1,3-BAPr ligand, a dinuclear complex with the formula of [Pt(SMe2)Ph2]2(1,3-BAPr) was also obtained and its structure was established by X-ray diffraction analysis. The linker group's impact on the N–Pt–N bite angle and the relative orientation of the two 7-azaindolyl rings was examined by using the crystal structural data. Intramolecular three-center four-electron PtII⋯H–C interactions have been established for all Pt(II) complexes with class I ligands by single-crystal X-ray diffraction and 1H NMR spectroscopic analyses. Complexes Pt(1,3-BAPr)Me2 and Pt(1,4-BABu)Me2 have been found to have a poor stability, compared to the 1,2-BAB and BAM analogues. Two geometric isomers of the Pt(BAHE)Ph2 complex have been identified by NMR spectra and the structure of one of the isomers has been determined by X-ray diffraction analysis, establishing that the BAHE ligand is most effective in blocking the 5th coordination site of the Pt(II) center.
Introduction
Since the demonstration of the Shilov chemistry,1 the Pt(II) mediated electrophilic CH4activation and the subsequent oxidation process have attracted much research interest due to their relevance in direct hydrocarbon functionalization.2 Extensive investigations on the key steps involved in the Shilov system have been conducted using a variety of Pt(II) model systems, which often contain chelating ligands such as diamine,3diimine,4,5β-diiminate,6tri(pyrazolyl)borate,7di(pyrid-2-yl)borate,8dipyridyl,9 and bis(diphenylphosphino)10 derivatives, and mechanistic details on the role of the Pt(II) center have been obtained. Despite significant advances in the mechanistic study,2,11 previously established Pt(II) systems are still not adequate for practical C–H activation and functionalization. Ligands have been elucidated to play a crucial role on the property and performance of a Pt(II) center in C–H activation. Both ligand steric and electronic properties have been shown to have a significant impact on the selectivity and reactivity of the C–H activation and functionalization process by the Pt(II) based systems, as demonstrated previously by a number of research groups.3–11
Recently, we have demonstrated several Pt(II) complexes with 7-azaindolyl derivative N,N-chelating ligands (N,N-L) that are very effective for arene C–H activation, including Pt(1,2-BAB)Me2 and Pt(BAM)Me2 (Chart 1), where 1,2-BAB = 1,2-bis(7-azaindol-1-yl)benzene and BAM = bis(7-azaindol-1-yl)methane (Chart 1), and Pt2(TTAB)Me4, where TTAB = 1,2,4,5-tetrakis(7-azaindol-1-yl)benzene.12,13 The 7-azaindolyl derivative ligands in these Pt(II) complexes were found to be able to partially block the 5th coordination site of the Pt(II) center, thus allowing not only the stabilization and isolation of rare Pt(II)12c and Pt(IV) species,12d but also regio- and diastereoselective ethylbenzene C–H activation.12c The different steric blocking effects imposed by BAM and 1,2-BAB ligands have been found to cause distinct regio- and diastereoselectivity by the Pt(1,2-BAB)Me2 and Pt(BAM)Me2 complexes in ethylbenzene C–H activation.12d In addition, unusual and strong intramolecular PtII⋯H–C interactions were also observed in the BAM Pt(II) complex.12 Recently, Puddephatt and coworkers10d–f have demonstrated the effect of the bite angle of N,N -chelating ligands on the reactivity of the PtMe2(N,N-L) complexes, by comparing complexes of di-2-pyridylamine or di-2-pyridyl ketone. We have shown recently that the chelate ring strain in a Pt(II) complex, Pt(N,N-NPA)Me2, can lead to a spontaneous roll-over intramolecular C–H activation, resulting in the formation of a self-assembled macrocycle Pt4Me4(N,C-NPA)4.14 These previous findings consistently support that the chelate ligand plays an important role in Pt(II) C–H activation chemistry, which stimulate our continued interest to further investigate the Pt(II) chemistry involving 7-azaindolyl derivative ligands. To examine the aliphatic linker effect, we have expanded the BAM ligand system to the 1,3-bis(7-azaindolyl)propane (BAPr) and 1,4-bis(7-azaindoly)butane (BABu) ligand systems. To compare with the related but rigid aromatic linker, we have synthesized 1,3-bis(7-azaindolyl)benzene (1,3-BAB). To further enhance the steric blocking effect, we have also synthesized 1,4-dihydronaphthalene-1,4-epoxide (BAHE). These new ligand systems allow us to conduct a systematic examination on the effect of the linker on the structure, the stability and ultimately the reactivity of Pt(N,N-L)R2 complexes based on bis(7-azaindolyl) chelating ligands. This report focuses on the syntheses, structures and stabilities of the Pt(II) complexes based on the new ligand systems. Two previously undisclosed complexes Pt(BAM)Ph2 and Pt(1,2-BAB)Ph2 are also included in this report for comparison purpose.
 |
| | Chart 1 | |
Experimental
All reactions were performed under N2 with the standard Schlenk techniques unless otherwise noted. All starting materials were purchased from Aldrich Chemical Co. and used without further purification. THF, Et2O, and hexanes were purified using the solvent purification system (Innovation Technology, Inc.). Deuterated solvents (Cambridge Isotopes) were used as received without further drying. NMR spectra were recorded on Bruker Avance 300, 400 or 500 MHz spectrometers. 1H and 13C NMR chemical shifts were referenced to the residual solvent peaks and have been reported in parts per million (ppm). High resolution mass spectra (HRMS) were obtained from a Waters/Micromass GC-TOF EI-MS spectrometer. [PtPh2(μ-SMe2)]n (n = 2 or 3)15a and [PtMe2(μ-SMe2)]215b were prepared by methods described in the literature. Bis(7-azaindol-1-yl)methane,16a1,2-bis(7-azaindol-1-yl)benzene (1,2-BAB)12b and 1,3-bis(7-azaindol-1-yl)benzene (1,3-BAB)16b were synthesized by our previously reported procedure.
Synthesis of 1,3-bis(7-azaindol-1-yl)propane (1,3-BAPr)
To a solid mixture of 7-azaindole (1.56 g, 13.3 mmol) and sodium hydride (60% dispersed in mineral oil) (0.62 g, 15.5 mmol) in a Schlenck flask was added quickly 25 mL of dry DMF. The resulting clear solution was stirred for 30 minutes at ambient temperature, and 1,3-dibromopropane (1.98 g, 9.8 mmol) was then added. The resulting brown suspension was stirred for 2 hours at ambient temperature and then heated at 110 °C for 18 hours. DMF was then removed by vacuum distillation and the residue was extracted by CH2Cl2 (50 mL × 3). After the removal of the solvent, the residue was purified by flash chromatograph on silica gel using hexanes/ethyl acetate (1/1) as the eluent. The 1,3-BAPr ligand was obtained as an colorless oil (0.84 g, 46% yield), which was solidified after the addition of hexanes and standing in a refrigerator for several days. 1H NMR for 1,3-BAPr (500 Hz, CDCl3, 25 °C): δ 8.35 (d; 3J = 5.0 Hz; 2H, 7-aza), 7.96 (d; 3J = 7.5 Hz; 2H, 7-aza), 7.34 (d; 3J = 3.5 Hz; 2H, 7-aza), 7.11 (dd; 3J1 = 5.0 Hz, 3J2 = 7.5 Hz; 2H, 7-aza), 6.49 (d; 3J = 3.5 Hz; 2H, 7-aza), 4.41 (t; 3J = 6.5 Hz; 4H, CH2), 1.91 (m; 3J = 6.5 Hz; 3H, CH2) ppm. 13C NMR: δ 148.21, 143.42, 129.48, 128.84, 121.40, 116.41, 100.35, 42.79, 31.69 ppm. Anal. calcd. for C17H16N4: C 73.89, H 5.84, N 20.27; found: C 73.83, H 5.99, N 20.22. The side product 1-allyl-7-azaindole was obtained as colorless oil (0.35 g, 17% yield). 1H NMR for 1-allyl-7-azaindole (300 Hz, CDCl3, 25 °C): δ 8.35 (d; 3J = 4.8 Hz; 1H, 7-aza), 7.94 (d; 3J = 7.8 Hz; 1H, 7-aza), 7.23 (d; 3J = 3.3 Hz; 1H, 7-aza), 7.08 (dd; 3J1 = 4.8 Hz, 3J2 = 7.8 Hz; 1H, 7-aza), 6.49 (d; 3J = 3.3 Hz; 1H, 7-aza), 6.15 (m; 1H, CH2CH=CHH′), 5.21 (d; 3J = 10.2 Hz; 1H, CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHH′), 5.10 (d; 3J = 17.1 Hz; 1H, CH2CHCHH′), 4.94 (m; 2H, CH2CHCHH′) ppm.
CHH′), 5.10 (d; 3J = 17.1 Hz; 1H, CH2CHCHH′), 4.94 (m; 2H, CH2CHCHH′) ppm.
Synthesis of 1,4-bis(7-azaindol-1-yl)butane (1,4-BABu)
To a solid mixture of 7-azaindole (0.75 g, 6.4 mmol) and sodium hydride (60% dispersed in mineral oil) (0.28 g, 7.0 mmol) in a Schlenck flask was added quickly 15 mL of dry DMF. The resulting clear solution was stirred for 30 minutes at ambient temperature, and 1,4-dibromobutane (0.69 g, 3.2 mmol) was then added. The resulting brown suspension was stirred for 2 hours at ambient temperature and then heated at 110 °C for 18 hours. DMF was then removed by vacuum distillation and the yellow residue was extracted by CH2Cl2 (30 mL × 3). After the removal of the solvent, the residue was purified by flash chromatograph on silica gel using hexanes/ethyl acetate (2/1) as the eluent to afford 1,4-BABu as a white solid (0.66 g, 71% yield). 1H NMR (500 Hz, CDCl3, 25 °C): δ 8.30 (dd; 3J = 5.0 Hz, 4J = 1.5 Hz; 2H, 7-aza), 7.92 (dd; 3J = 8.0 Hz, 4J = 1.0 Hz; 2H, 7-aza), 7.14 (d; 3J = 3.5 Hz; 2H, 7-aza), 7.06 (dd; 3J1 = 5.0 Hz, 3J2 = 8.0 Hz; 2H, 7-aza), 6.46 (d; 3J = 3.5 Hz; 2H, 7-aza), 4.35 (m; 4H, CH2), 1.91 (m; 4H, CH2) ppm. 13C NMR: δ 148.09, 143.37, 129.44, 128.56, 121.24, 116.29, 100.15, 44.65, 28.38 ppm. Anal. calcd. for C18H18N4: C 74.46, H 6.25, N 19.30; found: C 74.38, H 6.45, N 19.45.
Synthesis of 6,7-bis(7-azaindol-1-yl)-1,4-dihydronaphthalene-1,4-epoxide (BAHE)
6,7-Dibromo-1,4-dihydronaphthalene-1,4-epoxide (0.77 g, 2.5 mmol), 7-azaindole (0.75 g, 6.4 mmol), CuI (0.068 g, 0.35 mmol), 1,10-phenanthroline (0.15 g, 0.70 mmol), Cs2CO3 (4.10 g, 12.6 mmol) were mixed together in a Schlenck flask. The flask was degassed under high vacuum and refilled with N2. 4.0 mL of dry DMF was then added. The mixture was heated at 145–150 °C for 3 days. After cooling down to ambient temperature, the mixture was diluted with 100 mL of CH2Cl2 and filtered through a plug of silica gel. The filtrate was then concentrated and the residue was purified by flash chromatograph on silica gel using hexanes/ethylacetate (2:1) as the eluent to BAHE as a yellow residue (0.48 g, 50% yield). Light yellow crystals of BAHE were obtained from the recrystallization of the residue with THF/hexanes. 1H NMR (400 Hz, CDCl3, 25 °C): 8.25 δ (dd; 4J = 1.2 Hz, 3J = 4.8 Hz; 2H, 7-aza), 7.84 (dd; 4J = 1.2 Hz, 3J2 = 8.0 Hz; 2H, 7-aza), 7.64 (s; 2H), 7.14 (s; 2H), 7.08 (dd; 3J1 = 4.8 Hz, 3J2 = 8.0 Hz; 2H, 7-aza), 6.78 (d, br; 3J = 3.6 Hz; 2H, 7-aza), 6.24 (d; 3J = 3.6 Hz; 2H, 7-aza), 5.85 (s; 2H) ppm. 13C NMR: δ 149.94, 147.88, 143.27, 143.17, 130.98, 129.55, 129.32, 121.37, 121.18, 116.85, 101.92, 82.56 ppm. HRMS calcd. for C24H16N4O: 376.1324, found: 376.1361.
Besides the BAHE ligand, two side products 6-(7-azaindol-1-yl)-1,4-dihydronaphthalene- 1,4-epoxide (NAHE) (∼13% yield) and 2,3,6-tris(7-azaindol-1-yl)naphthalene (TAN) (∼8% yield) from the reaction. The characterization data of NAHE and TAN are shown below.
For NAHE, 1H NMR (400 Hz, CDCl3, 25 °C): δ 8.39 (br; 1H, 7-aza), 7.99 (d; 3J = 8.5 Hz, 1H, 7-aza), 7.71 (s; 1H), 7.48 (d; 3J = 5.5 Hz; 1H, 7-aza), 7.38 (d; 3J = 12.5Hz; 1H), 7.31 (d; 3J = 12.5 Hz; 1H), 7.16 (m; 1H, 7-aza), 7.09 (br; 2H); 6.64 (d; 3J = 5.5 Hz; 1H, 7-aza), 5.81 (s; 1H), 5.80 (s; 1H) ppm. HRMS calcd. for C17H12N2O: 260.0950; found: 260.0968.
For TAN, 1H NMR (400 Hz, CDCl3, 25 °C): δ 8.46 (dd; 3J = 4.8 Hz, 4J = 1.6 Hz; 1H,7-aza), 4.42 (d; 3J = 1.6 Hz; 1H), 8.29 (s; 1H), 8.28 (s; 1H), 8.26–8.16 (m; 3H), 8.10 (d; 3J = 8.8 Hz; 1H), 8.06 (dd; 3J = 8.0 Hz, 4J = 1.6 Hz; 1H, 7-aza), 7.88 (m; 1H, 7-aza), 7.86 (m; 1H, 7-aza), 7.76 (d; 3J = 3.6 Hz, 1H, 7-aza), 7.21 (dd; 3J1 = 4.8 Hz, 3J2 = 8.0 Hz; 7-aza), 7.15–7.06 (m; 2H), 6.98 (d; 3J= 3.6 Hz; 1H, 7-aza), 6.94 (d; 3J= 3.6 Hz; 1H, 7-aza), 6.74 (d; 3J= 3.6 Hz; 1H, 7-aza), 6.37 (d; 3J= 3.6 Hz; 1H, 7-aza), 6.34 (d; 3J= 3.6 Hz; 1H, 7-aza) ppm. 13C NMR: δ 148.83, 148.78, 148.09, 143.88, 143.77, 143.40, 142.99, 137.77, 133.50, 133.17, 132.52, 130.70, 129.66, 129.43, 129.19, 129.11, 129.02, 128.89, 128.27, 128.17, 127.91, 127.07, 125.44, 124.09, 122.55, 122.28, 121.64, 120.80, 120.76, 120.60, 118.78, 117.40, 116.89, 116.86, 116.09, 102.80, 101.65, 101.37, 100.79 ppm. HRMS calcd. for C31H26N6: 476.1749; found: 476.1758.
Synthesis of Pt(BAM)Ph2 (1)
BAM (0.124 g, 0.50 mmol) and [PtPh2(μ-SMe2)]n (n = 2 or 3) (0.206 g, 0.50 mmol based on Pt) were mixed in Et2O (20 mL) and stirred for 5 hours. The resulting white precipitate was allowed to settle and the clear solution was decanted. The solid was washed with Et2O and dried under vacuum. Recrystallization from THF afforded 1 in 85% yield. 1H NMR (400 Hz, CD2Cl2, 25 °C): δ 11.91 (d, satellite; 2J = 14.4 Hz, JPt–H = ∼11 Hz; 1H, CH2), 8.73 (dd, satellite; 4J = 1.5 Hz, 3J = 5.7 Hz, 3JPt–H = 24.6 Hz; 2H, 7-aza), 7.92 (dd; 4J = 1.2 Hz, 3J = 7.8 Hz; 2H, 7-aza), 7.56 (d; 3J = 3.6 Hz; 2H, 7-aza), 7.43 (dd, satellite; 4J = 1.2 Hz, 3J = 7.8 Hz, 3JPt–H = 72 Hz; 4H, Phenyl), 7.01 (dd; 3J1 = 5.4 Hz, 3J2 = 7.8 Hz; 2H, aza), 6.80 (m; 4H, Phenyl), 6.70 (m; 2H, Phenyl), 6.63 (d; 3J = 3.6 Hz; 2H, 7-aza), 6.47 (d, satellites; 2J = 14.4 Hz, JPt–H = ∼11 Hz; 1H, CH2) ppm. 13C NMR: δ 146.5, 144.7, 142.9, 138.2, 130.5, 129.1, 126.7, 124.4, 121.8, 117.5, 103.1, 68.2 ppm. Anal. calcd. for C27H22N4Pt·0.5THF: C 54.97, H 4.14, N 8.84; found: C 54.68, H 4.38, N 8.38.
Synthesis of Pt(1,2-BAB)Ph2 (2)
1,2-BAB (0.052 g, 0.17 mmol) and [PtPh2(μ-SMe2)]n (n = 2 or 3) (0.069 g, 0.17 mmol based on Pt) were mixed in 15 mL of Et2O and stirred for 2 hours. The resulting white precipitate was allowed to settle and the clear solution was decanted. The solid was washed with Et2O and dried under vacuum. Recrystallization from Et2O yielded light yellow crystals of 2 (73% yield). 1H NMR (300 Hz, CD2Cl2, 25 °C): δ 8.70 (dd, satellites; 3J = 5.4 Hz, 4J = 1.5 Hz; 2H; 7-aza), 7.88 (m; 4H, 7-aza and phenyl of 1,2-BAB), 7.59 (m; 2H, phenyl of 1,2-BAB), 7.20 (d zH; 3J = 3.9 Hz; 2H, 7-aza), 7.00 (m zH, satellite; JPt–H = 74 Hz; 6H, 7-azain and phenyl), 6.67 (m zH; 6H, phenyl), 6.56 (d; 3J = 3.9 Hz; 2H, 7-aza) ppm. 13C NMR: δ 148.6, 145.6, 141.7, 138.6, 138.0, 131.5, 131.2, 130.9, 130.3, 126.2, 123.7, 121.4, 117.6, 103.0 ppm. Anal. calcd. for C32H24N4Pt: C 58.27, H 3.64, N 8.50; found: C 57.88, H 3.94, N 8.33.
Synthesis of Pt(1,3-BAPr)Ph2 (3)
1,3-BAPr (0.055 g, 0.20 mmol) and [PtPh2(μ-SMe2)]n (n = 2 or 3) (0.082 g, 0.20 mmol based on Pt) were mixed in CH2Cl2 (15 mL) and stirred overnight at ambient temperature. The solvent was decanted, and the resulting yellow solid was then washed with Et2O (2 mL × 3). Yellow crystals of 3 were obtained from the slow evaporation of its methylene chloride solution (∼70% yield). 1H NMR (400 Hz, CD2Cl2, 25 °C): δ 8.76 (dd; 3J = 4.4 Hz, 4J = 1.6 Hz; 2H, 7-aza), 8.19 (m; 2J = 12 Hz; 2H, Naza-CHaHbCHaHbCHaHb-Naza), 7.63 (dd; 3J = 8.0 Hz, 4J = 1.2 Hz; 2H, 7-aza), 7.56 (d, satellite; 3J = 8.0 Hz, 3JPt–H = 70 Hz; 2H, ortho-H of PtPh2), 7.13 (d; 3J = 3.6 Hz, 7-aza), 6.95–6.72 (m; 8H, 2H from 7-aza & 2H from para-H of PtPh2 & 4H from meta-H of PtPh2), 6.27 (d; 3J = 3.6 Hz, 7-aza), 4.20 (m; 2H, Naza-CHaHbCHaHbCHaHb-Naza), 2.83–2.71 (m; 2H, Naza-CHaHbCHaHbCHaHb-Naza) ppm. 13C NMR: δ 146.722, 144.37, 139.18, 138.38 (satellite; JPt–C = 33.7 Hz), 129.59, 129.25, 126.60 (satellite; JPt–C = 81.7 Hz), 123.14, 121.76, 115.84, 101.51, 44.24, 31.70 ppm. Anal. calcd. for C29H26N4Pt: C 55.67, H 4.19, N 8.96; found: C 56.01, H 4.28, N 8.64.
Synthesis of Pt(1,4-BABu)Ph2 (4)
1,4-BABu (0.058 g, 0.20 mmol) and [PtPh2(μ-SMe2)]n (n = 2 or 3) (0.082 g, 0.20 mmol based on Pt) were mixed in CH2Cl2 (15 mL) and stirred overnight at ambient temperature. The solvent was decanted and the resulting yellow solid was then washed with Et2O (2 mL × 3). Yellow crystals of 4 were obtained from the slow evaporation of its methylene chloride/toluene (3:1) solution at room temperature (70% yield). 1H NMR (400 Hz, CD2Cl2, 25 °C): δ 9.10 (dd; 3J = 5.2 Hz, 4J = 1.2 Hz; 2H, 7-aza), 8.45 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza), 7. 90 (dd; 3J = 6.0 Hz, 4J = 1.2 Hz; 2H, 7-aza), 7.31–7.16 (m, satellite; 3JPt–H = 76 Hz; 4H from ortho-H of PtPh2 and 2H from 7-aza); 7.11 (dd; 3J1 = 5.2 Hz, 3J2 = 6.0 Hz; 2H, 7-aza), 6.75–6.66 (m; 6H, para- and meta-H of PtPh2), 6.52 (d; 3J = 3.6 Hz; 2H, 7-aza), 4.15–4.01 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza), 2.10–1.98 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza), 1.63–1.55 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza) ppm. 13C NMR: δ 145.26, 139.66, 138.11 (satellite; JPt–C = 35.2 Hz), 130.20, 129.80, 126.21 (satellite; JPt–C = 81.4 Hz), 124.25, 121.50, 116.12, 101.28, 43.26, 27.64 ppm. Anal. calcd. for C30H28N4Pt: C 56.33, H 4.41, N 8.76; found: C 56.41, H 4.37, N 8.28.
Synthesis of Pt2(1,3-BAPr)Ph4(SMe2)2 (5)
Complex 5 was obtained using the same method as described for 3 using 1,3-BAPr (0.028 g, 0.10 mmol) and [PtPh2(μ-SMe2)]n (n = 2 or 3) (0.082 g, 0.20 mmol based on Pt) in CH2Cl2 (15 mL). Pale yellow crystals of 5 were obtained from the slow evaporation of its methylene chloride solution (81% yield). 1H NMR (400 Hz, CD2Cl2, 25 °C): δ 8.76 (dd; 3J = 5.2 Hz, 4J = 1.2 Hz; 2H, 7-aza), 8.16 (t; 3J = 12.4 Hz; 2H, Naza-CHaHbCHaHbCHaHb-Naza), 7.63 (dd; 3J = 7.8 Hz, 4J = 1.2 Hz, 2H, 7-aza), 7.57 (d, satellite; 3J = 8.0 Hz, 3JPt–H = 61 Hz; 4H, ortho-H of PtPh2), 7.38 (d, satellite; 3J = 7.8 Hz, 3JPt–H = 72 Hz; 4H, ortho-H′ of PtPh2), 7.13 (d; 3J = 3.6 Hz, 7-aza), 6.97–6.78 (m; 14H, 2H from 7aza, 4H from para-H of PtPh2 and 8H from meta-H of PtPh2), 6.27 (d; 3J = 3.6 Hz; 2H, 7-aza), 4.20 (d; 3J = 12.4 Hz; 2H, Naza-CHaHbCHaHbCHaHb-Naza), 2.72–2.50 (m; 2H, Naza-CHaHbCHaHbCHaHb-Naza), 2.13 (s, satellite; 3JPt–H = 24 Hz; 12H) ppm. 13C NMR: δ 146.80, 144.75, 138.87, 138.69, 136.87, 136.64, 129.66, 129.16, 128.98, 128.75, 127.50, 126.81, 124.11, 123.01, 122.35, 122.14, 115.96, 101.81, 44.08, 31.83, 23.36, 20.80 ppm. Anal. calcd. for C45H48N4Pt2S2: C 49.17, H 4.40, N 5.10; found: C 49.36, H 4.23, N 5.20.
Synthesis of Pt(1,3-BAPr)Me2 (6)
Complex Pt(1,3-BAPr)Me2 was obtained using the similar method as described for 3 using 1,3-BAPr (0.055 g, 0.20 mmol) and [PtMe2(μ-SMe2)]2 (0.057 g, 0.10 mmol) in THF (10 mL) for 2 days. The solvent was decanted. The pale yellow solid was extracted with Et2O/hexanes (1:1) (5 mL × 3). The crystals of Pt(1,3-BAPr)Me2 were obtained by cooling the Et2O/hexanes (1:1) solution in a refrigerator for several days. Due to its poor stability, compound 6 was only characterized by single-crystal X-ray diffraction analyses.
Synthesis of Pt(1,4-BABu)Me2 (7)
Complex Pt(1,4-BABu)Me2 was obtained using the same method as described for 3 using 1,4-BABu (0.129 g, 0.44 mmol) and [PtMe2(μ-SMe2)]2 (0.127 g, 0.22 mmol) in THF (15 mL). Pale yellow crystals of 7 were obtained from cooling the Et2O/hexanes (1:1) solution in a refrigerator for several days. 1H NMR (400 Hz, CD2Cl2, 25 °C): δ 8.97 (d, satellite; 3J = 5.2 Hz, 3JPt–H = 40 Hz; 2H, 7-aza), 8.42 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza), 7. 90 (d; 3J = 8.0 Hz; 2H, 7-aza), 7.25 (d; 3J = 3.2 Hz; 2H, 7-aza), 7.01 (dd; 3J1 = 5.2 Hz, 3J2 = 8.0 Hz; 2H, 7-aza), 6.53 (d; 3J = 3.2 Hz; 2H, 7-aza), 4.07–4.02 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza), 1.89–1.81 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza), 1.48–1.42 (m; 2H, Naza-CHaHbCHaHbCHaHbCHaHb-Naza), 0.52 (s, satellites; 2JPt–H = 86.4 Hz) ppm.
Synthesis of PtPh2(1,3-BAB) (8)
1,3-BAB (0.40 mmol, 0.124 g) and [PtPh2(Me2S)]n (n = 2, 3) (0.164 g, 0.40 mmol based on Pt) were mixed in 20 mL of THF. After the solution was refluxed for 4 hours, the solvent was removed under vacuum. The residue was recrystallized from CH2Cl2/hexanes to afford 8 (25% yield). 1H NMR (500 MHz, CD2Cl2, 25 °C): δ 9.31 (dd; 3J = 5.0 Hz, 4J = 1.5 Hz; 2H, 7-aza), 8.22 (m; 1H, phenyl of 1,3-BAB), 8.04 (dd; 3J = 8.0 Hz, 4J = 1.5 Hz; 2H, 7aza), 7.97 (t; 3J = 8.0 Hz; 1H, phenyl of 1,3-BAB), 7.67 (m; 4H, 7-aza and phenyl of 1,3-BAB); 7.28 (dd; 3J = 8.0 Hz, 4J = 5.5 Hz; 2H, 7-aza), 6.76 (m, satellites; 4H, phenyl), 6.69 (d; 3J = 3.5 Hz; 2H, 7aza), 6.43 (m; 6H, phenyl). 13C NMR: δ = 147.4, 147.0, 138.7, 137.3, 136.0, 131.0, 130.4, 128.7, 125.8, 125.3, 123.4, 121.0, 120.7, 118.1, 102.9. Anal. calcd. for C32H24N4Pt: C 58.27, H 3.64, N 8.50; found: C 57.71, H 3.65, N 8.15.
Synthesis of Pt(BAHE)Ph2 (9)
BAHE (0.037 g, 0.10 mmol) and [PtPh2(μ-SMe2)]n (n = 2 or 3) (0.041 g, 0.10 mmol based on Pt) were mixed in THF (10 mL) and the mixture was stirred overnight at ambient temperature. The solvent was decanted, and the resulting solid was then washed with Et2O (4 mL × 3). The residue was dissolved by THF and filtered through a pad of Celite. The slow evaporation of the THF resulted in 9 as brown power (ca. 50% yield), which was recrystallized from a solvent mixture of CH2Cl2, THF and hexanes. 1H NMR (400 Hz, CD2Cl2, 25 °C): δ 8.72 (d; 3J = 5.5 Hz; 2H, 7-aza), 7.86 (d; 3J = 6.5 Hz; 2H, 7-aza), 7.47 (s; 2H); 7.29 (s; 2H), 7.17 (d; 3J = 3.5 Hz; 2H, 7-aza), 7.02 (dd; 3J1 = 5.5 Hz, 3J2 = 6.5 Hz; 2H, 7-aza), 6.96 (d, satellites; 3JPt–H = 80 Hz, 3J = 8.5 Hz; 4H, ortho-H of PtPh2), 6.70 (m; 4H, meta-H of PtPh2), 6.63 (m; 2H, para-H of PtPh2), 6.55 (d; 3J = 3.5 Hz; 2H, 7-aza), 5.96 (s; 2H) ppm. 13C NMR: δ 152.18, 128.77, 145.26, 142.88. 141.85, 138.35, 134.23, 131.57, 131.21, 130.08, 125.87, 123.38, 122.28, 121.16, 117.39, 102.80, 82.87 ppm. Anal. calcd. for C36H26N4OPt·0.25CH2Cl2 : C 58.29, H 3.58, N 7.50; found: C 58.37, H 3.99, N 7.39.
X-Ray crystallographic analyses
Single crystals of BAHE, 1–9a were mounted on glass fibers for data collection. Data for BAHE, 1, 2, 5–8 were collected on a Bruker Smart 1000 CCD X-ray diffractometer while data for 4 and 9a were collected on a Bruker Apex II single-crystal X-ray diffractometer with graphite-monochromated Mo Kα radiation, operating at 50 kV and 30 mA, at either 298 K or 180 K. Compound 9 produces a mixture of distinctly different crystalline solids due to the presence of isomers. The isomer 9a forms block-shaped crystals which can be picked out for X-ray study. Isomer 9b forms microcrystalline powders that are not suitable for single-crystal X-ray study. These two isomers form an intimate solid mixture that is difficult to separate in bulk quantity. No significant decay was observed for any of the crystals. Data were processed on a PC using Bruker Apex II software and corrected for absorption effects. The structural solution and refinements were performed using the Bruker SHELXTL software package (version 5.10). All structures were solved by direct methods. The quality of crystals of 5 is poor. One of the methyl groups on a SMe2 group in 5 is disordered over two sites with about 50% occupancy factor for each site. Because of the disordering, some of the S–C distances are considerably shorter than a typical S–C bond length. To address this problem, we used DFIX in the SHELXTL program to restrain the S–C bond length at 1.80 Å in the refinements. This approach improves the S–C bond lengths somewhat, but some of the S–C bond lengths are still shorter than typical. Attempts to obtain better crystals for 5 were made but without success. The data for structure 5 is from our best efforts. Crystals of 1 and 7 contain disordered solvent molecules which could not be fully modeled and refined. As a result, the solvent contributions were removed for these two molecules using the Squeeze algorithm in Platon program.17 The crystal data for 1 and 7 in Table 1 excluded contributions from solvent molecules in the lattice. All non-hydrogen atoms were refined anisotropically except the disorder methyl groups C(42), C(42A) and C(43), and the C(29) atom in structure 5. The positions of hydrogen atoms except those on the disordered methyl group in 5 were calculated, and their contributions in structural factor calculations were included. The crystal data for all complexes are summarized in Table 1. Important bond lengths and angles for all the compounds are listed in Table 2. The data for the free ligand BAHE is provided in the supporting materials.†
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9a
|
|
R
1 = ∑|F0| − |Fc|/∑|F0|.
w
R
2 = [∑w [(F02−Fc2)2]/∑[w(F02)2]]1/2, w = 1/[σ2(F02) + (0.075P)2], where P = [Max (F02,0) + 2Fc2]/3.
|
| Formula |
C27H22N4Pt |
C32H24N4Pt |
C29H26N4Pt |
C30H28N4Pt |
C45H48N4Pt2S2 |
C19H22N4Pt |
C20H24N4Pt |
C32H24N4Pt |
C36H26N4PtO |
|
FW
|
597.58 |
659.64 |
625.63 |
639.65 |
1099.17 |
501.59 |
515.52 |
659.64 |
725.70 |
| Space group |
P21/n |
P21/c |
P-1
|
P21/n |
P-1
|
P-1
|
P-1
|
P-1
|
P21/n |
|
a/Å |
12.238(3) |
15.404(3) |
8.2788(1) |
13.068(2) |
11.7088(15) |
7.9960(11) |
8.031(1) |
10.778(2) |
16.6466(3) |
|
b/Å |
11.930(3) |
11.527(2) |
10.2878(1) |
10.512(1) |
12.9165(17) |
9.6192(13) |
10.707(1) |
9.984(2) |
9.7584(2) |
|
c/Å |
18.202(4) |
15.536(3) |
15.4456(3) |
19.077(2) |
14.9862(19) |
11.9856(16) |
13.863(1) |
13.777(3) |
17.6630(3) |
|
α/° |
90 |
90 |
73.386(1) |
90 |
80.192(2) |
84.184(2) |
82.463(1) |
103.297(4) |
90 |
|
β/° |
93.953(6) |
112.390(4) |
89.311(1) |
109.140(2) |
83.251(3) |
72.328(2) |
85.130(1) |
96.241(4) |
104.846(1) |
|
γ/° |
90 |
90 |
69.110(1) |
90 |
63.096(2) |
87.362(2) |
83.618(1) |
115.883(3) |
90 |
|
V/Å3 |
2651.0(10) |
2550.7(8) |
1171.77(3) |
2475.8(5) |
1989.5(4) |
873.7(2) |
1171.4(2) |
1260.4(4) |
2773.47(9) |
|
Z
|
4 |
4 |
2 |
4 |
2 |
2 |
2 |
2 |
4 |
|
D
calc,/g cm−3 |
1.497 |
1.718 |
1.773 |
1.716 |
1.835 |
1.906 |
1.462 |
1.738 |
1.738 |
|
T/K |
293 |
294 |
180 |
180 |
180 |
180 |
180(2) |
298 |
180 |
|
μ/mm−1 |
5.312 |
5.530 |
6.013 |
5.694 |
7.167 |
8.037 |
5.997 |
5.595 |
5.097 |
| 2θmax/° |
54.00 |
54.00 |
52.00 |
54.00 |
56.00 |
54.00 |
52.00 |
56.00 |
54.36 |
| Reflns measured |
17999 |
17073 |
10077 |
16791 |
14309 |
6093 |
7432 |
8109 |
16310 |
| Reflns used (Rint) |
5766(0.033) |
5546(0.017) |
4573(0.021) |
5408(0.040) |
9042(0.044) |
3768 (0.025) |
4542 (0.017) |
4909(0.098) |
6130 (0.034) |
| Parameters |
289 |
334 |
307 |
316 |
470 |
219 |
228 |
334 |
379 |
|
R [I > 2σ(I)]: |
|
R
1
a
|
0.0251 |
0.0188 |
0.0200 |
0.0384 |
0.0502 |
0.0319 |
0.0190 |
0.0775 |
0.0321 |
|
w
R
2
b
|
0.0382 |
0.0340 |
0.0449 |
0.0835 |
0.0639 |
0.0621 |
0.0405 |
0.2071 |
0.0736 |
|
R (all data): |
|
R
1
a
|
0.0468 |
0.0316 |
0.0226 |
0.0642 |
0.1638 |
0.0420 |
0.0213 |
0.0791 |
0.0469 |
|
w
R
2
b
|
0.0401 |
0.0363 |
0.0461 |
0.0946 |
0.0758 |
0.0645 |
0.0407 |
0.2089 |
0.0794 |
| GOF on F2 |
0.723 |
0.925 |
1.025 |
0.998 |
1.188 |
0.924 |
0.979 |
1.092 |
1.047 |
Table 2 Selected bond lengths (Å) and angles (°) for complexes 1–8 and 9a
| Compound 1 |
Compound 2 |
Compound 3 |
Compound 4 |
Compound 5 |
| Pt(1)–C(16) |
1.983(4) |
Pt(1)–C(21) |
1.998(3) |
Pt(1)–C(24) |
2.000(3) |
Pt(1)–C(19) |
1.995(6) |
Pt(1)–C(21) |
2.035(7) |
| Pt(1)–C(22) |
1.994(3) |
Pt(1)–C(27) |
2.003(3) |
Pt(1)–C(18) |
2.014(3) |
Pt(1)–C(25) |
2.010(6) |
Pt(1)–C(15) |
2.026(11) |
| Pt(1)–N(2) |
2.125(3) |
Pt(1)–N(4) |
2.141(2) |
Pt(1)–N(1) |
2.146(3) |
Pt(1)–N(3) |
2.163(5) |
Pt(1)–N(2) |
2.125(7) |
| Pt(1)–N(4) |
2.130(3) |
Pt(1)–N(2) |
2.157(2) |
Pt(1)–N(3) |
2.164(3) |
Pt(1)–N(1) |
2.193(5) |
Pt(1)–S(1) |
2.353(3) |
| C(16)–Pt(1)–C(22) |
90.01(14) |
C(21)–Pt(1)–C(27) |
90.66(10) |
C(24)–Pt(1)–C(18) |
92.56(13) |
C(19)–Pt(1)–C(25) |
91.3(2) |
Pt(2)–C(27) |
1.962(8) |
| C(16)–Pt(1)–N(2) |
177.20(15) |
C(21)–Pt(1)–N(4) |
93.81(9) |
C(24)–Pt(1)–N(1) |
91.61(11) |
C(19)–Pt(1)–N(3) |
170.6(2) |
Pt(2)–C(33) |
2.013(9) |
| C(22)–Pt(1)–N(2) |
90.72(13) |
C(27)–Pt(1)–N(4) |
174.03(8) |
C(18)–Pt(1)–N(1) |
175.82(11) |
C(25)–Pt(1)–N(3) |
90.3(2) |
Pt(2)–N(4) |
2.124(8) |
| C(16)–Pt(1)–N(4) |
91.57(12) |
C(21)–Pt(1)–N(2) |
176.82(9) |
C(24)–Pt(1)–N(3) |
172.24(11) |
C(19)–Pt(1)–N(1) |
87.5(2) |
Pt(2)–S(2) |
2.357(3) |
| C(22)–Pt(1)–N(4) |
177.04(14) |
C(27)–Pt(1)–N(2) |
92.52(9) |
C(18)–Pt(1)–N(3) |
94.62(12) |
C(25)–Pt(1)–N(1) |
176.7(2) |
C(21)–Pt(1)–C(15) |
93.8(4) |
| N(2)–Pt(1)–N(4) |
87.82(11) |
N(4)–Pt(1)–N(2) |
83.01(7) |
N(1)–Pt(1)–N(3) |
81.24(10) |
N(3)–Pt(1)–N(1) |
90.40(18) |
C(21)–Pt(1)–N(2) |
174.5(4) |
| Compound 6 |
Compound 7 |
Compound 8 |
Compound 9a |
C(15)–Pt(1)–N(2) |
91.5(4) |
| Pt(1)–C(2) |
2.023(6) |
Pt(1)–C(19) |
2.033(3) |
Pt(1)–C(21) |
1.990(9) |
Pt(1)–C(25) |
2.009(4) |
C(21)–Pt(1)–S(1) |
86.1(3) |
| Pt(1)–C(1) |
2.025(5) |
Pt(1)–C(20) |
2.034(3) |
Pt(1)–C(27) |
1.996(10) |
Pt(1)–C(31) |
2.011(4) |
C(15)–Pt(1)–S(1) |
178.4(3) |
| Pt(1)–N(3) |
2.150(4) |
Pt(1)–N(1) |
2.160(2) |
Pt(1)–N(2) |
2.161(9) |
Pt(1)–N(1) |
2.132(3) |
N(2)–Pt(1)–S(1) |
88.5(2) |
| Pt(1)–N(1) |
2.159(5) |
Pt(1)–N(3) |
2.165(3) |
Pt(1)–N(4) |
2.165(8) |
Pt(1)–N(3) |
2.143(4) |
C(27)–Pt(2)–C(33) |
89.8(4) |
| C(2)–Pt(1)–C(1) |
89.9(3) |
C(19)–Pt(1)–C(20) |
88.13(13) |
C(21)–Pt(1)–C(27) |
89.3(4) |
O(1)–C(21) |
1.446(6) |
C(27)–Pt(2)–N(4) |
174.8(3) |
| C(2)–Pt(1)–N(3) |
93.3(2) |
C(19)–Pt(1)–N(1) |
91.72(11) |
C(21)–Pt(1)–N(2) |
170.4(4) |
O(1)–C(24) |
1.463(6) |
C(33)–Pt(2)–N(4) |
95.3(4) |
| C(1)–Pt(1)–N(3) |
176.5(2) |
C(20)–Pt(1)–N(1) |
177.54(12) |
C(27)–Pt(1)–N(2) |
86.1(4) |
C(25)–Pt(1)–C(31) |
88.49(16) |
C(27)–Pt(2)–S(2) |
92.4(3) |
| C(2)–Pt(1)–N(1) |
176.0(2) |
C(19)–Pt(1)–N(3) |
174.07(11) |
C(21)–Pt(1)–N(4) |
89.2(4) |
C(25)–Pt(1)–N(1) |
176.82(15) |
C(33)–Pt(2)–S(2) |
177.3(3) |
| C(1)–Pt(1)–N(1) |
93.0(2) |
C(20)–Pt(1)–N(3) |
88.13(11) |
C(27)–Pt(1)–N(4) |
176.3(3) |
C(31)–Pt(1)–N(1) |
89.98(15) |
N(4)–Pt(2)–S(2) |
82.5(2) |
| N(3)–Pt(1)–N(1) |
83.75(17) |
N(1)–Pt(1)–N(3) |
91.82(9) |
N(2)–Pt(1)–N(4) |
95.9(3) |
C(25)–Pt(1)–N(3) |
93.35(15) |
|
|
| |
|
|
|
|
|
C(31)–Pt(1)–N(3) |
174.14(16) |
|
|
| |
|
|
|
|
|
N(1)–Pt(1)–N(3) |
87.90(13) |
|
|
| |
|
|
|
|
|
C(21)–O(1)–C(24) |
95.0(3) |
|
|
Result and discussion
Syntheses and characterization of new ligands 1,3-BAr, 1,4-BABu, and BAHE
Ligands bis(7-azaindol-1-yl)methane (BAM), 1,2-bis(7-azaindol-1-yl)benzene (1,2-BAB), and 1,3-bis(7-azaindol-1-yl)benzene (1,3-BAB) were synthesized using previously reported procedures.12,16 To examine the effect of increasing chelate ring size and the interactions between a Pt(II) center and a longer and more flexible linker than the CH2group in the BAM ligand, two new ligands 1,3-bis(7-azaindol-1-yl)propane (1,3-BAPr) and 1,4-bis(7-azaindol-1-yl)butane (1,4-BABu) as BAM analogues were designed and synthesized. As tautomerism is a typical phenomenon for 7-azaindole, alkylation of 7-azaindole under basic conditions has the tendency to afford a mixture of N1, C3 and N7 functionalized isomers. For example, in the synthesis of the BAM ligand by the nucleophilic substitution reaction of CH2Br2 with 7-azaindole in the presence of KOH and a phase-transfer catalyst, NBu4Br, in refluxing toluene and water, in addition to BAM, other isomers were also isolated, albeit in low yields.16a Our initial attempts towards the syntheses of the 1,3-BAPr and 1,4-BABu ligands under the same conditions used for BAM failed to result in any product, presumably due to the reduced reactivity of 1,3-dibromopropane and 1,4-dibromobutane compared to that of CH2Br2. Since alkylation of indole analogues preferentially occurs at the N1 position rather than C3 position in high polar solvents such as DMF,18 to increase the yield and avoid the other isomers, the reaction of 1,3-dibromopropane or 1,4-dibromobutane with the sodium salt of 7-azaindole, generated in situ by deprotonation of 7-azaindole with NaH, was carried out in DMF at 110 °C (Scheme 1). 1,3-BAPr or 1,4-BABu were obtained in good yields.
 |
| | Scheme 1 | |
To examine the impact of an aromatic linker that is larger than the phenyl group in 1,2-BAB and has a donor atom, we attempted the synthesis of 6,7-bis(7-azaindol-1-yl)-1,4-dihydronaphthalene-1,4-epoxide (BAHE) using the procedure shown in Scheme 2. BAHE was obtained in ca 50% yield using the homogeneous C–N coupling method developed by the Buchwald group,19 where 6,7-dibromo-1,4-dihydronaphthalene-1,4-epoxide, prepared according to literature methods,20 and 7-azaindole were reacted in the presence of CuI, 1,10-phenanthroline, and Cs2CO3 in DMF at 145–150 °C under N2 atmosphere for 72 hours. BAHE was fully characterized by NMR spectroscopy, HRMS, and single crystal X-ray diffraction analyses (see ESI†). Notably, besides BAHE, the mono-7-azaindolyl substituted debromination product NAHE and a tris-7-azaindolyl substituted naphthalene derivative compound TAN were also isolated as the side products in 13% and 8% yield, respectively (Scheme 2). Both side products have been fully characterized by NMR and HRMS. The formation of TAN likely occurred via processes as illustrated in Scheme 3. The nucleophilic addition of 7-azaindole anion to BAHE initiated a tandem CC double bond migration and simultaneous ring-opening process, generating a dihydronaphthalene derived intermediate, which underwent subsequent protonation and dehydration, resulting in TAN. Similar nucleophilic addition induced 1,4-dihydronaphthalene-1,4-epoxide ring opening processes have been well studied toward producing highly functionalized dihydronaphthalene products.21
 |
| | Scheme 2 | |
 |
| | Scheme 3 | |
Syntheses and characterization of Pt(II) complexes
Complexes Pt(BAM)Ph2 (1) and Pt(1,2-BAB)Ph2 (2) (Chart 2) were obtained in good yields from the reactions of [PtPh2(μ-SMe2)]n (n = 2, 3) with the corresponding chelate ligand in a 1:1 [Pt]/[L] ratio. The monomeric PtPh2 complexes Pt(1,3-BAPr)Ph2 (3) and Pt(1,4-BABu)Ph2 (4) were obtained as stable yellow crystalline solids in good yields by ligand displacement of the bridging SMe2 in [PtPh2(μ-SMe2)]n (n = 2, 3) with either 1,3-BAPr or 1,4-BABu with a 1:1 [Pt]/[L] ratio in THF at ambient temperature, as shown in Scheme 4. Interestingly, when the reaction was carried out with a 2:1 [Pt]/[1,3-BAPr] ratio, a dinuclear complex Pt2(1,3-BAPr)Ph4(SMe2)2 (5) was isolated. Upon sonicating its CH2Cl2 solution, complex 5 was quickly converted to 3via the elimination of one equivalent of PtPh2(SMe2)2, an indication that compound 3 is thermodynamically favored while compound 5 is kinetically favored. Efforts were also made to synthesize the PtMe2 analogues of 3 and 4, Pt(1,3-BABr)Me2 (6) and Pt(1,4-BABu)Me2 (7). The ligand displacement reactions of [PtMe2(μ-SMe2)2] with 1,3-BAPr and 1,4-BABu both were found to be sluggish, and the desired compounds 6 and 7 were obtained in 40–60% yields after 2 days. Moreover, while both complexes displayed a poor stability, making it difficult to purify them, the decomposition of 6 occurred much more quickly, consistently resulting in the formation of Pt black and the free 1,3-BAPr ligand. Fortunately, some crystals of both 6 and 7 have been obtained by cooling their Et2O/hexanes (1:1) solutions, thereby allowing unambiguous examination of their structures. The PtPh2 complex Pt(1,3-BAB)Ph2 (8) (Chart 2) was synthesized in the same manner as for Pt(1,2-BAB)Ph2. However, the yield for 8 is much lower than that of Pt(1,2-BAB)Ph2, which can be attributed to the poor chelating ability of the 1,3-BAB ligand. Although 1,3-BAB is capable of chelating Pd(II) and Pt(II) in an N,C,N-tridentate mode, such cyclometallated products were not obtained from the reaction, presumably because the Pt–C bond in the [PtPh2(μ-Me2S)]n (n = 2 or 3) starting material is relatively strong, which cannot be cleaved at ambient temperature. Attempt to improve the yield of 8 by using higher temperature (e.g. refluxing toluene) led to partial decomposition with Pt(0) deposition with the remaining reaction mixture still being dominated by the unreacted starting material. The reaction of [PtMe2(μ-Me2S)]2 with 1,3-BAB encountered the same problem and consequently the isolation and characterization of Pt(1,3-BAB)Me2 was not achieved. The BAHE complex, Pt(BAHE)Ph2 (9) was obtained in modest yield (∼50%) using a procedure similar to that of 8. However, 1H NMR spectra indicated that compound 9 exists in two isomeric forms 9a and 9b, as shown in Chart 2.
![Reagents and conditions: i) [PtPh2(μ-SMe2)n] (n = 2, 3), THF, rt, overnight ([Pt]:[L] = 1:1); ii) [PtPh2(μ-SMe2)n] (n = 2, 3), rt, overnight, ([Pt]:[L] = 2:1); iii) sonication, CH2Cl2, rt.](/image/article/2008/DT/b813501k/b813501k-s4.gif) |
| | Scheme 4 Reagents and conditions: i) [PtPh2(μ-SMe2)n] (n = 2, 3), THF, rt, overnight ([Pt]:[L] = 1:1); ii) [PtPh2(μ-SMe2)n] (n = 2, 3), rt, overnight, ([Pt]:[L] = 2:1); iii) sonication, CH2Cl2, rt. | |
 |
| | Chart 2 | |
Structures of complexes 1, 3–7 with an aliphatic linker
To verify the binding modes and the intramolecular interactions between the linker groups in the chelate ligands and the Pt(II) centers in the complexes, single-crystal X-ray diffraction analyses were carried out for all complexes. The molecular structures of PtPh2 complexes 1, 3–5 are shown in Fig. 1–4, respectively. Compounds 1, 3 and 4 are mononuclear Pt(II) complexes with their ligands adopting the N,N-chelating mode to form either a 8-membered (1), a 10-membered (3) or 11-membered (4) chelate ring. Compound 5 is a dinuclear complex with the 1,3-BAPr ligand acting as a bridging ligand for the two Pt(II) centers and a SMe2 being bound to each Pt(II) center. The Pt(II) centers in all four complexes have a typical square planar geometry, with the PtPh2 groups being consistently cis to each other. Although some variations of Pt–N and Pt–C bond lengths are evident for these complexes as shown in Table 2, they are similar to those observed in Pt(BAM)Me2.12a The most notable difference between 1, 3 and 4 is the N–Pt–C and N–Pt–N bond angles. In 1, the average N–Pt–Ctrans angles is 177.1(2)° and the N–Pt–N angle is 87.82(11)°. In 3 and 4, however, the average N–Pt–Ctrans angles are 174.0(1)° and 173.7(2)°, respectively, a large deviation from linearity. Most striking is the small N–Pt–N angle of 81.24(10)° in 3, which is highly strained, compared to those of 1. In contrast, despite a larger chelate ring in 4 compared to that in 1, the N–Pt–N angle in 4 is 90.40(18)°, a nearly ideal chelating angle for a square planar Pt(II) center. Consistent with the greater chelate ring strain in 3 is the smaller dihedral angle between the two 7-azaindolyl rings (44.5°) defined by the diagram shown in Fig. 13 (see later), compared to those in 1 (94.3°) and 4 (109.7°) (Table 3). The large chelate ring strain in 3 appears to be imposed by the geometry of the propyl linker. The butyl linker in 4 is much more flexible, thus allowing the chelate ring to be more relaxed. The two Pt(II) square planes in 5 are parallel to each other with an approximate staggered configuration. Compared to the highly strained mononuclear complex 3, the dinuclear Pt(II) molecule 5 appears to be more sterically favored. Its facile conversion to 3 is likely driven by chelating effect.
Table 3 Dihedral angles between the two 7-azaindolyl rings in 1–4 and 6–9a
| Linker in the chelate ligand |
Compound |
Dihedral angle between two 7-azaindolyl rings/° |
N–Pt–N bond angle/° |
| CH2 |
Pt(BAM)Ph2 (1) |
94.3 |
87.82(11) |
| |
Pt(BAM)Me212a |
102.7 |
89.98(11) |
| CH2CH2CH2 |
Pt(1,3-BAPr)Ph2 (3) |
44.5 |
81.24(10) |
| |
Pt(1,3-BAPr)Me2 (6) |
46.6 |
83.75(17) |
| CH2CH2CH2CH2 |
Pt(1,4-BABu)Ph2 (4) |
109.7 |
90.40(18) |
| |
Pt(1,4-BABu)Me2 (7) |
100.5 |
91.82(9) |
|
1,2-phenyl
|
Pt(1,2-BAB)Ph2 (2) |
62.3 |
83.01(7) |
| |
Pt(1,2-BAB)Me212b |
82.5 |
85.8(3) |
| |
Pt(BAHE)Ph2 (9a) |
73.4 |
87.90(13) |
|
1,3-phenyl
|
Pt(1,3-BAB)Ph2 (8) |
140.2 |
95.9(3) |
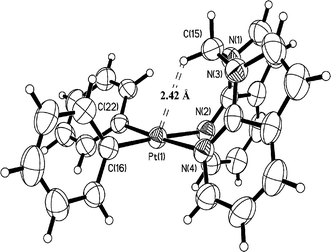 |
| | Fig. 1 Crystal structure of 1 with 50% ellipsoids. | |
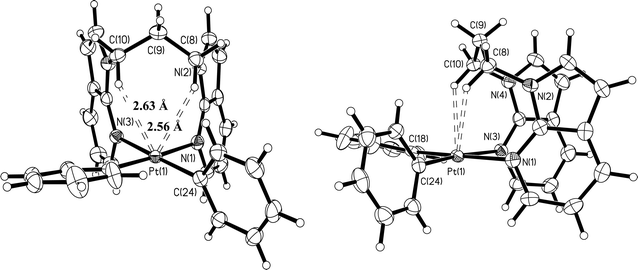 |
| | Fig. 2 Crystal structure of 3 with 50% ellipsoids: front view (left) and side view (right). | |
 |
| | Fig. 3 Crystal structure of 4 with 50% ellipsoids: front view (left) and side view (right). | |
 |
| | Fig. 4 Crystal structure of 5 with 50% ellipsoids. All carbon atoms are shown as ideal spheres for clarity. | |
The structures of Pt(1,3-BAPr)Me2 (6) and Pt(1,4-BABu)Me2 (7) are analogous to complexes 3 and 4 and are shown in Fig. 5 and 6, respectively. As shown by Table 3, the trend of the N–Pt–N angles and the dihedral angles between the two 7-azaindolyl planes for the PtMe2 complexes of the BAM, 1,3-BAPr and 1,4-BABu ligands is the same as that of the corresponding PtPh2 complexes, supporting that the linker in the chelate ligand dictates the chelate bond angle and the dihedral angle of the two 7-azaindolyl groups. The N–Pt–N angles of the PtMe2 complexes are all greater than those of the PtPh2 analogue, The dihedral angle between the two 7-azaindolyl rings follows a similar trend except that the 1,4-BABu complex 7 has a smaller dihedral angle than that of the PtPh2 complex 4. Therefore, the size of the R group bound to the Pt(II) center also has a significant effect, albeit much less than the linker group, on the chelate angle and the dihedral angle.
 |
| | Fig. 5 Crystal structure of 6 with 50% ellipsoids: front view (left) and side view (right). | |
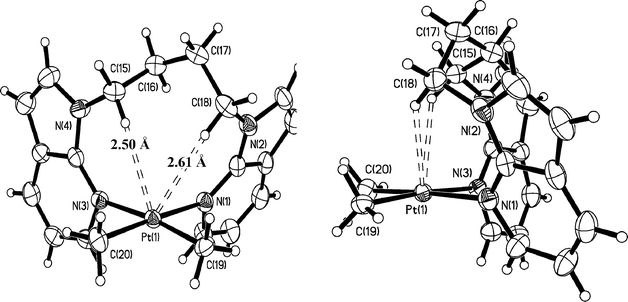 |
| | Fig. 6 Crystal structure of 7 with 50% ellipsoids: front view (left) and side view (right). | |
Intramolecular Pt⋯H interactions in 1, 3–7
As shown in Fig. 1–6, one common feature in structures 1, 3–7, is the partial blockage of the square planar Pt(II) 5th coordination site by the linker group. Due to the pronounced boat conformation adopted by the eight-membered chelate ring in 1, the H′ atom (Chart 1) of its methylene carbon is oriented toward the dz2 orbital of the Pt(II) center, with a short Pt⋯H′ separation distance of 2.42 Å, which is similar to that (2.44 Å) observed in Pt(BAM)Me2.12a In the chelating complexes 3, 4, 6 and 7, the analogous boat conformations give rise to both H′ atoms of two Cα atoms of their aliphatic linkers in close contact with the Pt(II) center, with the shortest Pt⋯H′ separation distances being 2.56 Å in 3, 2.58 Å in 4, 2.61 Å in 6, and 2.49 Å in 7, respectively. In addition to the short intramolecular Pt⋯H distances, a short intermolecular Pt⋯H distance with a 7-azaindolyl ring is also observed in 3 (3.02 Å) and 7 (2.92 Å), as shown in Fig. 7. The crystals of 4 and 6 do not show such intermolecular interactions. For the dinuclear Pt2 complex 5, due to similar geometrical constraints, the two H′(Cα) atoms display even shorter Pt⋯H′ separation distances (2.31 Å and 2.43 Å).
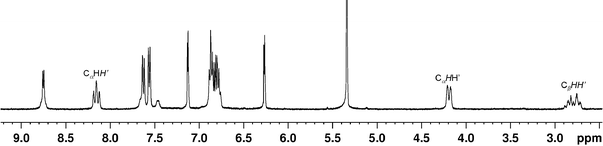
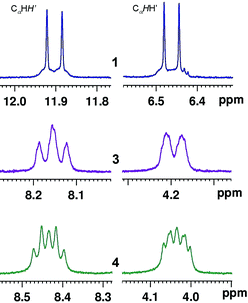
We have reported previously that the short Pt⋯H′ separation distances in BAM Pt(II) complexes can induce PtII⋯H–C interactions. While the BAM CH2hydrogen atoms in these complexes displayed typical “AB” patterns, in the 1H NMR spectra, dramatic δH downfield shifts (Δδ > 4.5 ppm) were observed for the H′ atoms that are oriented toward the dz2 orbitals of Pt(II) centers.12 The origin of these lowfield resonances was tentatively ascribed to a three-center four-electron PtII⋯H–C electrostatic interaction.22–23 The spectral studies of the above new Pt(II) complexes were informative for us to understand such a phenomenon. If these Pt(II) complexes retain the same structures in solution, each of the CH2groups in the linkers in 1 and 3–7 should display an “AB” pattern in the 1H NMR spectra. Indeed, the “AB” pattern was observed and well resolved for complex 1 where the CHH′ atoms display two distinct chemical shifts with Δδ = 5.44 ppm (Fig. 8), which is almost the same as that (5.45 ppm) observed12a in Pt(BAM)Me2, in agreement to the nearly identical Pt⋯H separation distances in 1 and Pt(BAM)Me2. Similar distinct “AB” patterns were also observed for the CαHH′ protons in the 1,3-BAPr complexes 3, and the 1,4-BABu complexes 4 and 7, as illustrated in Fig. 8 and 9. In comparison with 1, the elongated Pt⋯H′ separation distances in these complexes resulted in noticeably reduced Δδ for the CαHH′ atoms in 3 (3.99 ppm), 4 (4.41 ppm) and 7 (4.36 ppm). The two CαHH′ atoms in 5 also displayed an “AB” patterns with a Δδ value observed as 3.96 ppm. Note also that, the β-CHH′ atoms in the linkers in 3–7 also displayed an “AB” pattern in the 1H NMR spectra, indicating that these complexes have the same structural features in solution as those observed in the solid state.
 |
| | Fig. 7 Diagrams showing intermolecular Pt⋯H interactions in 3 (top) and 7 (bottom). | |
 |
| | Fig. 8 Partial 1H NMR spectra of 1, 3 and 4 in CD2Cl2 at ambient temperature, showing the two distinct chemical shifts and the “AB” pattern of the CαHH′ protons. | |
Despite a number of known examples of platinum alkyl cations with β-agostic PtII⋯H–C interactions,25 previous studies have demonstrated that typical neutral square planar Pd(II) and Pt(II) complexes usually display only the three-center four-electron PdII/PtII⋯H–C electrostatic interactions utilizing the electron pair in the dz2 orbital,24 with the Pd/Pt⋯H separation distances being > 2.2 Å and a characteristic downfield chemical shift by the proton. In contrast, agostic interactions are intramolecular three-center two-electron interactions, characterized by short M⋯H separation distances (1.8–2.2 Å) as well as an upfield 1H chemical shift for the proton involved.23 Agostic interactions are often encountered in coordinatively unsaturated MoII, RuII, OsII, RhIII, IrIII and PtII complexes due to the requirement of an empty orbital on the metal center.26 Therefore, the δ downfield shifts by CαHH′ atoms in our Pt(II) complexes can be unequivocally attributed to three-center four-electron PtII⋯H–C interactions rather than agostic interactions.
Structures of complexes 2, 8–9a with an aromatic linker
The structures of Pt(1,2-BAB)Ph2 (2), Pt(1,3-BAB)Ph2 (8) and Pt(BAHE)Ph2 (9a) are shown in Fig. 10–12. The common feature for these three molecules is that the linker group between two 7-azaindolyl rings is a phenyl at either 1,2-positions or 1,3-positions. The structures of 2 and 9a are closely related since both contain a 1,2-phenyl linker. The average N–Pt–Ctrans angles in 2, 8 and 9a are 175.4(9)°, 173.4(4)° and 175.5(2)°, respectively, and the N–Pt–N angles are 83.01(7)°, 95.9(3)° and 87.90(13)°, respectively. The most significant difference between the three structures is the dihedral angle between the two 7-azaindolyl groups, 62.3° for 2, 140.2° for 8, and 73.4° for 9a. The structure of 2 resembles that of Pt(1,2-BAB)Me2, except that the PtMe2 analogue has a much greater dihedral angle than 2, as shown in Table 3. The large dihedral angle in 8 is an indication of the poor chelating ability of the 1,3-BAB ligand, consistent with the poor stability and the low yield of compound 8. The impact of various linker groups on the dihedral angle of the two 7-azaindolyl rings in the PtPh2 complexes is illustrated by Fig. 13. The other significant difference is the steric blockage of the Pt 5th coordination site by the linker in the three molecules. The 1,2-phenyl linker in 2 and 9a has a much smaller dihedral angle with the Pt square plane (∼33° and 42°, respectively) than the 1,3-linker in 8 (∼48°). Hence, the 1,2-phenyl linker is much more effective in capping the 5th coordination site than the 1,3-phenyl linker. The BAHE ligand in 9a is especially effective in sterically blocking access to the 5th coordination site due to the presence of the furan ring, as shown by the space filling diagram in Fig. 12. We have demonstrated recently that the effective blockage of the 5th coordination site is critical in achieving effective regio- and diastereoselective C-H activation.12 Hence, compound 9a is a very promising molecule for use in regio- and stereoselective C-H activation, which will be investigated and reported in due course. The oxygen atom in 9a forms an intermolecular hydrogen bond with a 7-azaindolyl group, causing a bi-layer-like extended structure shown in Fig. 12. The structure of the isomer 9b was not determined by X-ray diffraction analysis due to the lack of suitable crystals. 1H NMR spectra confirmed the presence of 9b and that the ratio of 9a : 9b is ∼4.5:1. Isomer 9b is not favored presumably because of the greater steric repulsion between the oxygen atom and the PtPh2 moiety in 9b than that between the ethylene and the PtPh2 in 9a.
 |
| | Fig. 10 The side view of structures of 2 (left) and 9a (right) with 50% ellipsoids. | |
 |
| | Fig. 11 Crystal structure of 8 with 50% ellipsoids: front view (left) and side view (right). | |
 |
| | Fig. 12 Left: The space-filling diagram of 9a showing the effective blocking of the Pt atom by the BAHE ligand; Right: A diagram showing intermolecular H bonds involving the oxygen atom in the crystal lattice of 9a. | |
 |
| | Fig. 13 A diagram showing the orientation and the dihedral angle between the two 7-azaindolyl rings in the PtPh2 complexes with various linkers. The phenyl and the linker groups are removed for clarity. | |
The stability of the Pt(N,N-L)R2 complexes
Pt(II) complexes with a large chelate-ring size are not common.9 The bis(7-azaindol-1-yl) derivative ligands appear to have the unusual ability/tendency to form Pt(II) chelate complexes despite the unfavorable large chelate ring sizes. Nonetheless, the chelate ligands in these Pt(II) complexes do appear to have great impact on the stability of the complex. For example, among the PtMe2 complexes, a distinct difference in stability was observed for Pt(BAM)Me2, 6 and 7: the 1,3-BAPr and 1,4-BABu complexes 6 and 7 have a much greater tendency to decompose at ambient temperature than the BAM complex with complex Pt(1,3-BAPr)Me2 (6) being the least stable. The chelate ring strain in 6 may be the main cause of its instability. The PtPh2 complexes in general are all stable at ambient temperature with the exception of Pt(1,3-BAB)Ph2 (8), which appears to have a low stability in solution, presumably caused by the incompatible N,N-chelating angle of 1,3-BAB with a square planar Pt(II) ion. The greater instability of the PtMe2 complexes versus their PtPh2 analogues can be attributed to the greater trans influence of the methyl group, and the weaker Pt–C bond, compared to the PtPh2 analogues.
Conclusions
The syntheses of new 7-azaindolyl derivative ligands 1,3-BAPr, 1,4-BABu and BAHE have been accomplished, enabling a systematic examination on the impact of the linker groups in the chelate ligands on the structure and the stability of a series of Pt(II) complexes. Our investigation has established that regardless of the linker's size and the geometry, the bis(7-azaindol-1-yl) chelate ligands in general favor the formation of mononuclear chelate complexes with a Pt(II) center. Ligands with aromatic linkers such as 1,2-BAB or BAHE are most effective in blocking the 5th coordination site of the Pt(II) center. In comparison, ligands with an aliphatic linker such as BAM, 1,3-BAPr and 1,4-BABu are in general much less effective in blocking the 5th coordination site. The aromatic 1,3-BAB ligand is the least effective in steric blocking and as a chelate ligand. Strong intramolecular Pt⋯H interactions have been established in Pt(II) complexes with ligands bearing aliphatic linkers and confirmed to be three-center four-electron PtII⋯H–C interactions, by NMR and crystal structural data. PtMe2 complexes with 1,3-BAPr and 1,4-BABu ligands have been found to have a low stability. The BAHE ligand can bind to the Pt(II) center with two different geometries, forming two geometric isomers, with isomer 9a being the dominant one. 9a also appears to be most promising for use in regio- and diastereoselective C–H activation due to the full blockage of the 5th coordination site of the Pt(II) center by the linker.
Acknowledgements
We thank Dr. Rui-Yao Wang for his assistance in some of the crystal structural work, and the Natural Sciences and Engineering Council of Canada for financial support.
References
-
(a) N. F. Goldshlegger, M. B. Tyabin, A. E. Shilov and A. A. Shteinman, Zh. Fiz. Khim., 1969, 43, 2174 CAS;
(b) N. F. Goldshlegger, V. V. Eskova, A. E. Shilov and A. A. Shteinman, Zh. Fiz. Khim., 1972, 46, 1353 CAS.
-
(a) For recent comprehensive reviews on cationic Pt(II)-mediated C–H bond activation studies, see: J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 Search PubMed;
(b)
K. I. Goldberg, and A. S. Goldman, Activation and Functionalization of C–H Bonds, American Chemical Society, Washington, DC, 2004 Search PubMed;
(c) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS.
-
(a) G. A. Luinstra, L. Wang, S. S. Stahl, J. A. Labinger and J. E. Bercaw, J. Organomet. Chem., 1995, 504, 75 CrossRef CAS;
(b) S. S. Stahl, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1996, 118, 5961 CrossRef CAS;
(c) M. W. Holtcamp, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1997, 119, 848 CrossRef CAS.
-
(a) L. Johansson, M. Tilset, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2000, 122, 10846 CrossRef CAS;
(b) J. Procelewska, A. Zahl, R. van Eldik, H. A. Zhong, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 2002, 41, 2808 CrossRef CAS;
(c) H. A. Zhong, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2002, 124, 1378 CrossRef CAS;
(d) A. F. Heyduk, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2003, 125, 6366 CrossRef CAS;
(e) A. F. Heyduk, T. G. Driver, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2004, 126, 15034 CrossRef CAS;
(f) T. G. Driver, M. W. Day, J. A. Labinger and J. E. Bercaw, Organometallics, 2005, 24, 3644 CrossRef CAS;
(g) J. S. Owen, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2006, 128, 2005 CrossRef CAS.
-
(a) L. Johansson, O. B. Ryan and M. Tilset, J. Am. Chem. Soc., 1999, 121, 1974 CrossRef CAS;
(b) L. Johansson and M. Tilset, J. Am. Chem. Soc., 2001, 123, 739 CrossRef CAS;
(c) L. Johansson, O. B. Ryan, C. Rømming and M. Tilset, J. Am. Chem. Soc., 2001, 123, 6579 CrossRef CAS;
(d) B. J. Wik, M. Lersch and M. Tilset, J. Am. Chem. Soc., 2002, 124, 12116 CrossRef CAS;
(e) B. J. Wik, M. Lersch, A. Krivokapic and M. Tilset, J. Am. Chem. Soc., 2006, 128, 2682 CrossRef CAS.
-
(a) U. Fekl, W. Kaminsky and K. I. Goldberg, J. Am. Chem. Soc., 2001, 123, 6423 CrossRef CAS;
(b) U. Fekl, W. Kaminsky and K. I. Goldberg, J. Am. Chem. Soc., 2003, 125, 15286 CrossRef CAS;
(c) U. Fekl and K. I. Goldberg, J. Am. Chem. Soc., 2002, 124, 6804 CrossRef CAS;
(d) A. T. Luedtke and K. I. Goldberg, Inorg. Chem., 2007, 46, 8496 CrossRef CAS;
(e) S. M. Kloek and K. I. Goldberg, J. Am. Chem. Soc., 2007, 129, 3460 CrossRef CAS;
(f) S. M. Kloek and K. I. Goldberg, J. Am. Chem. Soc., 2007, 129, 3460 CrossRef CAS.
-
(a) D. D. Wick and K. I. Goldberg, J. Am. Chem. Soc., 1997, 119, 10235 CrossRef CAS;
(b) D. D. Wick and K. I. Goldberg, J. Am. Chem. Soc., 1999, 121, 11900 CrossRef CAS;
(c) U. Fekl, R. Van Eldik, S. Lovell and K. I. Goldberg, Organometallics, 2000, 19, 3535 CrossRef CAS;
(d) M. P. Jensen, D. D. Wick, S. Reinartz, P. S. White, J. T. Templeton and K. I. Goldberg, J. Am. Chem. Soc., 2003, 125, 8614 CrossRef CAS.
-
(a) J. R. Khusnutdinova, L. L. Newman, P. Y. Zavalij, Y. -F. Lam and A. N. Vedernikov, J. Am. Chem. Soc., 2008, 130, 2174 CrossRef CAS;
(b) J. R. Khusnutdinova, P. Y. Zavalij and A. N. Vedernikov, Organometallics, 2007, 26, 2402 CrossRef CAS;
(c) E. Khaskin, P. Y. Zavalij and A. N. Vedernikov, J. Am. Chem. Soc., 2006, 128, 13054 CrossRef CAS;
(d) A. N. Vedernikov, S. A. Binfield, P. Y. Zavalij and J. R. Khusnutdinova, J. Am. Chem. Soc., 2006, 128, 82 CrossRef CAS;
(e) A. N. Vedernikov, J. C. Fettinger and F. Mohr, J. Am. Chem. Soc., 2004, 126, 11160 CrossRef CAS;
(f) E. Khaskin, P. Y. Zavalij and A. N. Vedernikov, Angew. Chem., Int. Ed., 2007, 46, 6309 CrossRef CAS.
-
(a) G. S. Hill, L. Manojlovic-Muir, K. W. Muir and R. J. Puddephatt, Organometallics, 1997, 16, 525 CrossRef CAS;
(b) E. M. Prokopchuk, H. A. Jenkins and R. J. Puddephatt, Organometallics, 1999, 18, 2861 CrossRef CAS;
(c) J. G. Hinman, C. R. Baar, M. C. Jennings and R. J. Puddephatt, Organometallics, 2000, 19, 563 CrossRef CAS;
(d) C. M. Ong, T. J. Burchell and R. J. Puddephatt, Organometallics, 2004, 23, 1493 CrossRef CAS;
(e) F. Zhang, C. W. Kirby, D. W. Hairsine, M. C. Jennings and R. J. Puddephatt, J. Am. Chem. Soc., 2005, 127, 14196 CrossRef CAS;
(f) F. Zhang, E. M. Prokopchuk, M. E. Broczkowski, M. C. Jennings and R. J. Puddephatt, Organometallics, 2006, 25, 1583 CrossRef CAS;
(g) F. Zhang, M. E. Broczkowski, M. C. Jennings and R. J. Puddephatt, Can. J. Chem., 2005, 83, 595 CrossRef CAS;
(h) C. M. Thomas and J. C. Peters, Organometallics, 2005, 24, 5858 CrossRef CAS.
-
(a) J. R. Khusnutdinova, L. L. Newman, P. Y. Zavalij, Y.-F. Lam and A. N. Vedernikov, J. Am. Chem. Soc., 2008, 130, 2174 CrossRef CAS;
(b) A. V. Pawlikowski, A. D. Getty and K. I. Goldberg, J. Am. Chem. Soc., 2007, 129, 10382 CrossRef CAS;
(c) J. Procelewska, A. Zahl, G. Liehr, R. van Eldik, N. A. Smythe, B. S. Williams and K. I. Goldberg, Inorg. Chem., 2005, 44, 7732 CrossRef CAS;
(d) K. L. Arthur, Q. L. Wang, D. M. Bregel, N. A. Smythe, B. A. O'Neill, K. I. Goldberg and K. G. Moloy, Organometallics, 2005, 24, 4624 CrossRef CAS;
(e) D. M. Crumpton-Bregel and K. I. Goldberg, J. Am. Chem. Soc., 2003, 125, 9442 CrossRef CAS;
(f) D. M. Crumpton and K. I. Goldberg, J. Am. Chem. Soc., 2000, 122, 962 CrossRef CAS;
(g) B. S. Williams, A. W. Holland and K. I. Goldberg, J. Am. Chem. Soc., 1999, 121, 252 CrossRef CAS;
(h) K. I. Goldberg, J.-Y. Yan and E. M. Breitung, J. Am. Chem. Soc., 1995, 117, 6889 CrossRef CAS;
(i) K. I. Goldberg, J.-Y. Yan and E. L. Winter, J. Am. Chem. Soc., 1994, 116, 1573 CrossRef CAS;
(j) J. C. Thomas and J. C. Peters, J. Am. Chem. Soc., 2001, 123, 5100 CrossRef CAS;
(k) W. V. Konze, B. L. Scott and G. J. Kubas, J. Am. Chem. Soc., 2002, 124, 12550 CrossRef CAS;
(l) J. C. Thomas and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 8870 CrossRef CAS.
-
(a) R. A. Periana, J. D. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS;
(b) A. Sen, Acc. Chem. Res., 1998, 31, 550 CrossRef CAS;
(c) V. R. Ziatdinov, J. Oxgaard, O. A. Mironov, K. J. H. Young, W. A. Goddard, III and R. A. Periana, J. Am. Chem. Soc., 2006, 128, 7404 CrossRef CAS.
-
(a) D. Song and S. Wang, Organometallics, 2003, 22, 2187 CrossRef CAS;
(b) S.-B. Zhao, D. Song, W.-L. Jia and S. Wang, Organometallics, 2005, 24, 3290 CrossRef CAS;
(c) S.-B. Zhao, G. Wu and S. Wang, Organometallics, 2006, 25, 5979 CrossRef CAS;
(d) S.-B. Zhao, G. Wu and S. Wang, Organometallics, 2008, 27, 1030 CrossRef CAS.
-
(a) D. Song, W.-L. Jia and S. Wang, Organometallics, 2004, 23, 1194 CrossRef CAS;
(b) D. Song, K. Sliwowski, J. Pang and S. Wang, Organometallics, 2002, 21, 4978 CrossRef CAS.
- S.-B. Zhao, R.-Y. Wang and S. Wang, J. Am. Chem. Soc., 2007, 129, 3092 CrossRef CAS.
-
(a) J. D. Scott and R. J. Puddephatt, Organometallics, 1983, 2, 1643 CrossRef CAS;
(b) D. Song and S. Wang, J. Organomet. Chem., 2002, 648, 302 CrossRef CAS.
-
(a) D. Song, H. Schmider and S. Wang, Org. Lett., 2002, 4, 4049 CrossRef CAS;
(b) Q. Wu, J. A. Lavigne, Y. Tao, M. D'Iorio and S. Wang, Chem. Mater., 2001, 13, 71 CrossRef CAS.
-
(a) A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34;
(b)
PLATON - A Multipurpose Crystallographic Tool, Ultrecht University, Ultrecht, The Netherlands, A. L. Spek, 2006 Search PubMed.
- S. Nunomoto, Y. Kawakami, Y. Yamashita, H. Takeuchi and S. Eguchi, J. Chem. Soc. Perkin Trans., 1990, 111 Search PubMed.
-
(a) A. Kiyomori, J. F. Marcoux and S. L. Buchwald, Tetrahedron Lett., 1999, 40, 2657 CrossRef CAS;
(b) A. Klapars, J. C. Antilla, X. H. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727 CrossRef CAS;
(c) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS;
(d) J. C. Antilla, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 11684 CrossRef CAS.
- H. Hart, A. Bashir-Hashemi, J. Luo and M. A. Meador, Tetrahedron, 1986, 42, 1641 CrossRef CAS.
-
(a) M. Lautens, K. Fagnou and D. Yang, J. Am. Chem. Soc., 2003, 125, 14884 CrossRef CAS;
(b) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS.
-
(a) F. A. Cotton, T. LaCour and A. G. Stanislowski, J. Am. Chem. Soc., 1974, 96, 754 CrossRef CAS;
(b) F. A. Cotton and A. G. Stanislowski, J. Am. Chem. Soc., 1974, 96, 5074 CrossRef CAS;
(c) F. A. Cotton and V. W. Day, J. Chem. Soc., Chem. Commun., 1974, 415 RSC;
(d) F. A. Cotton and R. L. Luck, Inorg. Chem., 1989, 28, 3210 CrossRef CAS.
-
(a) M. Brookhart and M. L. Green, J. Organomet. Chem., 1983, 250, 395 CrossRef CAS;
(b) R. H. Crabtree, E. M. Holt, M. Lavan and S. M. Morehouse, Inorg. Chem., 1985, 24, 1986 CrossRef CAS.
-
(a) J. M. Casas, L. R. Falvello, J. Forniés and A. Martín, Inorg. Chem., 1996, 35, 6009 CrossRef CAS;
(b) A. Albinati, F. Lianza, P. S. Pregosin and B. Müller, Inorg. Chem., 1994, 33, 2522 CrossRef CAS;
(c) T. W. Hambley, Inorg. Chem., 1998, 37, 3767 CrossRef CAS;
(d) T. Kawamoto, I. Nagasawa, H. Kuma and Y. Kushi, Inorg. Chem., 1996, 35, 2427 CrossRef CAS;
(e) A. Albinati, P. S. Pregosin and F. Wombacher, Inorg. Chem., 1990, 29, 1812 CrossRef CAS;
(f) A. Albinati, C. G. Anklin, F. Ganazzoli, H. Rüegg and P. S. Pregosin, Inorg. Chem., 1987, 26, 503 CrossRef CAS;
(g) A. Albinati, C. Arz and P. S. Pregosin, Inorg. Chem., 1987, 26, 508 CrossRef CAS.
-
(a) N. Carr, L. Mole, A. G. Orpen and J. L. Spencer, J. Chem. Soc., Dalton Trans., 1992, 2653 RSC;
(b) J. L. Spencer and G. S. Mhinzi, J. Chem. Soc., Dalton Trans., 1995, 3819 RSC.
-
(a) S. Niu and M. B. Hall, Chem. Rev., 2000, 100, 353 CrossRef CAS;
(b) C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.