Luminescent rare earth nanomaterials for bioprobe applications
Jie
Shen
,
Ling-Dong
Sun
and
Chun-Hua
Yan
*
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications & PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, Peking University, Beijing. E-mail: yan@pku.edu.cn; Fax: +86-10-6275-4179; Tel: +86-10-6275-4179
First published on 6th August 2008
Abstract
Inorganic fluorescent nanoparticles (NPs) have initiated an extensive upsurge in biological application research. Just as quantum dots are regarded as a vigorous reinforcement of the organic dye family, rare earth (RE) fluorescent NPs, as another phosphors branch, also possess unique optical characteristics. The advantages of RE NPs in photostability and colorimetric purity make them suitable for bioprobe applications. Since the preparation technologies have been well developed, it is favourable to prompt the research in the interdisciplinary field of biology and material sciences. Herein, we summarize the synthesis and performance, together with bioprobe applications of RE oxide, sulfoxide, vanadate, phosphate, fluoride, and sodium RE fluoride nanomaterials. The prospects of these promising materials as applied in the biological field is described to draw readers′ attention and to attract more research interest.
 Jie Shen Jie Shen |
 Ling-Dong Sun Ling-Dong Sun |
 Chun-Hua Yan Chun-Hua Yan | Chun-Hua Yan received his PhD from Peking University (PKU) in 1988. He later became Lecturer (1988), Associate Professor (1989), Professor (1992), and Cheung Kong Professor of Chemistry (1999). His research fields are rare earth functional nanomaterials and molecular-based materials. |
1. Introduction
Fluorescence analysis is one of the most important techniques in biology and biomedicine due to its non-invasive mode and high sensitivity. Generally speaking, a biomolecule probe is grafted with certain markers to generate a detectable fluorescent signal. As a prerequisite for bio-analysis, neither the bioactivity of the host probe nor the optical property of the marker is damaged when the biomolecule is labled with the fluorescent marker. The marker-labeled composite probes can be investigated conveniently when they are performing biological functions. Various instruments (i.e.fluorescence microscope, plate reader, and biochip scanner) will support the research for qualitative/quantitative purposes. For in vitro detection, ideal fluorescent probes should have good monochromaticity, high luminescent efficiency, large absorption cross section, vigorous photo-bleaching resistance, and stable chemical/physical characteristics. In addtion, biocompatibility is not negligible for in vivo bioassay. To satisfy these requisites, organic dyes and fluorescent proteins are commonly used as optical bioprobes.1 Organic dyes have a high quantum efficiency, and ultra-sensitive detection has been demonstrated even down to single molecule level.2 Furthermore, steric hindrance is negligible because the molecules are too small to obstruct the biological interactions or events. But to serve as optical markers, the rapid photobleaching of dyes limits the available detection time,3 and the short fluorescent lifetimes and broad spectral features do not benefit reducing the background interference to increase the signal to noise ratio. The subsidiary marker system relies on fluorescent proteins, whose manipulation can be combined with genetic techniques.4 These engineered proteins can be endogenously expressed by organisms, but always suffer from weak luminescence.5 The above mentioned limitations ensure a foothold for other species of fluorescent markers.Over the past decade, inorganic luminescent NPs, quantum dots (QDs) as a typical representation, have attracted a great deal of attention from biologists.6 QDs are distinguished from all other materials by their characteristics of unique size- and composition-dependent luminescence which can be tuned precisely.7,8 As a kind of fluorescent marker, QDs are endowed with many advantages such as narrow emission bandwidth, considerable photo-stability, single-source-excitation but multiplex detection, and comparable size with that of biomolecules (below 10 nm). Since the first report of colloidal QDs as bioprobes in 1998,9,10 papers have avalanched concerning immunoassay, cell imaging, single-molecule detection, and polynucleotide diagnostics.11–13 However, the imperfections of QDs lie in hydrophobicity, toxicity, and high synthesis expenditure.
The demand for new markers is still on-going, and rare earth (RE) nanomaterials are proposed as attractive candidates. The RE elements are composed of the lanthanide (Ln) series (from lanthanum to lutetium), yttrium, and scandium. With abundant f-orbital configurations, Ln ions can exhibit sharp fluorescent emission viaintra-4f or 4f–5d transitions. Their monochromaticity and resistance to photobleaching is undoubtedly useful in bioprobe applications, together with other infusive properties such as large Stoke’s shift and long fluorescence lifetime. Fluorescent Ln chelates have been used in immunoassays prior to RE NPs,14–16 which promoted the development of the time-resolved fluorescence detection methodology.17 However, Ln ions in chelates may be quenched by water molecules or hydroxyl groups. Recently, RE inorganic nanomaterials have been proposed to be more suitable for optical markers, because rigid crystal lattices provide a steady microenvironment for the emitter, Ln dopants. This Perspective reviews the development of current RE luminescent NPs combined with their potential in biological applications. Meanwhile, we introduce our recent endeavours on this theme and give a general prospect.
2. Luminescent RE NPs
Conventional RE bulk phosphors play a vital role in lighting sources and solid-state lasers. Now, growing endeavours are devoted to their nanophase synthesis for new applications. It has been reported that the RE NPs inherit the unique characteristics of their bulk compound: (i) these NPs have a much longer fluorescence lifetime than QDs and organic species, up to millisecond magnitude, which make it possible to substantially diminish the spontaneous background fluorescence of the biological specimen via time-gated detection;18 (ii) the luminescence of Ln ions does not involve valence electron transitions, which may be conducive to the stability of compounds against photooxidation;19 (iii) multicolor luminescence can be easily realized by varying the Ln dopant and host matrix. The atomic like emission lines of Ln ions are entirely isolated without overlap. Moreover, unlike the size-dependent emission of QDs, the fluorescence wavelength of Ln ions is not sensitive to particle size. Rigorous synthesis is thus avoided while the spectral purity of the products is ensured; and (iv) up-conversion (UC) luminescence from Er3+, Tm3+, Ho3+ and Pr3+, etc, a potential new hotspot in the bioprobe field, can be realized besides common down-conversion behavior.20For the purpose of bioprobe applications, a large amount of synthetic strategies on RE nanomaterials have been described. These studies covered several series of Ln doped NPs, including RE oxides, sulfoxides, vanadates, phosphates, fluorides, and sodium RE fluorides. The emission spectra extend from the visible to the near infrared (NIR) region (450–1650 nm) via an up/down-conversion mechanism. However, exploration of their biological application is still less developed. In the following sections, a few key RE nanomaterials will be discussed in detail. Some sub-micrometer sized RE phosphors are also involved, although the strict definition of nanomaterial confines artificial compounds to those between 1 and 100 nm in diameter only.
2.1 RE oxides and oxysulfides
RE oxides and sulfoxides are widely used in illumination and in the tricolour display industry. For instance, Y2O3:Eu, Gd2O3:Eu, and Y2O2S:Eu always take up the host of red phosphors. Their excitation spectra mainly lie in the ultraviolet (UV) region below 300 nm, which are assigned to the charge transfer band (CTB) of Eu3+–O2− pairs. This energy gathering mode is extremely effective and the quantum yield (QY) in bulk materials approximates to 100%. The sulfoxide matrix has an additional advantage in cathode ray and X ray absorptivity (i.e., Gd2O2S:Tb). Altering these traditional phosphors to low dimensions may be a shortcut to create novel bioprobes.Pure Eu2O3 emits weak luminescence due to strong concentration quenching, and Gd2O3 is an excellent host compound to disperse Eu(III) dopant. ca. 120 nm spherical Gd2O3 NPs were doped with 20% Eu3+ to target phenoxybenzoic acid (PBA) by a microarray immunoassay (Fig. 1).22 The Gd2O3:Eu NPs were synthesized by spray pyrolysis to reach a high doping concentration and avoid phase segregation. After encapsulation of the bare particle by poly(L-lysine), anti-PBA IgG was covalently conjugated to the surface. In the model microarray assay, PBA-labeled BSA protein (bovine serum albumin) was introduced into strip patterns as capture antigen, and BSA–fluorescein was anchored into the spacing regions as internal standard. The capture antigens competed with free PBA molecules for binding sites on the Gd2O3:Eu surface, then immobilized NPs were evaluated by a microplate reader equipped with a 488 nm Ar+ laser. Kennedy, Nichkova et al. also reported a modification of Gd2O3:Eu NPs with IgG through a physical adsorption method.23 The rigid particle exhibited a radial shell after anti-rabbit IgG coating as observed by transmission electron microscopy (TEM). Besides the similar fluorescent imaging experiment, scanning electron microscopy was employed to analyse the planar morphology of microarrays. Two methodologies both confirmed the specific labeling of NPs.
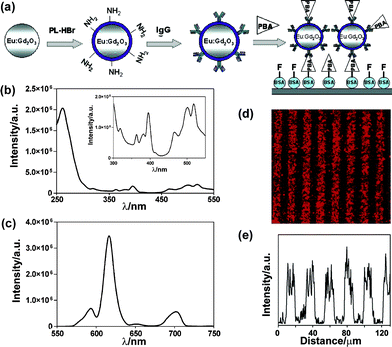 | ||
| Fig. 1 (a) Scheme of the competitive immunoassay. (b) The excitation spectra of Gd2O3:Eu NPs with λem= 615 nm and (c) the emission spectra with λex= 260 nm. The inset in (b) shows the excitation peaks of Eu3+. (d) Fluorescence image of Gd2O3:Eu labeled microarrays and (e) the corresponding fluorescence intensity profile. Reprinted with permission from M. Nichkova, D. Dosev, S. J. Gee, B. D. Hammock and I. M. Kennedy, Anal. Chem., 2005, 77, 6864. © 2005 American Chemical Society. | ||
In contrast to the ordinary energy transition mode of “down-conversion”, the mechanism of up-conversion is a process that converts two or three low energy photons to a high energy one which gives out shorter wavelength emissons. The UC luminescence provides several advantages for use in bioassays: (i) large anti-Stoke’s emission of visible light under NIR illumination; (ii) multicolor fluorescence through the up-conversion process with single wavelength excitation (980 nm); (iii) free of background autofluorescence from biological specimens; (iv) mild attenuation decrease and low photo-induced damage while 980 nm wavelength light penetrates organic tissues; and (v) low cost to equip NIR diode laser source or high-power xenon lamp. Since a sulfoxide matrix has a lower phonon vibrational energy compared with a RE oxide lattice, it is a good candidate for UC fluorescent materials. Using solid state reactions, sub-micrometer sized UC phosphors Y2O2S:Yb,Er(Tm) can be synthesized.24 In brief, the process was co-precipitation of RE ions plus a high temperature fluidised bed treatment. The co-doped Yb3+ can increase the absorption efficiency of 980 nm NIR light and sensitize Er3+/Tm3+ luminescence. When the excitation energy is transferred from Yb3+ to adjacent emitters, two- or three-photon are processed, which show 2 or 3 orders of emission intensity dependent excitation power, takes place for Er3+ or Tm3+, and green/red or blue emissions are observed.
A former review article by Corstjens et al.24 has summarized the application of oxysulfide UC markers. Some highlights are refined here. Tanke and co-workers reported satisfactory sensitivity for DNA detection using 200 nm Y2O2S:Yb,Er@SiO2 particles.25 Because of no background interference, the assay detection limit was even four times lower than that of the cyanin 5 (Cy5) system (Fig. 2). Zijlmans et al.26 utilized Y2O2S:Yb,Er(Tm) particles to detect prostate-specific antigen in human prostate tissue sections. Different from standard immunohistochemistry methods, background autofluorescence was completely diminished in the microscope image. Corstjens et al.24 used two kind of UC phosphors (doped with Er and Tm, respectively) to capture IgM and IgG from cytomegalovirus-positive serum. The assay was sensitive enough to screen for a positive signal from 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold dilution serum samples. Some other attempts were devoted to rapid immuno-chromatography. UC markers can analyse single-strand DNA, drug molecules and pathogens by the lateral-flow method.27–29
000-fold dilution serum samples. Some other attempts were devoted to rapid immuno-chromatography. UC markers can analyse single-strand DNA, drug molecules and pathogens by the lateral-flow method.27–29
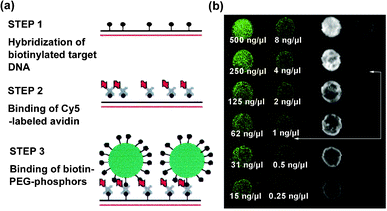 | ||
| Fig. 2 (a) Experimental procedure for DNA detection. (b) Fluorescence outputs from UC phosphors (left) and Cy5 dyes (right). The data refer to DNA concentration and arrows indicate detection limit. Reprinted with permission from F. Rijke, H. Zijlmans, S. Li, T. Vail, A. K. Raap, R. S. Niedbala and H. J. Tanke, Nat. Biotechnol., 2001, 19, 273. © 2001 Nature Publishing Group. | ||
Not only in oxysulfides, but also Yb/Er in an Y2O3 matrix displays UC ability. Lim et al.30 described an UC imaging case in vivo. Y2O3:Yb,Er particles with high Er3+ concentration favored red emission, while less Er3+ doped particles mainly unveiled green fluorescence. For tracing the digestive system of the nematode Caenorhabditis elegans, 150 nm naked particles were directly transferred to the growth medium used to feed the worms. Both NIR laser and electron radiation were used to illuminate the UC phosphors in the worm's intestines. Further tracking showed the worm could excrete its swallowed particles immediately when new foodstuff was available (Fig. 3). Neither degradation of phosphors nor damage to worms were observed, and this suggested the biocompatibility of Y2O3:Yb,Er.
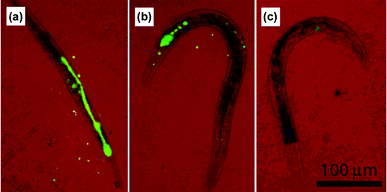 | ||
| Fig. 3 The excretion behavior of C. elegans in food abundant medium for (a) 0 h, (b) 1 h, and (c) 2 h. False color images with red representing the bright field and green standing for the Y2O3:Yb,Er emission. Reprinted with permission from S. F. Lim, R. Riehn, W. S. Ryu, N. Khanarian, C. Tung, D. Tank and R. H. Austin, Nano Lett., 2006, 6, 169. © 2006 American Chemical Society. | ||
These applications of RE oxide and sulfoxide markers achieved preliminary success, but all these compounds are not nanomaterials according to the rigid definition. A sub-micrometer sized particle is inaccessible and hardly dispersible after all; further improvement is desired for versatile bioprobes. Some retrieval steps like bead-milling can not thoroughly solve this problem unless researchers bring a revolution in synthesis methodology for nanosized particles.31,32
Roux's group extended the polyol method in order to prepare Y2O3:Eu and Gd2O3:Eu NPs.33 This design relied on two reasons: hydrolysis of RECl3 in diethylene glycol avoided particle agglomeration, and the elevated temperature benefited getting rid of the water component during the reaction. Using a similar protocol, green fluorescent Gd2O3:Tb NPs were synthesized.34 The NPs were decorated with streptavidin or oligonucleotides to label different microbeads. Pyrolysis of organometallic precursors in high-boiling-point solvents is a versatile methodology to synthesize metal and metal oxide NPs. Our group developed a general route to prepare the whole series of RE2O3nanocrystals including CeO2.35 RE benzoylacetonate complex was used as the precursor and oleic acid/oleylamine as capping ligands, thus uniform hydrophobic nanoparticles were obtained. The products were readily dispersed in non-polar solvents. By varying the ratio of capping ligands, the morphology can be adjusted to nanopolyhedra, nanoplate, and nanodisk. Gao's group synthesized monodisperse Ln oxysulfide NPs by a one-step reaction (Ln = Sm, Eu, Gd).36 During the thermolysis process, the diethyldithiocarbamate ligands of the Ln chelate precursor provided sulfur for Ln2O2S growth. Meanwhile, the oxygen acted as the oxidant. This mild method does not need the time and energy consumed by the sulfuration process on a fluidised bed, offering a new preparation route for oxysulfide NPs.
In conclusion, wet chemical techniques improve the dispersibility and size distribution of Ln oxide and oxysulfide NPs. Hydrophobic products can be obtained with subsequent surface modification. Thus, compared with non-solution routes, wet synthesis methodologies are more feasible towards bioprobe research.
2.2 RE oxy-acid salt
Quite a lot of commercial RE phosphors belong to solid oxy-acid compounds. The available host materials include borate, silicate, aluminate besides commonly used vanadate and phosphate. This section concerns only the latter two sorts. Ln ions have low absorption coefficient due to both spin and Laporte forbidden f–f transitions. This limitation can be overcome by the indirect excitation of dopants. Two examples are red phosphor YVO4:Eu as well as green phosphor LaPO4:Ce,Tb. Compared with the weak and narrow excitation line of Eu3+, the broad absorption ranged at 200–350 nm is attributed to the VO43−groups. The excited vanadate subsequently transfers its energy to adjacent emitters via thermally activated migration, and the QY of bulk YVO4:Eu could reach ca. 70%. For LaPO4:Ce,Tb phosphors, the Ce3+ acts as activator which has a strong 4f–4f5d absorption between 250–290 nm in phosphate matrix, and the energy is non-radiatively transferred to the emitter, the Tb3+ ion. The highest QY for its bulk material is ca. 80%. Such an efficient host-sensitized energy transfer process makes YVO4:Eu and LaPO4:Ce,Tb reveal good emission performances, and the corresponding nanosized crystals are proposed to be useful in biological imaging.Derived from pure RE vanadate, Riwotzki and Haase37 first synthesized phosphate-diluted vanadate NPs YPxV1−xO4:Eu, which were employed as model compounds for the luminescence study of YVO4:Eu NPs. Very recently, Liu's group aimed at the YP0.75V0.25O4 matrix to produce multicolor NPs by mixing the fluorescence from Ln dopants and the YP0.75V0.25O4host into the final tunable color.38 Furthermore, Ln-doped phosphate and vanadate NPs exhibited various spectra for the transition probability which was closely related to the crystal field.20,39 Other researches focused on the NIR fluorescence from Nd3+, Er3+, Pr3+ and Ho3+ emitters,19,40–44 enlarging the variety of RE optical markers.
However, all these reported YVO4:Eu NPs showed low QY of only around 15% at room temperature, which may be basically ascribed to surface quenching. The size of YVO4:Eu NPs is close to the exciton diffusion length in its bulk crystal, thus the energy transferred to surface europium sites is probably quenched by coordinated groups. As indirect evidence, an anticipated upgrade in QY was observed by dispersing YVO4:Eu NPs in D2O instead of H2O.48 Riwotzki and Haase attributed the energy dissipation mainly to non-radiative recombination of surface vanadate groups, supported by temperature dependent luminescence spectroscopy and luminescence lifetime measurements. They also proposed a core/shell structure YVO4:Eu@YPO4 to solve this problem.37
Another point is toxicity of vanadium compounds.50 Although YVO4:Eu NPs have little chance to release free vanadate anions and their main applications are concentrated with bioassays in vitro, rigorous surface passivation is still necessary for biological safety. To enwrap REVO4 NP in a shell for this purpose, low toxicity REPO4 is a good choice. Successful surface modification methods can be referenced from QD research.12 Anyway this issue is solvable.
Since the core/shell strategy has been successfully applied to QDs to improve their fluorescence, Haase and co-workers were inspired to epitaxially grow a LaPO4 shell to passivate the CePO4:Tb surface.53 The inner CePO4:Tb cores were prepared in organic solvents following their previous route, then another portion of phosphoric acid and La3+ ions were added for the growth of the phosphate shell. High-resolution TEM (HRTEM) images indicated that the products were all single crystal NPs. No CePO4 was mixed with the LaPO4 outer layer, as confirmed by X-ray photoelectron spectroscopy. This core/shell structure was favorable to suppress the surface energy loss channel. The energy diffusion was completely confined to the core region while only inert La3+ existed in the protective shell, therefore a high QY of 70% was achieved. A similar method was also used for LaPO4:Eu@LaPO4 NPs.54 Boilot utilized sodium tripolyphosphate (Na5P3O10) and Ln nitrate to prepare trigonal rhabdophane LaPO4:Ce,Tb·xH2O NPs in aqueous conditions,55 in which Na5P3O10 played the role of complexing agent as well as anion source. The residual polyphosphate stabilized the as-synthesized NPs to form a transparent colloidal solution directly. To prevent the oxidation of Ce3+, they actualized the CePO4:Tb·xH2O@LaPO4·xH2O core/shell structure later.56 Although the formation of Ce4+ quenching sites was inhibited, the promotion of QY was not efficient enough (increased from 30 to 35%). This was ascribed to various factors, such as low crystallinity below a 90 °C environment, incomplete shell coverage, inappropriate crystal phase, excess structure water and so on. Even though the core/shell strategy is rarely utilized in Ln doped NPs at present, it is useful to explore more efficient nanosized phosphors with a core/shell structure for future applications.
 | ||
| Fig. 4 (a) Scheme of guanidine-labeled YVO4:Eu NPs. (b) Fluorescence spectrum including excitation lines of Eu(III). (c) Wide-field image and (d) time-gated image of Na+ channels blocked with Gua–NPs. Reprinted with permission from E. Beaurepaire, V. Buissette, M. Sauviat, D. Giaume, K. Lahlil, A. Mercuri, D. Casanova, A. Huignard, J. Martin, T. Gacoin, J. Boilot and A. Alexandrou, Nano Lett., 2004, 4, 2079. © 2004 American Chemical Society. | ||
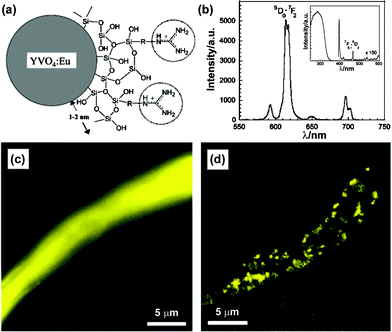
Here, we are glad to introduce our unpublished results on the bioapplications of YVO4:Eu and LaPO4,Ce,Tb NPs. A straightforward hydrothermal method was employed to synthesize high quality zircon type YVO4:Eu and monazite type LaPO4:Ce,Tb NPs in the presence of phosphorus containing poly-acrylic acid (PPA). PPA acted as chelating agent and surface stabilizer here, whose phosphono and carboxyl groups exhibited coordinative ability. By controlled release of RE3+ from PPA chelates, the co-precipitation process was restrained and the slow growth resulted in well-defined morphology. TEM and HRTEM images showed the products were single crystals with sizes of ca. 20 nm for YVO4:Eu and ca. 6 nm for LaPO4:Ce,Tb NPs. The colloid NPs exhibit high stability in aqueous and buffer solutions as reserved for more than one year. Further biotinhydrazide modification was conducted according to the traditional method, with activating carboxyl groups of PPA by N-hydroxysulfosuccinmide sodium salt (Sulfo-NHS) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC).60 Then these biotin-labeled NPs were successfully applied in FRET assay, microarray detection, and fluoroimmuno-assay, respectively.61,62 In FRET studies, the non-radiative energy transfer from excited biotin–LaPO4:Ce,Tb NPs (donors) to avidin-labeled gold NPs (acceptors) was substantiated.61 The FRET efficiency studies of changing the diameter of gold NPs and the donor/acceptor ratio clearly indicated that the resonance energy transfer process mainly took place at the surface of the RE NPs.
For simulation of biochips, biotin-labeled YVO4:Eu NPs were used to detect trace amounts of avidin in microarrays. Avidin protein was immobilized on an aldehyde slide from 0.5 to 0.016 ng for each spot with a 2-fold dilution order. And these spots were arranged into six 10 × 10 subarrays with spot spacing of 450 μm. After labeling with biotin–YVO4:Eu NPs, fluorescent patterns were visualized by the naked eye under a 254 nm UV lamp. The fluorescence intensity of each subarray matched well to the different avidin amounts. Dual color microarray patterns were also performed by using YVO4:Eu (red) and LaPO4:Ce,Tb (green) markers (Fig. 5). More than this semi-quantitative analysis, the above RE markers were tested by quantitative fluoroimmunoassays. In a sandwich immunoassay model, avidin and biotin–YVO4:Eu NPs were substituted for the traditionally used enzyme-linked-avidin to recognize the biotin-labeled second antibody (detection anti-body). The fluorescence output exhibited good linearity with the analyte concentrations viafluorometry ELISA reader. Moreover, we used a vanadate fluorescent marker for the preliminary cell image tests. PPA capped GdVO4:Eu NPs were directly incubated with adherent macrophages for 2 h, then the cells were thoroughly washed and fixed. Cell images were obtained from a point-by-point spectrum mapping route. The location of the characteristic emission of GdVO4:Eu (from 613 to 621 nm) was coincident with the cell boundary, displaying excellent spatial resolution and signal/noise ratio (Fig. 6). An extension of this research is labeling target molecules in distinct cellular compartments. The size and surface chemistry of RE NPs will be focus points then, which evaluate the material biocompatibility and lead the bioapplication studies.
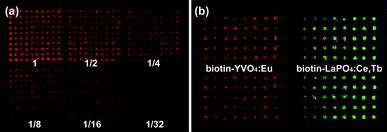 | ||
| Fig. 5 (a) Avidin microarrays of 2-fold dilution series labeled with biotin–YVO4:Eu NPs. (b) A two-color microarray imaging strategy using red YVO4:Eu and green LaPO4:Ce,Tb markers. | ||
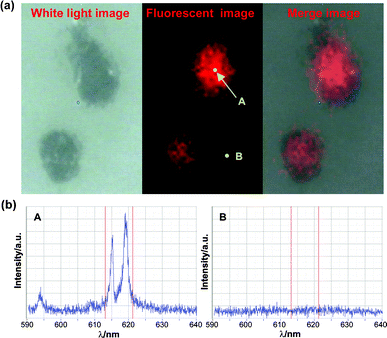 | ||
| Fig. 6 (a) Fluorescence mapping image of adherent macrophages probed by GdVO4:Eu@PPA NPs and (b) the corresponding fluorescence spectra. | ||
The crucial point for Y(Gd)VO4:Eu and LaPO4:Ce,Tb NPs is that their effective excitation is located in the UV region. It is proved that irreversible damage is brought to creatures under long-term UV irradiation. Furthermore, short wavelength light has weak organism penetrability, and high power UV source equipment is not popular. Therefore, their application as bioprobes is temporarily limited. Even though Y(Gd)VO4:Eu and LaPO4:Ce,Tb NPs have the above drawbacks, they are still worthy of studying as they are totally harmless in in vitro bioassays.
2.3 RE fluoride and sodium RE fluoride
REF3 and NaREF4 are two similar RE based phosphors. The host compounds themselves have low absorbency, and the emitter can be excited directly. Besides generally studied Eu3+ or Tb3+ doped compounds, the main application of RE fluoride is UC fluorescent materials. Especially, hexagonal phase NaYF4 is one of the most efficient matrices for UC emission due to its low phonon vibration energy.63 Aimed at biological applications, NaREF4 turns out to be the major focus in UC phosphor research field now. Nanosized phosphors with Ho, Er, Tm dopants have been described.64–66UC fluorescent bioprobes possess special superiorities as stated in the former section. While RE oxide/oxysulfide markers are limited by their complex synthesis, wet chemical routes show convenience and variability in preparation of RE fluoride and relative phosphors. The size criterion of bioprobes is also satisfied by REF3/NaREF4. Due to the importance of UC fluorescent markers, this section will take more space to discuss sodium RE fluoride nanomaterials.
Veggel et al.71 obtained water-soluble and bio-functionalized fluoride NPs from a one-pot reaction. They grafted biotin onto hetero-bifunctional spacer molecules in advance to control NP precipitation. To further eliminate the surface nonspecific adsorption, triethylene glycol methyl ester was inserted among the former ligands. The as-prepared LaF3:Eu NPs could specifically bind to avidin cross-linked agarose beads. With a prevalent route, Veggel's group modified citrate-stabilized LaF3:Ln NPs with silica shells, and decorated the particle with biotin after APTMS modification.72 These kind of markers exhibited NIR emission when utilizing Nd3+ dopants. Wang et al. synthesized LaF3:Eu NPs by adopting chitosan as a capping agent in aqueous solution. The biocompatibility was validated by the human colon HT-29 cell line, and no influence on cell viability was observed during LaF3:Eu@chitosan incubation.73 Core/shell strategy was practiced for fluoride NPs by Lin et al.74 The CeF3:Tb@LaF3 NPs prepared in 200 °C diethylene glycol exhibited an expected luminescence enhancement. As subsequent work, they stabilized CeF3:Tb NPs with a SiO2-NH2 shell, and followed the conventional avidin/biotin biotarget protocol.75 Wang and Li reported non-enzymatic glucose detection with glucose-labeled LaF3:Ce,Tb NPs as donor and reactive rhodamine B (RhB) as acceptor in FRET assay. To capture the donor, RhB was derivatised with boronic acid for conjugation with ortho-dihydroxy of glucose.76 If free glucose molecules competed with glucose–LnF3 for binding with the RhB, the FRET efficiency would be markedly reduced.
Haase and co-workers obtained cubic-phase NaYF4:Yb,Er (Tm) NPs for the first time via the organic solvent route.78 Two stock solutions which contained cationic (Na+ and Re3+) and anionic-educt (F−), respectively, were mixed and heated for nanocrystal growth. UC fluorescence of these compounds was 107 times stronger referring to formerly reported YbPO4:Er and LuPO4:Yb,Tm NPs. Yi et al.79 reported co-precipitation of RECl3 and NaF in aqueous conditions with the aid of ethylenediamine tetraacetic acid (EDTA), and the crude deposits annealed at 400 °C to form NaYF4:Yb,Er NPs. Certain antibody proteins were labeled on NaREF4@SiO2-NH2 for immunoassay, and the as-synthesized probes showed specific recognizability revealed by NIR scanned images.
It is well-known that hexagonal (β-phase) NaYF4:Ln exhibit much stronger UC fluorescence than the cubic (α-phase) modality.66 To facilitate an α-to-β phase transition,80annealing treatment at 400–600 °C did not ensure the phase purity of product, and sometimes led to undesired changes on NP morphology and monodispersity. Alternatively, a simple solvothermal route was developed to avoid the two typical problems described above.81 The as-synthesized hexagonal NaYF4:Yb,Er particles had a uniform size of ca. 50 nm, and they were dispersible in ethanol. Li and co-workers subsequently applied these UC NPs in FRET assay for avidin.82 Two kinds of polyelectrolyte loading opposite charges were used to stabilize NPs according to the layer-by-layer (LbL) assembly method. The outmost polymer layer also provided amino groups for biotin labeling. The analyte avidin acted as a bridge to combine biotin–NaYF4:Yb,Er NPs (donor) and biotin–gold NPs (acceptor), then 7 nm gold NPs with an absorption band at 540 nm quenched the UC fluorescence. Associated with magnetic NPs, UC markers were also competent for ultra-sensitive assays of single-strand DNA (ssDNA).83 The target ssDNA was first hybridized with its complementary sequence labeled on Fe3O4 NPs, and then its overhanging region was matched to a NaYF4:Yb,Er labeled probe strand (Fig. 7). By magnetic separation, the UC fluorescence from the sandwich composite was correlative with the concentration of target ssDNA. This method was free of ssDNA amplification by polymerase chain reactions, and the detection limit was around 10 nM.
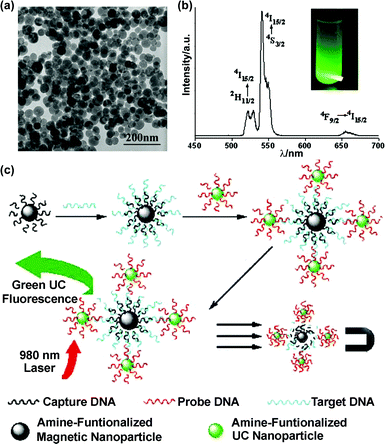 | ||
| Fig. 7 (a) TEM image of 50 nm β-NaYF4:Yb,Er NPs. (b) The UC fluorescence spectrum and visual image of a solution sample. (c) Scheme of ssDNA sandwich assay.83 Reprinted with permission from L. Y. Wang and Y. D. Li, Chem. Commun., 2006, 2557. © 2006 Royal Society of Chemistry. | ||
There were some other synthesis strategies available besides solvothermal methods. Chen et al.84 adopted RE oleate and NaF to synthesize β-NaYF4 nanoplates with liquid/solid hetreogeneous reaction. The nucleation and growth process was localized at the two-phase interface, and an interface transfer mechanism was proposed to explain the narrow size distribution of products. Liet al.85,86 performed a more complicated three-phase system to prepare uniform α and β phase NaYF4 NPs, which included RE aliphatate salt solid phases, ethanol–aliphatic acid liquid phases and water–ethanol solution phases. Inheriting previous LaF3 research, we have demonstrated a general synthesis strategy for monodisperse NaREF4 (RE = Pr to Lu, Y) NPs viapyrolysis of metal trifluoroacetate multi-precursors.87 High-quality α or β phase NaYF4:Yb,Er(Tm) NPs were prepared in one step by varying the ratio of CF3COONa/RE(CF3COO)3 and the solvent composition. We also studied the process by which α-NaYF4 precursors changed to β-NaYF4 NPs under extreme treatment (330 °C, oleic acid/1-octadecene). The formation of large β-phase nanoplates was accompanied by the consuming of small α-phase particles. The detailed mechanism was discussed in our recent paper.88
An important advantage for RE based UC markers is that single excitation (980 nm larser) activates multicolor emissions from different dopants or the same dopant but of various concentrations or structures.89 Previous multicolor fluorescence of NaREF4 was visualized through filters,78,90,91 rather than reflecting the entire spectrum. However, the multicolor emissions of our UC NPs is naked-eye compatible. Digital images of fluorescence and the corresponding spectra (displayed in Fig. 8) were gathered from cyclohexane solution under 980 nm diode laser (50 mW) excitation. The UC fluorescence colors of green, red, and yellow were based on different sized α or β phase NaYF4:Yb,Er NPs and NaYF4:Yb,Er@α-NaYF4 core/shell NPs.92 Generally speaking, the fluorescence from 4H11/2 to 4S3/2 and 4I15/2 (green) is weak in α-NaYF4, but is largely promoted in hexagonal phase NPs. The size of α-NaYF4 NPs also has an effect on the transition probability, just like smaller NPs prefer red fluorescence. Interestingly, the α-NaYF4 shell coating increased not only the emission intensity but also the green/red ratio for both α and β phase NaYF4:Yb,Er NPs.92 In Yi and Chow's previous research, β-NaYF4:Ln@β-NaYF4 enhanced UC efficiency with 7.4 and 29.6 times for Yb/Er and Yb/Tm doped NPs, respectively,93 and our results complemented this core/shell strategy.
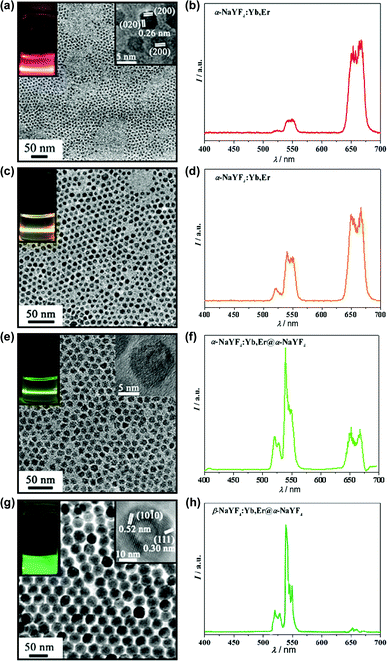 | ||
| Fig. 8 The TEM images, visual fluorescence images (a, c, e, g), and corresponding UC emission spectra of NaYF4:Yb,Er series (b, d, f, h). | ||
NaYF4:Ln NPs mentioned in most cases were protected by hydrophobic ligands, and should be rendered with water-stability before using as bioprobes. Here are some practical methods to obtain aqueous NaYF4. Yi and Chow obtained β-NaYF4:Ln NPs in pure oleylamine at 330 °C. The surface oleylamine was afterwards exchanged by glycol-600-diacid in ethanol/hexane 1:1 mixture.94 Amphiphilic polymer, octylamine/isopropylamine grafted poly(acrylic acid), was also used to exchange the surfactant. With polymer encapsulation, the UC NPs were also tuned to be water-soluble.93 Zhang's group used polyvinylpyrrolidone (PVP) as chelating agent and ethylene glycol as solvent.90 The yielded α-NaYF4:Yb,Er NPs were dispersable in either polar or non-polar solvents, such as water, DMF, DMSO, chloroform, hexaneetc. The universal solubility was due to the amphiphilic surfactant PVP absorbed on NPs. For biocompatibility and further labeling, a silica shell was coated directly onto the particle by the hydrolysis of tetraethoxysilane. No additional surfactants were needed in contrast with ordinary silicatization methods. Liet al.95 provided a different approach in that the oleic acid at the NaYF4:Ln surface was oxidized in situ by the Lemieux–von Rudloff reagent (KMnO4/NaIO4 mixture). Converted from carbon–carbon double bonds, the newly formed overhanging carboxylic groups stabilized the particle in water. Then the streptavidin-labeled UC probes were operated as donors in ssDNA FRET assay. Referring to the literature route,96 we transferred hydrophobic β-NaYF4:Ln NPs to a water phase using the surfactant cetyl trimethylammonium bromide (CTAB), then addition of tetraethylorthosilicate (TEOS) plus APTMS formed the SiO2–NH2 shell. The uniform core/shell particles exhibit good dispersibility and fluorescence output in aqueous solution. Further research is on the way.
The NIR excitability of UC fluorescent markers definitely facilitates imaging research in vivo, since the largest obstruction to fluorescence assays, autofluorescence of organic tissues, can be eliminated with utilization of UC fluorescent markers. Multicolored and multisized UC markers may generate a revolutionary development in the near future.
2.4 RE bifunctional composite
Magnetic substances play a crucial role in magnetic resonance imaging (MRI) besides micro-manipulation technology, and recently the combination of fluorescent/magnetic properties has grown quickly in the nanomaterial research field. This section will envisage the potential of RE compounds as dual-mode probes.MRI is based on the difference relaxation rate of water protons exposed to a strong magnetic field. Some magnetic species could accelerate the relaxation of local water protons so as to enhance the signal contrast. Detailed information on MRI can be found elsewhere.97 In pre-surgical diagnosis, MRI is always adopted to uncover deep pathological changes. This X-ray-free technology enables surgeons to obtain 2-dimensional anatomic structures non-invasively, but it cannot participate in surgical operation due to its rigorous running parameters. On the other hand, fluorescence imaging methodology is good at real-time visualization of biological specimens. Several groups have successfully applied QDs as markers in vivo.98–101 Therefore it is possible to use dual-mode traceable probes for both pre-operative/intraoperative detection when they possess optical and magnetic functions simultaneously.102,103 This duplex method will increase the precision of diagnosis and therapy. Up to now, some magnetic/optical composite NPs have emerged, such as Mn2+ doped QDs,104,105 Fe3O4/dye complexes,106,107 FePt(Fe3O4, γ-Fe2O3)/QD heterojunctions,108–110 and γ-Fe2O3(Fe3O4)/QD@SiO2 nanobeads.111,112 Ln doped fluorescent nanomaterials also joined the trend of dual-functional combination. Chen and co-workers precipitated a NaYF4:Yb,Er shell onto Fe3O4 cores in an aqueous system, where the core particles were first synthesized from ferrous and ferric salts.113 In the presence of a magnetic field, the orientational arrangement of the NPs was observed via NIR-diode-equipped fluorescent microscopy. Kennedy's group reported Fe2O3:Co,Nd/Gd2O3:Eu core/shell particles prepared by spray pyrolysis.114 They encapsulated magnetic cores with 20 nm thick Gd2O3:Eu through a second pyrolysis step where Fe2O3:Co,Nd NPs served as seeds. The composite particles displayed a paramagnetic response ensuring specific separation, and the emitted red luminescence belongs to the Eu3+ dopants.
Ln cations with unpaired f-electrons should exhibit paramagnetic behavior at room temperature. The current commercial MRI contrast agents, Magnevist and Dotarem are Gd3+ chelates which have 7 unpaired f-electrons.115 Many explorations on dual-model probes still have recourse to gadolinium diethylenetriamine pentaacetic acid complex (Gd-DTPA, the active ingredient of Magnevist).116 Recent research found that Gd3+ in rigid lattices also influences the relaxation of water protons under a magnetic field.117 Suzuki et al.118 claimed that the GdPO4@dextran NPs are comparable to Gd-DTPA (Magnevist) in T1-weighted MR imaging. Roux and co-workers reported a bifunctional probe composed of Gd2O3 core and a polysiloxane shell with organic fluorophore dopants. After polyethylene glycol (PEG) modification, this probe was rendered with excellent circulating ability in animal vessel system. Both fluorescence and MR imaging uncovered the renal excretion of composite NPs, which were administrated by intravenous injection.119
Based on our experience on RE luminescence materials, we bring forward a convincing strategy. Homogeneous magnetic/optical functionalized NPs could be easily prepared by co-doping Eu3+/Tb3+ ions into Gd3+ host materials. Compared with heterostructured nano-composites, this mono-structured marker will be superior either from a synthesis feasibility or functional homogenization point of view.
3. Conclusions and outlook
The preparation, properties, and bioapplications of several key RE functional nanomaterials have been generally reviewed. Concerning these nanomaterials, elaborate control of morphology and performance is most important as their optical inherence is unique for bioassays. Although they still need to be improved to satisfy the requirements of fluorescent bioprobes, RE NPs are expected to fulfill special functions and complement the currently used and studied bioprobes. For instance, RE phosphors with long fluorescence lifetimes are more preferred in time-resolved fluorescence detection, and RE nanomaterials is a new rising field for multiphoton excitation imaging.Currently, bioprobe applications of Ln doped NPs is developing in a steady manner, and related research involve immunoassay, DNA detection, FRET assay, microarray imaging, cell targeting, etc. The ultimate aim for us is also gradually becoming unambiguous. First, optical performance is the most essential point to pursue, such as QY, photostability, fluorescent colorimetric purity and detection sensitivity. Second, the size/morphology and surface properties of NPs should be addressed. Uniform spherical particles below 50 nm are perfect as bioprobes, because small size is more accessible for biomolecules and easier to disperse. Furthermore, ultra-small particles (at least below 10 nm) with a neutral charged surface can exhibit efficient renal clearance while they are intravenously administered into model animals, so as to depress the accumulation of NPs in vivo and reduce their chronic toxicity.119,120 The small NPs also can make use of defective vascular architecture in tumor tissue to extravasate from vasculature for a specific target,121 which is known as the enhanced permeability and retention effect. Simultaneously, the hydrophilic surface of NPs with prolific chemically reactive groups (e.g. –COOH, –NH2, –SH) are desirable. Several papers report the formation of aqueous colloids of Ln doped NPs by a one-step synthesis, which is undoubtedly more convenient for further bioapplications. Third, improving the biocompatibility will be the last crucial procedure before we put Ln doped NPs into practice. Bioassay in vivo is increasingly accepted by academia and the safety of bioprobes must be ensured. It is believed that RE elements with similar behavior to calcium ions in organisms may have less toxicity than components in QDs, but toxicity evaluation research on Ln doped NPs is still less involved. Appropriate surface modifications should be adopted. Encapsulation of particles with a SiO2 shell is an effective way to obstruct the exposure of RE ions. Further, grafting PEG will enhance NP resistance to protein adsorption and their escape from the reticulo-endothelial system to promote elimination. The operating mode of probes should also be considered in combination with biocompatibility. For biological tissues, low power NIR irradiation is relatively less harmful than irradiation in visible and UV regions, thus Ln doped UC phosphors will be vastly developed. However, the study on the biological effect of nanomaterials in vivo is still inadequate, and research should be administrated prudently.
QDs overwhelmed the expeditions of nanotechnology towards biology in the past, and now, functional RE NPs have the opportunity to establish new hotspots in this interdisciplinary field. One promising avenue is based on the UC property of Ln species, which is a potential improvement for multiphoton microscopic methodologies. IR excited fluorescence through a two-photon process has special originality in biological microimaging due to the advantages mentioned in the former section.122 However, the microscope requires costly pulsed lasers to generate two-photon absorption of organic dyes or QDs. It was reported that the excitation density for the UC process of Ln doped phosphors was at least three orders of magnitude lower than that of the two-photon absorption process for dyes or QDs.123 Thus much cheaper 980 nm NIR diode lasers can be substituted for pulsed laser equipment if Ln doped UC markers are employed, and the output signal will be enormously enhanced if keeping to theoretical expectation. Another strategy is to develop magnetic/optical bifunctional RE probes, which may regenerate the clinic contrast agents. The currently used contrast agent, Gd-DTPA, is non-specificly dispersed over the whole body, and the different relaxation of water protons was detected in various tissues. If the magnetic/optical probes are labeled with bioactive molecules to recognize pathological regions, surgical treatment could be purposefully conducted at pre-operative and intra-operative periods with MR and optical information, respectively. Although we should be chary of the in vivo applications, this multiplex detection can improve the diagnosis precision, and is especially helpful to distinguish tumor boundaries during intra-operation. The above two ideas are not fancinesses, but more endeavour is needed to make them practical.
Acknowledgements
This work is supported by the NSFC (20221101, 20671005, and 20423005), NSFC & RGC (20610068), MOE of China (NCET-06-0010), and the MOST of China (2006CB601104).Notes and references
- D. Altschuh, S. Oncul and A. P. Demchenko, J. Mol. Recognit., 2006, 19, 459 CrossRef CAS.
- S. Weiss, Science, 1999, 283, 1676 CrossRef CAS.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41 CrossRef CAS.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509 CrossRef CAS.
- P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2005, 44, 2642 CrossRef CAS.
- M. Seydack, Biosens. Bioelectron., 2005, 20, 2454 CrossRef CAS.
- A. L. Efros, Sov. Phys. Semicond., 1982, 16, 772 Search PubMed.
- A. I. Ekimov and A. A. Onushchenko, Sov. Phys. Semicond., 1982, 16, 775 Search PubMed.
- M. Bruchez, Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435 CrossRef CAS.
- F. Pinaud, X. Michalet, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Iyer and S. Weiss, Biomaterials, 2006, 27, 1679 CrossRef CAS.
- T. K. Christopoulos and E. P. Diamandis, Anal. Chem., 1992, 64, 342 CrossRef CAS.
- X. Yang, W. Chang and Y. Ci, Anal. Chem., 1994, 66, 2590 CrossRef CAS.
- H. Mikola, H. Takalo and I. Hemmila, Bioconjugate Chem., 1995, 6, 235 CrossRef CAS.
- R. C. Morton and E. P. Diamandis, Anal. Chem., 1990, 62, 1841 CrossRef CAS.
- E. Beaurepaire, V. Buissette, M. Sauviat, D. Giaume, K. Lahlil, A. Mercuri, D. Casanova, A. Huignard, J. Martin, T. Gacoin, J. Boilot and A. Alexandrou, Nano Lett., 2004, 4, 2079 CrossRef CAS.
- J. W. Stouwdam, M. Raudsepp and F. C. J. M. van Veggel, Langmuir, 2005, 21, 7003 CrossRef CAS.
- S. Heer, O. Lehmann, M. Haase and H. U. Güdel, Angew. Chem., Int. Ed., 2003, 42, 3179 CrossRef CAS.
- J. Feng, G. Shan, A. Maquieira, M. E. Koivunen, B. Guo, B. D. Hammock and I. M. Kennedy, Anal. Chem., 2003, 75, 5282 CrossRef CAS.
- M. Nichkova, D. Dosev, S. J. Gee, B. D. Hammock and I. M. Kennedy, Anal. Chem., 2005, 77, 6864 CrossRef CAS.
- M. Nichkova, D. Dosev, R. Perron, S. J. Gee, B. D. Hammock and I. M. Kennedy, Anal. Bioanal. Chem., 2006, 384, 631 CrossRef CAS.
- P. L. A. M. Corstjens, S. Li, M. Zuiderwijk, K. Kardos, W. R. Abrams, R. S. Niedbala and H. J. Tanke, IEE Proc. Nanobiotechnol., 2005, 152, 64 CrossRef CAS.
- F. Rijke, H. Zijlmans, S. Li, T. Vail, A. K. Raap, R. S. Niedbala and H. J. Tanke, Nat. Biotechnol., 2001, 19, 273 CrossRef.
- H. J. M. A. A. Zijlmans, J. Bonnet, J. Burton, K. Kardos, T. Vail, R. S. Niedbala and H. J. Tanke, Anal. Biochem., 1999, 267, 30 CrossRef CAS.
- P. L. A. M. Corstjens, M. Zuiderwijk, M. Nilsson, H. Feindt, R. S. Niedbala and H. J. Tanke, Anal. Biochem., 2003, 312, 191 CrossRef CAS.
- R. S. Niedbala, H. Feindt, K. Kardos, T. Vail, J. Burton, B. Bielska, S. Li, D. Milunic, P. Bourdelle and R. Vallejo, Anal. Biochem., 2001, 293, 22 CrossRef CAS.
- P. Corstjens, M. Zuiderwijk, A. Brink, S. Li, H. Feindt, R. S. Niedbala and H. Tanke, Clin. Chem. (Washington, D. C.), 2001, 47, 1885 CAS.
- S. F. Lim, R. Riehn, W. S. Ryu, N. Khanarian, C. Tung, D. Tank and R. H. Austin, Nano Lett., 2006, 6, 169 CrossRef CAS.
- K. Kuningas, T. Rantanen, U. Karhunen, T. Lövgren and T. Soukka, Anal. Chem., 2005, 77, 2826 CrossRef CAS.
- K. Kuningas, T. Rantanen, T. Ukonaho, T. Lövgren and T. Soukka, Anal. Chem., 2005, 77, 7348 CrossRef CAS.
- R. Bazzi, M. A. Flores, C. Louis, K. Lebbou, W. Zhang, C. Dujardin, S. Roux, B. Mercier, G. Ledoux, E. Bernstein, P. Perriat and O. Tillement, J. Colloid Interface Sci., 2004, 273, 191 CrossRef CAS.
- C. Louis, R. Bazzi, C. A. Marquette, J. L. Bridot, S. Roux, G. Ledoux, B. Mercier, L. Blum, P. Perriat and O. Tillement, Chem. Mater., 2005, 17, 1673 CrossRef CAS.
- R. Si, Y. W. Zhang, L. P. You and C. H. Yan, Angew. Chem., Int. Ed., 2005, 44, 3256 CrossRef CAS.
- F. Zhao, M. Yuan, W. Zhang and S. Gao, J. Am. Chem. Soc., 2006, 128, 11758 CrossRef CAS.
- K. Riwotzki and M. Haase, J. Phys. Chem. B, 2001, 105, 12709 CrossRef CAS.
- F. Wang, X. Xue and X. Liu, Angew. Chem., Int. Ed., 2008, 47, 906 CrossRef CAS.
- K. Riwotzki and M. Haase, J. Phys. Chem. B, 1998, 102, 10129 CrossRef CAS.
- G. A. Hebbink, J. W. Stouwdam, D. N. Reinhoudt and F. C. J. M. van Veggel, Adv. Mater., 2002, 14, 1147 CrossRef CAS.
- J. W. Stouwdam, G. A. Hebbink, J. Huskens and F. C. J. M. van Veggel, Chem. Mater., 2003, 15, 4604 CrossRef CAS.
- O. Lehmann, H. Meyssamy, K. Kömpe, H. Schnablegger and M. Haase, J. Phys. Chem. B, 2003, 107, 7449 CrossRef CAS.
- Y. Sun, H. Liu, X. Wang, X. Kong and H. Zhang, Chem. Mater., 2006, 18, 2726 CrossRef CAS.
- J. F. Liu and Y. D. Li, Adv. Mater., 2007, 19, 1118 CrossRef CAS.
- F. C. Palilla, A. K. Levine and M. Rinkevics, J. Electrochem. Soc., 1965, 112, 776 CrossRef CAS.
- V. Buissette, D. Giaume, T. Gacoin and J. P. Boilot, J. Mater. Chem., 2006, 16, 529 RSC.
- H. Meyssamy, K. Riwotzki, A. Kornowski, S. Naused and M. Haase, Adv. Mater., 1999, 11, 840 CrossRef CAS.
- A. Huignard, T. Gacoin and J. P. Boilot, Chem. Mater., 2000, 12, 1090 CrossRef CAS.
- A. Huignard, V. Buissette, G. Laurent, T. Gacoin and J. P. Boilot, Chem. Mater., 2002, 14, 2264 CrossRef CAS.
- S. S. Soares, F. Henao, M. Aureliano and C. G. Merino, Chem. Res. Toxicol., 2008, 21, 607 CrossRef CAS.
- K. Riwotzki, H. Meyssamy, A. Kornowski and M. Haase, J. Phys. Chem. B, 2000, 104, 2824 CrossRef CAS.
- K. Riwotzki, H. Meyssamy, H. Schnablegger, A. Kornowski and M. Haase, Angew. Chem., Int. Ed., 2001, 40, 573 CrossRef CAS.
- K. Kömpe, H. Borchert, J. Storz, A. Lobo, S. Adam, T. Möller and M. Haase, Angew. Chem., Int. Ed., 2003, 42, 5513 CrossRef.
- O. Lehmann, K. Kömpe and M. Haase, J. Am. Chem. Soc., 2004, 126, 14935 CrossRef CAS.
- V. Buissette, M. Moreau, T. Gacoin, J. P. Boilot, J. Y. C. Ching and T. L. Mercier, Chem. Mater., 2004, 16, 3767 CrossRef CAS.
- V. Buissette, M. Moreau, T. Gacoin and J. P. Boilot, Adv. Funct. Mater., 2006, 16, 351 CrossRef CAS.
- F. Meiser, C. Cortez and F. Caruso, Angew. Chem., Int. Ed., 2004, 43, 5954 CrossRef CAS.
- D. Casanova, D. Giaume, T. Gacoin, J. P. Boilot and A. Alexandrou, J. Phys. Chem. B, 2006, 110, 19264 CrossRef CAS.
- D. Casanova, D. Giaume, M. Moreau, J. L. Martin, T. Gacoin, J. P. Boilot and A. Alexandrou, J. Am. Chem. Soc., 2007, 129, 12592 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press Inc., San Diego, 1996, 90–100 Search PubMed.
- J. Q. Gu, J. Shen, L. D. Sun and C. H. Yan, J. Phys. Chem. C, 2008, 112, 6589 CrossRef CAS.
- J. Shen, L. D. Sun and C. H. Yan, Nanoscience and Nanotechnology for Biological/Biomedical/Chemical Sensing, ECI, Hong Kong, 2007 Search PubMed.
- K. W. Krämer, D. Biner, G. Frei, H. U. Güdel, M. P. Hehlen and S. R. Lüthi, Chem. Mater., 2004, 16, 1244 CrossRef.
- R. X. Yan and Y. D. Li, Adv. Funct. Mater., 2005, 15, 763 CrossRef CAS.
- G. S. Yi and G. M. Chow, J. Mater. Chem., 2005, 15, 4460 RSC.
- C. H. Liu and D. P. Chen, J. Mater. Chem., 2007, 17, 3875 RSC.
- J. W. Stouwdam and F. C. J. M. van Veggel, Nano Lett., 2002, 2, 733 CrossRef CAS.
- V. Sudarsan, S. Sivakumar, F. C. J. M. van Veggel and M. Raudsepp, Chem. Mater., 2005, 17, 4736 CrossRef CAS.
- Y. W. Zhang, X. Sun, R. Si, L. P. You and C. H. Yan, J. Am. Chem. Soc., 2005, 127, 3260 CrossRef CAS.
- X. Sun, Y. W. Zhan, Y. P. Du, Z. G. Yan, R. Si, L. P. You and C. H. Yan, Chem.–Eur. J., 2007, 13, 2320 CrossRef CAS.
- P. R. Diamente, R. D. Burke and F. C. J. M. van Veggel, Langmuir, 2006, 22, 1782 CrossRef CAS.
- S. Sivakumar, P. R. Diamente and F. C. J. M. van Veggel, Chem.–Eur. J., 2006, 12, 5878 CrossRef CAS.
- F. Wang, Y. Zhang, X. P. Fan and M. Q. Wang, Nanotechnology, 2006, 17, 1527 CrossRef CAS.
- Z. L. Wang, Z. W. Quan, P. Y. Jia, C. K. Lin, Y. Luo, Y. Chen, J. Fang, W. Zhou, C. J. O'Connor and J. Lin, Chem. Mater., 2006, 18, 2030 CrossRef CAS.
- D. Y. Kong, Z. L. Wang, C. K. Lin, Z. W. Quan, Y. Y. Li, C. X. Li and J. Lin, Nanotechnology, 2007, 18, 057601.
- L. Y. Wang and Y. D. Li, Chem.–Eur. J., 2007, 13, 4203 CrossRef CAS.
- N. Menyuk, K. Dwight and F. Pinaud, Appl. Phys. Lett., 1972, 21, 159 CAS.
- S. Heer, K. Kömpe, H. Güdel and M. Haase, Adv. Mater., 2004, 16, 2102 CrossRef CAS.
- G. S. Yi, H. C. Lu, S. Y. Zhao, Y. Ge, W. J. Yang, D. P. Chen and L. H. Guo, Nano Lett., 2004, 4, 2191 CrossRef CAS.
- R. E. Thomas, H. Insley and G. M. Hebert, Inorg. Chem., 1966, 5, 1222 CrossRef CAS.
- J. H. Zeng, J. Su, Z. H. Li, R. X. Yan and Y. D. Li, Adv. Mater., 2005, 17, 2119 CrossRef CAS.
- L. Y. Wang, R. X. Yan, Z. Y. Huo, L. Wang, J. H. Zeng, J. Bao, X. Wang, Q. Peng and Y. D. Li, Angew. Chem., 2004, 116, 6080 ( Angew. Chem., Int. Ed. , 2005 , 44 , 6054 ) CrossRef.
- L. Y. Wang and Y. D. Li, Chem. Commun., 2006, 2557 RSC.
- Y. Wei, F. Q. Lu, X. R. Zhang and D. P. Chen, Chem. Mater., 2006, 18, 5733 CrossRef CAS.
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 437, 121 CrossRef CAS.
- L. Y. Wang and Y. D. Li, Chem. Mater., 2007, 19, 727 CrossRef CAS.
- H. X. Mai, Y. W. Zhang, R. Si, Z. G. Yan, L. D. Sun, L. P. You and C. H. Yan, J. Am. Chem. Soc., 2006, 128, 6426 CrossRef CAS.
- H. X. Mai, Y. W. Zhang, L. D. Sun and C. H. Yan, J. Phys. Chem. C, 2007, 111, 13730 CrossRef CAS.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642 CrossRef CAS.
- Z. Q. Li and Y. Zhang, Angew. Chem., Int. Ed., 2006, 45, 7732 CrossRef CAS.
- (a) J. C. Boyer, F. Vetrone, L. A. Cuccia and J. A. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444 CrossRef CAS; (b) J. C. Boyer, L. A. Cuccia and J. A. Capobianco, Nano Lett., 2007, 7, 847 CrossRef CAS.
- H. X. Mai, Y. W. Zhang, L. D. Sun and C. H. Yan, J. Phys. Chem. C, 2007, 111, 13721 CrossRef CAS.
- G. S. Yi and G. M. Chow, Chem. Mater., 2007, 19, 341 CrossRef CAS.
- G. S. Yi and G. M. Chow, Adv. Funct. Mater., 2006, 16, 2324 CrossRef CAS.
- Z. G. Chen, H. L. Chen, H. Hu, M. X. Yu, F. Y. Li, Q. Zhang, Z. G. Zhou, T. Yi and C. H. Huang, J. Am. Chem. Soc., 2008, 130, 3023 CrossRef CAS.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C. H. Shin, J. G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688 CrossRef CAS.
- P. A. Rinck, Magnetic Resonance in Medicine: The Basic Textbook of the European Magnetic Resonance Forum, Blackwell Scientific Publications, Oxford, 1993 Search PubMed.
- S. Kim, Y. T. Lim, E. G. Soltesz, A. M. D. Grand, J. Lee, A. Nakayama, J. A. Parker, T. Mihajevic, R. G. Laurence, D. M. Dor, L. H. Cohn, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2004, 22, 93 CrossRef.
- X. H. Gao, Y. Y. Cui, R. M. Levenson, L. W. K. Chung and S. M. Nie, Nat. Biotechnol., 2004, 22, 969 CrossRef CAS.
- H. Kobayashi, Y. Hama, Y. Koyama, T. Barrett, C. A. S. Regino, Y. Urano and P. L. Choyke, Nano Lett., 2007, 7, 1711 CrossRef CAS.
- W. B. Cai and X. Y. Chen, Small, 2007, 3, 1840 CrossRef CAS.
- M. F. Kircher, U. Mahmood, R. S. King, R. Weissleder and L. Josephson, Cancer Res., 2003, 63, 8122 CAS.
- K. Vuu, J. W. Xie, M. A. McDonald, M. Bernardo, F. Hunter, Y. T. Zhang, K. Li, M. Bednarski and S. Guccione, Bioconjugate Chem., 2005, 16, 995 CrossRef CAS.
- S. Santra, H. Yang, P. H. Holloway, J. T. Stanley and R. A. Mericle, J. Am. Chem. Soc., 2005, 127, 1656 CrossRef CAS.
- S. Wang, B. R. Jarrett, S. M. Kauzlarich and A. Y. Louie, J. Am. Chem. Soc., 2007, 129, 3848 CrossRef CAS.
- O. Veiseh, C. Sun, J. Gunn, N. Kohler, P. Gabikian, D. Lee, N. Bhattarai, R. Ellenbogen, R. Sze, A. Hallahan, J. Olson and M. Q. Zhang, Nano Lett., 2005, 5, 1003 CrossRef CAS.
- J. H. Lee, Y. W. Jun, S. I. Yeon, J. S. Shin and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 8160 CrossRef.
- (a) H. W. Gu, R. K. Zheng, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2004, 126, 5664 CrossRef CAS; (b) J. H. Gao, B. Zhang, Y. Gao, Y. Pan, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2007, 129, 11928 CrossRef CAS.
- K. W. Kwon, B. H. Lee and M. Shim, Chem. Mater., 2006, 18, 6357 CrossRef CAS.
- (a) K. W. Kwon and M. Shim, J. Am. Chem. Soc., 2005, 127, 10269 CrossRef CAS; (b) S. T. Selvan, P. K. Patra, C. Y. Ang and J. Y. Ying, Angew. Chem., Int. Ed., 2007, 46, 2448 CrossRef CAS.
- D. K. Yi, S. T. Selvan, S. S. Lee, G. C. Papaefthymiou, D. Kundaliya and J. Y. Ying, J. Am. Chem. Soc., 2005, 127, 4990 CrossRef CAS.
- V. S. Maceira, M. A. C. Duarte, M. Spasova, L. M. L. Marzán and M. Farle, Adv. Funct. Mater., 2006, 16, 509 CrossRef CAS.
- H. C. Lu, G. S. Yi, S. Y. Zhao, D. P. Chen, L. H. Guo and J. Cheng, J. Mater. Chem., 2004, 14, 1336 RSC.
- D. Dosev, M. Nichkova, R. K. Dumas, S. J. Gee, B. D. Hammock, K. Liu and I. M. Kennedy, Nanotechnology, 2007, 18, 055102 CrossRef.
- M. Oudkerk, P. E. Sijens, E. J. R. Vanbeek and T. J. A. Kuijpers, Invest. Radiol., 1995, 30, 75 CrossRef CAS.
- (a) J. C. Frias, K. J. Williams, E. A. Fisher and Z. A. Fayad, J. Am. Chem. Soc., 2004, 126, 16316 CrossRef CAS; (b) W. J. M. Mulder, R. Koole, R. J. Brandwijk, G. Storm, P. T. K. Chin, G. J. Strijkers, C. M. Donegá, K. Nicolay and A. W. Griffioen, Nano Lett., 2006, 6, 1 CrossRef CAS; (c) E. A. Anderson, S. Isaacman, D. S. Peabody, E. Y. Wang, J. W. Canary and K. Kirshenbaum, Nano Lett., 2006, 6, 1160 CrossRef CAS; (d) L. Prinzen, R. J. J. H. M. Miserus, A. Dirksen, T. M. Hackeng, N. Deckers, N. J. Bitsch, R. T. A. Megens, K. Douma, J. W. Heemskerk, M. E. Kooi, P. M. Frederik, D. W. Slaaf, M. A. M. J. Zandvoort and C. P. M. Reutelingsperger, Nano Lett., 2007, 7, 93 CrossRef CAS.
- F. Evanics, P. R. Diamente, F. C. J. M. van Veggel, G. J. Stanisz and R. S. Prosser, Chem. Mater., 2006, 18, 2499 CrossRef CAS.
- H. Hifumi, S. Yamaoka, A. Tanimoto, D. Citterio and K. Suzuki, J. Am. Chem. Soc., 2006, 128, 15090 CrossRef CAS.
- J. L. Bridot, A. C. Faure, S. Laurent, C. Rivière, C. Billotey, B. Hiba, M. Janier, V. Josserand, J. L. Coll, L. V. Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076 CrossRef CAS.
- (a) H. S. Choi, W. H. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS; (b) W. H. Liu, H. S. Choi, J. P. Zimmer, E. Tanaka, J. V. Frangioni and M. G. Bawendi, J. Am. Chem. Soc., 2007, 129, 14530 CrossRef CAS.
- (a) M. E. Åkerman, W. C. W. Chan, P. Laakkonen, S. N. Bhatia and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12617 CrossRef CAS; (b) X. H. Gao, Y. Y. Cui, R. M. Levenson, L. W. K. Chung and S. M. Nie, Nat. Biotechnol., 2004, 22, 969 CrossRef CAS.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434 CrossRef CAS.
- F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |