Activation of late transition metal catalysts for olefin polymerizations and olefin/CO copolymerizations
Timo M. J. Anselment, Sergej I. Vagin and Bernhard Rieger*
WACKER-Lehrstuhl für Makromolekulare Chemie, Technische Universität München, Lichtenbergstr. 4, 85748, Garching, Germany
First published on 9th June 2008
Abstract
The late transition metal catalysts for homo- and copolymerizations of olefins expand the polymer properties and the usable monomer feedstock of early transition metal catalysts. A critical step for a polymerization is the activation of catalyst precursors and the generation of the actual active site. This perspective discusses possible activation protocols for two well-known catalyst systems, the α-diimine Ni or Pd olefin polymerization catalysts and the Pd catalysts for alkene/CO copolymerization. The general concept of catalyst activation is outlined and similarities as well as differences between single-component catalysts and in situ activated systems are highlighted.
 Timo M. J. Anselment Timo M. J. Anselment |
 Sergej I. Vagin Sergej I. Vagin |
 Bernhard Rieger Bernhard Rieger | Timo Anselment started his PhD with Prof. B. Rieger in 2007. Sergej Vagin received his PhD degree from Ivanovo State University of Chemistry and Technology (Russia) in 2000 and joined Prof. Rieger's group in 2006. Bernhard Rieger obtained his PhD in chemistry at Ludwig-Maximilians-Universität, Munich in 1988. In 2006 he became head of the WACKER Chair of Macromolecular Science at TUM. |
1. Introduction
The late transition metal-catalyzed polymerization reactions are interesting systems to address problems faced in catalytic polymerization. These methods of creating polyolefin homo- or copolymers lead to different polymer structures and unique properties other than the previously existing early transition metal polymerization reactions, even in well-researched systems like ethylene polymerization. This is partially a consequence of the different reactivities of early and late transition metal complexes. The variability is increased with different ligand systems, which have a large influence on the obtained polymers. Several good reviews for an overview about these reactions are available.1–8As early transition metal metallocene or Ziegler catalysts are highly sensitive to polar reagents due to their strong Lewis acid character, copolymerization reactions with polar comonomers are only possible if the polar functional groups are protected9 or a high amount of protecting Lewis acid is introduced to the reaction system.10 Late transition metal complexes are usually much more tolerant to the presence of polar functionalities or solvents and possess a significantly smaller Lewis acid character, resulting in weaker coordination of polar substances. The strength of coordination of Lewis bases (functional groups, solvent molecules, counterions) is one major factor which influences the activity of catalysts.11 The weaker coordination can lead, as described further below, to interesting observations in these polymerization reactions, originating from the reversibility of this coordination. Additionally, the high tendency for β-hydride elimination in Ni or Pd catalyst systems has a significant influence on the polymer structure, copolymer composition and molecular weight. Because of the β-hydride elimination, usually only low molecular weight oligomers are obtained (e.g. in the Shell higher olefin process (SHOP)2) without special precautions. In the case of polymerization, it can lead to a variety of branched structures up to hyperbranched polyethylene oils without the addition of higher olefin comonomers.1,3 Nevertheless, addition of higher 1-olefins or functionalized comonomers further expands the variety of polymers obtained from metallocene and other polymerization methods due to their unique polymerization properties.1,3
The following review focuses on the activation of late transition metal polymerization catalysts. Firstly, the case of Ni(II) or Pd(II) α-diimine complexes with substituted diazadien (DAD) ligands of the type [ArN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)C(R)NAr] for olefin homo- and copolymerizations and, secondly, Pd(II) complexes for the alternating and non-alternating co- and terpolymerization of CO and 1-olefins. For these two examples the relevant steps and the variety of possible activation methods are discussed.
C(R)C(R)NAr] for olefin homo- and copolymerizations and, secondly, Pd(II) complexes for the alternating and non-alternating co- and terpolymerization of CO and 1-olefins. For these two examples the relevant steps and the variety of possible activation methods are discussed.
1.1 General aspects of catalytic polymerization reactions with late transition metals
The commonly accepted general layout of a homogenous catalytically-active cationic late transition metal complex 1 for polymerization reactions (Fig. 1) consists of a metal center serving as the active site and coordinated ligands which serve different purposes. | ||
| Fig. 1 General structure of a cationic polymerization catalyst (see text for details). | ||
A metal M coordinated alkyl-, aryl- or hydrogen-group R is the starting point for chain growth by migratory insertion of monomer units into this bond. Further ligands L1,2 (monodentate or chelating) provide stability and solubility for the complex. They also control the steric and electronic environment as well as the electronic configuration of the electrophilic Lewis acid metal. For a polymerization, it is crucial that a coordination site for monomer molecules (□; Fig. 1) is present cis respective to R, which allows monomer units to repeatedly coordinate to the metal and be incorporated by migratory insertion. As active cationic catalysts with coordinative unsaturation are highly reactive electron deficient moieties, stabilization is needed to increase the catalyst life time. This is usually observed by coordination of monomer units, solvent molecules, etc. Coordination of functionalized groups present in R (backbiting) is also possible, but the binding strength has to be low enough to allow exchange with monomer units under polymerization conditions.
All these factors together determine the activity of the polymerization catalyst, i.e. the chain growth rate, by the migratory insertion as well as the termination and reactivation rates on the metal center. It will depend on the exchange kinetics between coordinated groups and monomer units, the reaction mechanism and the electronic and steric environment of the metal. Additionally, chain transfer agents like hydrogen can be applied to control the polymer growth in certain catalyst systems, similar to metallocene catalyzed polymerizations.12
1.2 Activation of polymerization catalysts
Two concepts to generate active homogenous polymerization catalysts are possible. The common case consists of a catalyst precursor which has to be activated in situ by a cocatalyst before the addition of monomers. The second possibility uses SCCs (single-component catalysts) which directly initiate polymerization in the presence of monomer under reaction conditions.The definition of the activity for polymerization catalysts which is found in the literature is usually misleading and problematic. In a theoretic graph where the intrinsic activity A is depicted against the reaction time t the problems become obvious (Fig. 2). In the literature several meanings for the activity of a catalyst are found. Firstly, it represents the actual intrinsic activity of a catalyst at a certain time during the reaction. Secondly, it is sometimes confusingly mixed with the productivity of a catalyst; in this respect productivity per time is the average activity of a catalyst. Thus the term activity is also used for a description of the productivity. In both cases the changes of the intrinsic activity are neglected.
 | ||
| Fig. 2 Typical activation curves for two polymerization catalysts (monomer concentration or pressure not considered). Am = maximal intrinsic activity; AAv = average activity; P = productivity as the integral of the average activity over t. | ||
The realistic behaviour of a catalyst is, on the other hand, very sensitive to external influences. Firstly, the activation is usually not instantaneous and the activity reaches a maximum, the intrinsic maximal activity, after a certain time. Longer reaction times usually lead to a decrease of the activity. The reasons for this behaviour include factors like incomplete activation, transport limitations, different reactivity of intermediates, catalyst poisoning or deactivation, catalyst occlusion into polymer particles, changes in the homogeneity of the reaction medium, different heat transport characteristics over time, etc.
Together these factors can only be considered after a thorough kinetic investigation of the polymerization process. Nevertheless, the productivity per time or average activity (i.e. the yield of polymer per time, catalyst amount and pressure or concentration of the monomer in [kg[polymer] mol[catalyst]−1 bar[monomer]−1 h−1] or [kg[polymer] mol[catalyst]−1c[monomer]−1 h−1]) is a useful way to get an informative basis for further investigations. The influence of monomer concentration or pressure on the catalyst activity or productivity is not always accounted for and therefore literature units without [c[monomer]−1] or [bar[monomer]−1] can be found.
It must be emphasized that direct comparison of productivities is dangerous. Wrong evaluation of polymerization results can lead to mistakes, as highly reactive and sensitive catalysts with a high intrinsic activity can show a lower over-time productivity due to faster deactivation. This way a long time measurement can lead to false interpretations, e.g. concerning the catalyst structure-to-activity relationships. In principle the reaction time, as well as the reaction conditions have to be adjusted to a specific catalyst.
A comparison of published results in metallocene catalysis shows a strong dependence on the polymerization conditions and experimenters (due to different reaction protocols), and shows that results can contradict or vary strongly. For example, in the Cp2TiCl2 ethylene polymerization system, productivities from 4.3 to 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 488 × 105 gPE molTi−1 h−1 were reported under different conditions, and comparison of substitution effects leads to possible false interpretations.13
488 × 105 gPE molTi−1 h−1 were reported under different conditions, and comparison of substitution effects leads to possible false interpretations.13
The defined character of the reaction mixture allows a relatively easy identification of intermediates, thus facilitating an investigation of the reaction mechanism. Additionally, it sometimes can be useful to add small amounts of cocatalysts like methylalumoxane (MAO) or trimethylaluminium (TMA) to prevent deactivation of the system by reaction with impurities or to reactivate non-reactive intermediates.
2. Activation of Ni(II) and Pd(II) α-diimine polymerization catalysts
The polymerization reactions of olefins, described by Brookhart et al.14 in 1995 with the metal–alkyl complexes (DAD)M(alkyl)2 (M = Ni, Pd), previously known from the work of tom Dieck et al.,15 led to extensive research on this remarkable catalyst system which is used by DuPont under the trademark Versipol®.16 The interesting characteristics and polymerization results of this system were reviewed in 2000 by Ittel et al.1 Since then, the second generation of these catalysts has been developed by Rieger et al.,17,18 introducing hydrogen stable, easily modifiable catalysts for controlled ethylene polymerizations.2.1 Reaction mechanism
The reaction mechanism for the Ni and Pd catalyzed polymerization of ethylene with DAD complexes was studied by low temperature NMR experiments of diethyl ether and ethylene adducts.14,19 The initially synthesized, extremely reactive and highly sensitive [(DAD)NiMe(OEt2)][BAr′4] (BAr′4 = B[3,5-C6H3(CF3)2]4) complexes were followed by the palladium analogues, which can be investigated more easily because of their increased stability.20The reaction mechanism was investigated in detail, both experimentally14,19,21,22 and theoretically (see Fig. 3).23 In short, it is divided into the activation/initiation, propagation/isomerisation and chain transfer/termination stages. After generation of the active catalyst resting state 3,migratory insertion and the competitive isomerisation lead to chain propagation with intermediate alkyl complexes 4 stabilized by β-agostic bonds.19,22,24 Depending on the system properties and the resulting propagation/isomerisation rate, polymers ranging from linear to highly branched are obtained. The termination by chain transfer reproduces an active catalyst resting state 3 to re-enter the polymerization cycle.
![Reaction mechanism scheme for the polymerization of ethylene with the α-diimine (DAD) catalyst system; catalyst type e.g. [(DAD)MMe(OEt2)][BAr′4].](/image/article/2008/DT/b802913j/b802913j-f3.gif) | ||
| Fig. 3 Reaction mechanism scheme for the polymerization of ethylene with the α-diimine (DAD) catalyst system; catalyst type e.g. [(DAD)MMe(OEt2)][BAr′4]. | ||
Details of this mechanism are excellently reviewed by Brookhart et al.,20 and lead to the conclusion that a bulky ligand structure is required for polymerization instead of the very common oligomerization reactions.2 Thus in these catalysts, the bulky substituents on the 2,6-substituted aryl functionalities block the axial positions of the planar complexes, retarding associative chain transfer, and preform the reaction space for the polymer growth. The general trend that Ni catalysts are more active than Pd catalysts was observed, as well as a dependence of the degree of branching on ethylene pressure for Ni; Pd catalysts usually show constant high branching.1 In the latter case, the polymer topology (linear with short branches; hyperbranched) is dependent on the ethylene pressure due to changes of the branch length.3
The variability of this catalyst system is enlarged further by many possible copolymerization reactions. Thus the late transition metal catalysts allow copolymerization with acrylates,1,25–27 propene1,14 and higher 1-olefin1,14,28 comonomers with interesting characteristics. For example, acrylate units are incorporated only at the end of branches,1,25–27 and for higher olefin copolymers the maximum length and degree of branching is reduced by chain straightening.28 Also, the synthesis of block copolymers is possible as the catalysts show living behaviour under certain reaction conditions.29
The Ni catalysts of the second generation diimines can be easily modified by substitution on the terphenyl ligands. It was shown that this can lead to a cone-shaped reaction space for the propagation around the metal center.17,18 Isomerization and the resulting polymer branching can be controlled by the substitution and the steric demand of the ligands. Associative chain transfer is also effectively prevented by the terphenyl units pointing in the axial direction.
In general it was demonstrated that this highly active and versatile catalyst system enables the possibility to create polymers with controllable new architectures and properties.
2.2 Precursor synthesis
The versatile DAD polymerization catalysts were thoroughly reviewed by Ittel et al.1 The ligands are easily accessible by condensation of a 1,2-diketone with alkyl- or arylamines and thus provide a means of facile modification by the substitution of these two building blocks. Nevertheless, most research described in the literature employs very similar diimine ligands with a simple design (Fig. 4). The substituents in the 2,6-position of the aryl functionalities are usually Me or iPr groups and at the backbone of the DAD ligand mostly Me or H substituents are used. The DAD ligands employed in the literature mentioned in this perspective are mostly of this type or the second generation diimines with o-terphenyl substituents. The non-coordinating anion usually found in the literature is BAr′4−, although SbF6−, B(C6F5)4− and other counterions have been reported. | ||
| Fig. 4 Structures of commonly used α-diimine ligands. | ||
The actual substitution of the employed diimines is not considered here if no direct influence to the activation is observed. Interested readers are referred to the literature regarding the effects of differently substituted diimines on polymer architecture and reactivity.1,18,23a,30
The precursor synthesis is mostly based on the work of tom Dieck et al.15 and was attuned for the catalytic purpose. Several typical methods to prepare precursor complexes for activation methods, which are described later in this article, exist (Fig. 5).
 | ||
| Fig. 5 Overview of possible precursor synthesis methods, common precursors highlighted. | ||
One basic complex is (DAD)NiBr2 which can be synthesized by reaction of NiBr2 with DAD or the substitution of (DME)NiBr2 (DME = dimethoxyethane) in presence of the DAD ligand. This complex can be converted into the (DAD)Ni(alkyl)2 complexes by reaction with alkylating agents like MeMgX.15c A similarly suited starting compound is Ni(acac)2 (acac = acetylacetonate). Here the substitution with DAD in the presence of alkylating MeMgX or Me2Mg is possible, which forms the (DAD)NiMe2 precursor.14,15c Alternatively, one acac ligand can be abstracted with [Ph3C][B(C6F5)4] or [Ph3C][SbCl6] in the presence of DAD to form the complex (DAD)Ni(acac) which is important for the synthesis of the second generation catalyst precursors with very bulky DAD ligands.18 These methods are similar to those described below for the in situ activation of Ni(acac)2 with MAO in the presence of DAD ligand.31 The (DAD)NiMe2 complex is relatively unstable and decomposes around 0 °C with increasing instability for higher alkyl substituents.
The similar Pd precursors can be synthesized by various methods. The usual starting compound is (COD)PdMeCl (COD = 1,5-cyclooctadiene), which can be obtained, for example, by alkylation of (COD)PdCl2 with SnMe48 or CuMe2Li.32 Again, the (COD)PdMeCl can be converted to the dialkyl species with MeLi or Me2Mg or be substituted to (DAD)PdMeCl,14 which is an especially useful precursor activated by halide abstraction (see below). The (COD)PdMe2 precursor can also be converted into the DAD complex.16 The second generation Pd–DAD catalyst precursors were obtained by substitution of (C6H5CN)2PdCl2 with DAD and methylation with SnMe4 providing (DAD)PdMeCl. This modification is necessary as the ligands are too bulky for the usual method.18 Additionally, the synthesis of a halogen-free precursor with a η3-methallyl ligand was reported as an alternative to the bis-alkylated compounds. It can be synthesized by the addition of a DAD ligand to μ-(Br)2-bis-[Ni(II)(η3-methallyl)] in the presence of TlPF6.33
2.3 Activation of diimine catalyst precursors
2.3.1.1 In situ activation methods for M(II) precursors. In the common activation recipe, the active catalyst can be generated in situ by the reaction of the (DAD)NiBr2 precursor with methylalumoxanes in the presence of olefins14 or by a Ni(II) precursor in the presence of free DAD ligand and MAO (Fig. 6). The activation is believed to follow the same mechanism as in the metallocene polymerization for which it has been reviewed in detail.11
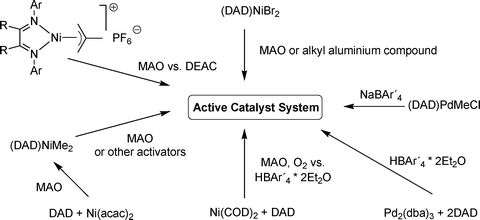 | ||
| Fig. 6 Examples of in situ activation methods for α-diimine catalysts. | ||
In the case of the dihalogenated precursor (DAD)MX2 this is achieved by a methylation with MAO or TMA contained in it. The actual mechanism for the generation of the cationic active species is complex for metallocenes as several intermediates can be generated depending on the Al/M ratio as well as the TMA content (free or coordinated) in the MAO, and the same is expected for the (DAD)MX2 system. Similar to this, the activation of dialkylmetallocenes and (DAD)M(alkyl)2 complexes is also achieved with MAO by abstraction of a methyl group.
Thus overall, MAO is assumed to act as a methylating agent and Lewis acid by abstracting chloride or methyl groups from the catalyst precursor. Furthermore, the active cationic metal species is stabilized by the formation of complex intermediates11 and ion pairs. These have, in addition to the stabilization, a huge influence on the Lewis acidity of the metal and consequently on the activity and productivity of the complex. Free or added TMA can usually increase and prolong the activity or productivity of the catalyst by reactivation of decay products. The activation itself is, as a consequence of this equilibrium between all intermediates, not quantitative and side reactions can lead to deactivation.11
The transfer of the metallocene activation methods to the α-diimine ligand system was studied by a direct comparison of different activation methods in terms of activity and polymer structure at low temperatures and low ethylene pressure. In this report, the activation of the (DAD)NiBr2 precursors was tested with MAO (Al/Ni mole ratio = 1000:1) and diethylaluminium chloride (DEAC) (Al/Ni mole ratio = 20:1) and the results showed that MAO activation leads to a greater productivity than activation with DEAC. On the other side, DEAC can be used at a much lower Al/Ni mole ratio. The resulting polymers show a strong dependence on the catalyst structure in terms of branching and melting temperature of the polymers, although DEAC activation generally yields more branched polymers. This could be a result of different cation–anion interactions and associated ion-pairs with different reactivities. Additional activation methods derived from metallocenes were carried out with the bisalkylated (DAD)NiMe2 complex and the activating agents MAO, DEAC, TMA, B(C6F5)3, [CPh3][B(C6F5)4] and HBF4, again at low temperature and ethylene pressure. All methods provided active polymerization catalysts but pronounced activity differences. It was shown that B(C6F5)3 (10 eq.) is more active than MAO (1000 eq.). Again, significant differences of the polymer branching were observable. Unfortunately, the molecular weights of the obtained polyethylene (PE) samples were not analyzed and compared.34
An example for a possible screening method was reported for the activation of Ni(acac)2 in the presence of a DAD ligand with MAO. This case allows the generation of halide free, in situ activated catalyst systems. Since no easily removable ligand like DME or COD is present, the substitution of one acac ligand is not possible by the addition of the DAD ligand. Here MAO is needed, which is thought to participate in the ligand substitution. Thereby the order of component addition is important, as the MAO will react with DAD without Ni(acac)2 present and prevent coordination to the metal. This activation was studied by 1H NMR spectroscopy, which showed the formation of the active diamagnetic Ni(II) species from the paramagnetic Ni(acac)2. The alkylated precursor (DAD)NiMe2 could be identified in the spectrum and methylated Ni species with coordinated anionic alumoxane moieties were proposed. Thus in this case, MAO serves as a co-reactant to coordinate the DAD ligand as well as for activation of the intermediates by methylation and subsequent methyl group abstraction. The best activities were obtained with an excess of the DAD ligand.31
Activation of the second generation of diimine–Ni catalysts shows that the steric environment of the metal has an influence on the suitable cocatalysts.18 Reactions with triisobutylaluminium (TIBA), MAO and TMA were carried out at a fixed Al/M ratio. TMA yields the best results, followed by MAO. The polymerization with the sterically demanding TIBA shows almost no activity for the catalyst. The amount of cocatalyst is also important as a second alkylation would result in Ni–dialkyls which are very labile and decompose to Ni(0) very easily.15
Substituted (η3-allyl)-Ni(II) complexes with DAD ligands are also active olefin polymerization catalysts. The η3-methallyl complex 5 (Fig. 7) with a counterion X (X = e.g. PF6−, BAr′4−) is activated in situ with MAO or Et2AlCl. This last cocatalyst can significantly reduce the Ni/Al ratio needed for the polymerization.33
![MAO or DEAC activated polymerization of ethylene with a [(DAD)Ni(η3-methallyl)][PF6] catalyst.](/image/article/2008/DT/b802913j/b802913j-f7.gif) | ||
| Fig. 7 MAO or DEAC activated polymerization of ethylene with a [(DAD)Ni(η3-methallyl)][PF6] catalyst. | ||
This discussion shows that the stabilization of the active species is a critical point. It should be balanced to ensure a high catalytic activity accompanied with a long lifetime of the catalyst to obtain a high over-time productivity of the polymerization catalysts.
Another in situ activation method of M(II) precursors is the halogenide ligand abstraction. The salt NaB(Ar′)4 will cause the precipitation of NaCl and provides a bulky non-coordinating counterion at the same time.16b This principle is exploited in various other reactions that lead to active single-component catalysts, as described below.
2.3.1.2 In situ activation protocols for M(0) precursors. The literature also mentions the in situ activation of M(0) precursors.1,16b During these reactions the metal precursors are activated by ligand exchange and oxidation. Therefore the metal compound is required to have easily exchangeable ligands which can be substituted by free DAD. Furthermore, the presence of an oxidising agent and a source of a non-coordinating counterion are needed.
An example is the reaction of Ni(COD)2 with MAO and oxygen in the presence of DAD. Accordingly, H(OEt2)2+B(Ar′)4−35 can be employed with this precursor both as the oxidating agent and the source of the counterion. Additional reactions can be found in the literature and include metal compounds as Pd2(dba)3 (dba = dibenzylidenacetone)1,16b (Fig. 6).
These principles can also be employed for catalyst screening processes due to their simple nature and easy ligand exchangeability.
2.3.2.1 Ether adducts. The stabilization of active catalyst intermediates with donor molecules is a useful tool to isolate directly active single-component catalysts. The first example was the protonolysis of α-diimine Pd(II) and Ni(II) dimethyl precursors15 with H(OEt2)2+B(Ar′)4−, which will result in the release of methane and the highly reactive ether coordinated cationic metal complex (19 for M = Pd) (Fig. 8).14,19,21
![Synthesis of the highly reactive ether-substituted cationic catalysts [(DAD)MMe(OEt2)][B(Ar′)4].](/image/article/2008/DT/b802913j/b802913j-f8.gif) | ||
| Fig. 8 Synthesis of the highly reactive ether-substituted cationic catalysts [(DAD)MMe(OEt2)][B(Ar′)4]. | ||
The use of non-coordinating counterions prevents the replacement of the very labile ether ligand. A facile ligand exchange makes these complexes ideally suitable for NMR studies at low temperatures. For example, they will react with olefins at −80 to −110 °C and generate the alkyl olefin complexes. Chain growth can be studied, as at low temperatures (−80 to −30 °C) the insertion can be monitored first by the disappearance of the Ni–Me signal and later by the rate of olefin consumption. These alkyl/olefin adducts represent the general catalyst resting state. After the first insertion of ethylene or propylene, agostic intermediates can be observed at −120 °C.19 These products could be isolated for the palladium system.21 It was also shown that the exchange of ethylene units is dependent on the ethylene pressure which indicates the aforementioned associative olefin addition mechanism.14
2.3.2.2 Reaction with polar functionalized olefins. A convenient way to obtain air and temperature-stable single-component catalysts was reported by the reaction of the (DAD)PdMeCl with equimolar amounts of acrylates in the presence of NaBAr′4.25
Low temperature NMR studies with the highly reactive [(DAD)PdMe(OEt2)][BAr′4] catalyst 6 (Fig. 9) show that after reaction with an equimolar amount of acrylate the π-olefin complex 7 is obtained and the acrylate is not coordinated to the metal by the oxygen atom.
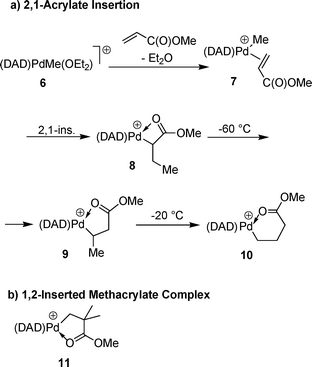 | ||
| Fig. 9 Insertion reaction of acrylates; anionic counterion BAr′4−. (a) Mechanism for the usual 2,1-insertion of acrylates; (b) product of the 1,2-insertion of MMA. | ||
After this coordination reaction, the insertion of the acrylate unit into the Pd–alkyl bond follows to give 8. The regioselectivity of this step depends on the used acrylate. 2,1-Insertion occurs with methylacrylate while, on the other hand, tert-butylacrylate is inserted in a 1,2 fashion. Isomerization of 8 leads in the end to the six-membered air and temperature stable chelate complex 10 which is stabilized by the coordination of the carboxyl group of the acrylate. The remarkable stability makes the compound an interesting SCC for homo- and copolymerizations of 1-olefins and/or acrylates.25 Because of the relatively strong coordination of the carboxylate group to the metal, competitive replacement with 1-olefins like ethylene is not complete at low olefin pressures (71% activated at 2 bar of ethylene pressure). Complete involvement of acrylate insertion products in polymerization reactions at 29 bar of ethylene or over long experiment times was confirmed by comparison with polymerization reactions catalyzed by ether adducts.26
Recent studies showed that methacrylates can be incorporated by 1,2-insertion because of increased steric bulk. Because of the high energy needed for the activation of methylmethacrylate (MMA), no olefin/MMA copolymerizations were possible, but block copolymers of the type alkene-block-alkene/carbon monoxide could be synthesized, and the catalyst is an active ethylene polymerization SCC.36
A similar method of activating the catalysts by incorporation of polar functionalized olefins resulting in coordinatively stabilized complexes is the reaction with acrolein dimethylacetal (ADMA) (Fig. 10). The synthesis is carried out with 12, NaBAr′4 and ADMA which yields the air stable complex 13, where one ether group is coordinated to the Pd center. 13 is active for the homopolymerization of ethylene and the copolymerization of ethylene and ADMA.37

2.3.2.3 Acetonitrile adducts. Acetonitrile adducts are also a common form to synthesize air- and temperature-stable active Pd(II) catalysts. For example, these are obtained by the displacement of a COD ligand in [(COD)PdMe(MeCN)][BAr′4] by DAD. Another possible synthesis is by halide abstraction and coordination of MeCN from (DAD)PdMeCl.16,26
2.3.2.4 Zwitterionic (DAD)Ni(butadiene)–borate adduct formation. Also derived from early transition metal-catalyzed polymerization reactions,38,39 complexes of the type (DAD)Ni(butadiene) 14 could be activated for olefin polymerization by the addition of B(C6F5)3 (Fig. 11), yielding a zwitterionic complex of type 15.
 | ||
| Fig. 11 SCC generation by addition of a borane to a (DAD)Ni(butadiene) precursor. | ||
In this reaction the borane adds regioselectively to the C4 carbon of the butadiene ligand, preserving the cisoid configuration of the butadiene fragment. Thus an anti-configured η3 allyl ligand is obtained. These complexes of the type of 15 are active SCCs for the polymerization of ethylene.40
3. 1-Olefin/carbon monoxide copolymerizations
3.1 General aspects and mechanism
The first metal-catalyzed copolymerizations of ethylene and carbon monoxide were reported in the 1940s and 1950s where Co41 and Ni42 salts were used as catalysts, though relatively harsh conditions had to be applied and low degrees of polymerization were observed. Since that time, a continuous improvement of catalytic systems for CO/ethylene copolymerization has been sustained,43 until a real breakthrough occurred after the discovery of the chelated diphosphine palladium(II) catalysts by Drent 44Up to now, a vast number of publications have been devoted to this topic, describing different catalytic systems in which also other metals, e.g. rhodium,45 were tested. Palladium-based catalysts typically display the best results in terms of high polymerization degrees and productivities. With these catalysts, the modification of chelating and supplementary ligands (e.g. weak coordinating ligands, solvent molecules etc.) at the catalytic metal center became the main tool for the optimization of CO/alkene copolymerizations. A wide range of chelating bidentate and polydentate ligands with P, O, N or other donor atoms was prepared and utilized for various catalytic systems.1,46 Depending on the nature of the olefin and the desired copolymer structure, a few generalized catalytic systems were developed to achieve the best yields. For example, systems based on palladium(II) complexes with bidentate chelating nitrogen (N⁁N) ligands and weakly or non-coordinating anions were found to effectively catalyze a perfectly alternating and regioselective CO/styrene copolymerization.43,46–48 Bidentate chelating phosphine ligands (P⁁P) are mostly applied in Pd(II)-catalyzed alternating copolymerization and terpolymerization of CO with ethene, propene or other 1-alkenes possessing “isolated” double bonds.46,49,50 Neutral palladium complexes of certain (P⁁O)-chelating bidentate phosphine–sulfonate ligands were found to catalyze the formation of non-alternating CO/ethylene copolymers with excess insertion of ethylene, etc.51,52
As for the previously described olefin polymerizations, besides the nature of the metal and chelating ligand, additional features such as polymerization media, catalyst activators or cocatalysts, temperature and concentrations play an important role in determining the effectiveness of the catalytic system and the structure and properties of the polymeric end-product.
The abundant literature data unambiguously demonstrate that the polymer chain propagation cycle in the CO/olefin copolymerization, as shown in Fig. 12, is accepted for all known catalytic systems. It is independent of the nature of ligands. Thus, alternating insertion of CO into M–alkyl bonds and of alkene into M–acyl bonds is the “driving force” of the propagation step. As shown before,53–55 insertion of CO into a Pd–acyl bond (double insertion of CO) is thermodynamically unfavorable, and the alternation in the copolymerization is controlled by competitive insertion of CO and alkene into the Pd–alkyl bond. In diphosphine palladium complexes, ethylene coordination to Pd–alkyl species, which could lead to a double ethylene incorporation, is prevented by a strong coordination of CO. Additionally, the chelation of the carbonyl oxygen in β-ketoalkylpalladium species and the formation of a five-membered ring hinders the coordination of alkene to Pd–alkyl species in the growing polymer chain. Together, these factors are the main reasons leading to the strictly alternating copolymers with a very low degree of misinsertion.56
 | ||
| Fig. 12 Schematic presentation of the catalytic cycle in CO/ethylene copolymerization with diphosphine palladium catalyst; counterion X− is omitted; P⁁P = chelating diphosphine ligand. | ||
3.2 Activation of the catalyst precursors
 | ||
| Fig. 13 Examples of SCC compounds and precursors. | ||
Unfortunately, end-group analysis of these polymers to gain more information on the initiation and termination steps was not possible due to the high molecular weights. It was, however, shown that these complexes do not polymerize pure ethylene in CH2Cl2, and the formation of a Pd–acyl species was proposed as the first step, required to enter the copolymerization cycle. The compounds are extraordinarily stable and easy to prepare.57
Similarly, monocationic (dppp)Pd complexes, e.g.17 (dppp = bis(diphenylphosphino)propane) with a 5-ethoxy-4,7-methylenehexahydroinden-6-yl moiety (derived from the dicyclopentadiene DCp; henceforth referred as DCp–OEt, see Fig. 13) catalyze the CO/propene copolymerization in CH2Cl2 or pure propylene.58
The highest molecular weight and yield of alternating copolymers prepared in this way were achieved in pure propylene with SbF6− as counterion, which is understandable. Indeed, an increase in the concentration of propylene should assist its competition with CO in the coordination to the Pd–center. This would increase the equilibrium concentration of the Pd–acyl(propene) species and accelerate the copolymer chain propagation. The lower coordination ability of SbF6−, compared to BF4−, is also favorable. The activity of such a catalytic system for the CO/propene copolymerization is practically equal to the activity of [(dppp)Pd(MeCN)2](BF4)2 in the presence of methanol as activator (see below), a well-known system generally used for the copolymerization of CO with alkenes.50 Preparation of diphosphine–palladium complexes with this DCp–OEt moiety proceeds smoothly from the dimeric μ-(Cl)2-bis-[Pd(DCp–OEt)] 18 upon abstraction of the chloride by silver salts in the presence of the chelating diphosphine.58
Other SCC examples utilizing the DCp–OEt moiety were prepared in a similar way by the reaction of the binuclear chloropalladium precursor 18 with substituted sodium o-diarylphosphinobenzenesulfonates, exemplified in Fig. 13 with complex 19.52 The resulting neutral complexes supported the non-alternating CO/ethylene copolymerization in CH2Cl2 without additional activation, though at elevated temperatures (110 °C).
The (DCp–OEt)–Pd derivatives described above are closely related to the 6-methoxycyclooctadecen-5-ylpalladium complexes (COD–OMe)–Pd, which were intensely studied as pre-activated catalysts for CO/styrene copolymerization under very mild conditions, e.g. at room temperature and atmospheric pressure in CH2Cl2.59,60 Interestingly, formation of the alkoxy precursor 18, in comparison to μ-(Cl)2-bis-[Pd(COD–OMe)] from the corresponding diene derivatives is remarkably facilitated due to the steric strain in complex 18.58,61
As an interesting side note, it was found that the nickel analogue (P⁁O)Ni(DCp–OEt) catalysts were not active for ethylene/CO copolymerization as expected due to the high affinity of Ni for CO but supported the homopolymerization of ethylene.62
Methylpalladium complexes of general structure 1 can also be prepared from the (P⁁P)PdMe2 precursor by reaction with H(OEt2)2+B(Ar′)4− as described above for diimine Ni/Pd complexes. Similar to that case, the fourth coordination position in Pd–diphosphine complexes is occupied by a diethyl ether molecule, as could also be seen from single crystal X-ray analysis.66
Quantitative conversion of different methyl–Pd complexes with CO into the acetyl Pd–COCH3 derivative at low temperatures was demonstrated and studied spectroscopically by several research groups. A subsequent exposure of Pd–acetyl derivatives to ethylene or cycloalkenes allowed the monitoring of the insertion of the latter into the Pd–acyl bond. This leads to the formation of palladium–(3-oxopropyl) derivatives,66–69 stabilized by chelation via the oxygen atom of the carbonyl group to give a five-membered ring which is known as one of the resting states in CO–alkene copolymerization cycle.
The aryl substituted analogues to the alkyl–Pd catalysts proved to be active as well. It has already been mentioned that [(P⁁P)Pd(o-(aminomethyl)phenyl)][X] 16 (X = e.g. PF6−) undergoes a CO insertion into the Pd–C bond. Another example of an active species with an aryl–palladium bond was proposed for the (dppp)(OAc)2Pd activation by B(C6F5)3 in the CO/ethene/propene terpolymerization.70 According to NMR studies, an aryl transfer from borane to palladium occurs during the activation via a relatively complex mechanism. The fourth coordination position at the Pd center is suggested to be occupied by hydroxo-tris(perfluorophenyl)borates acting as a counterion (see Fig. 14). End-group analysis of the resulting polymers indicates that the chain initiation can occur by insertion of an ethylene monomer into the Pd–aryl bond of the (C6F5)–Pd species. The activity of this multicomponent system was found to depend on the initial concentration of the activating agent, which is partially consumed during the copolymerization, and a complete activation of all metal centers is questionable.
 | ||
| Fig. 14 Formation of active species from (dppp)Pd(OAc)2 in the presence of B(C6F5)3. | ||
It is worthwhile to note here that aryl–halogen complexes of diphosphine-chelated palladium are relatively easy to prepare,71 and it is reasonable to assume that SCC species can be generated from these compounds, e.g.via halide abstraction and stabilization of the formed cationic complexes by a weak ligand. Surprisingly, no reports on the application of such systems in CO/alkene copolymerization could be found.
Activation of (dppp)Pd(OAc)2 and of other diphosphine palladium complexes for the CO/ethylene copolymerization can also be carried out with tert-butylalumoxanes, e.g. [(tBu)AlO]n (n = 6, 7, 9).72 Similar to the borane activation, the catalytic effectiveness was found to depend on the ratio of Pd and alumoxane as well as on the structure of the latter. It has been shown that at least two equivalents of alumoxane clusters per (dppp)Pd(OAc)2 are necessary to achieve the optimal performance of the corresponding catalytic system. This requisition is explained by the two-step activation of the Pd–acetate complex (see Fig. 15).
 | ||
| Fig. 15 Proposed steps in activation of (dppp)Pd(OAc)2 by alumoxanes. | ||
The first stage is the metathesis reaction of (dppp)Pd(OAc)2 and [(tBu)AlO]n to give (dppp)Pd(OAc)(tBu) and [(tBu)n−1AlnOn(OAc)]. The formed alumoxane derivative does not react further with (dppp)Pd(OAc)2 or (dppp)Pd(OAc)(tBu) whereas another [(tBu)AlO]n molecule abstracts the acetate from (dppp)Pd(OAc)(tBu) resulting in a catalytically active species for the synthesis of high molecular weight CO/ethene copolymers. Short copolymerization time (15 min) allowed isolation of CO/ethene co-oligomers for their end-group analysis. Surprisingly, only ethyl- and vinylketone end-groups were detected in such oligomers and no tert-butyl incorporation was observed. The reasonable explanation, therefore, is that the abstraction of the acetate from (dppp)Pd(tBu)(OAc) leads to a cationic (dppp)Pd(tBu)+ species, which, under the applied polymerization conditions (aprotic, non-coordinating solvent, 60 °C), is not as stable as the CH3–Pd analogue and undergoes very fast β-hydride elimination to a palladium hydride species prior to CO coordination and insertion. A palladium hydride in its turn can insert ethylene and enter the copolymerization cycle.
Chloropivaloylpalladium compounds (P⁁P)Pd{C(O)tBu}(Cl) can also be activated for CO/ethene copolymerization by chloride abstraction with the aforementioned alumoxanes. End-group analysis of copolymers prepared with these catalytic systems revealed the presence of tert-butylketone terminal fragments.72 This is consistent with the previously described examples where the polymer chain initiation occurs via the ethylene monomer insertion into the Pd–C bond of cationic acyl–palladium species.
The following method of Pd catalyst activation for the CO/alkene copolymerization is, perhaps, the most common one, and modifications of this protocol can lead to remarkable changes in the polymerization results. The method comprises the use of diphosphine chelated palladium(II) acetate in the presence of strong acids with low coordinating ability, e.g.p-toluenesulfonic, tetrafluoroboric or trifluoromethanesulfonic acids, in methanol.73 Generation of dicationic diphosphine–palladium species, which can coordinate CO and ethylene under these conditions, was considered to be essential, and two possible ways of polymer chain initiation by such systems were proposed from polymer end-group analysis. These are, firstly the formation of palladium hydride and secondly of palladium methoxycarbonyl species in methanol, both able to insert ethylene, which were suggested to independently occur under such conditions.43 Both of these species can give keto-ester terminated polymers (ethylketone fragment on one side of the linear polymer chain and a methoxycarbonyl fragment on the other side) if the polymer chain is terminated via solvolysis (methanolysis) and protonolysis.43,68,74 Indeed, the polymeric keto-esters are typically the main copolymerization products under such conditions although other terminal groups are also possible and their amount strongly depends on the experimental settings.
Alternatively to the in situ preparation of (P⁁P)Pd2+ from Pd(OAc)2, the use of Pd complexes with weak coordinating solvent molecules, e.g. acetonitrile, together with the non-coordinating counter-ions such as quaternary borates, allows their activation by methanol without excess of strong acid. On the other hand, the presence of acids delays the catalyst decomposition.43
The role of methanol as both a catalyst initiator and a chain transfer agent leads to a remarkable dependence of the activity as well as of the copolymer structure on the methanol concentration. As shown for the CO/propene copolymerization by [(dppp)Pd(MeCN)2][BF4]2, there is no reaction in the absence of methanol. The addition of ca. 150 equivalents of MeOH respective to Pd results in an optimal activity of the catalytic system with the highest polymer Mw.75 Further increase of the methanol concentration lowers the Mw of the obtained polymers and leads to a drop of activity at high methanol concentrations. A similar trend was also observed when water was used as the activating agent. Incomplete activation of the catalyst at low activator concentrations and the decomposition or deactivation of the catalyst at very high activator concentrations are the reasons for the observed copolymerization tendencies concerning the activator amount as described above. Besides water and methanol, a variety of alcohols can be applied to activate the dicationic palladium catalysts or to increase the activity of the monocationic complexes towards CO/alkene copolymerization. For example, trifluoroethanol was shown to diminish the deactivation problems for (N⁁N)-chelated methylpalladium catalysts during CO/vinylarene copolymerization.64 Activation of [Pd(dppp)(MeCN)2][BF4]2 for the CO/propene copolymerization using ethylene glycol monomethyl ether (Me-EG), poly(ethylene glycol) 1000 or polyvinyl alcohol was also achieved. The incorporation of these alcohols into the polymer structure was demonstrated by different techniques, thus presenting a promising approach towards new block copolymer materials.76 Although the experiments above were not aimed at the verification of the activation mechanism under the applied conditions, some interesting conclusions in this respect could be drawn. It was observed, for example, that, in case of activation by Me-EG and quenching by H2O, solely the polymer with keto-ester end-groups was formed and in nearly equimolar amount respective to the used catalyst, at that. This led to a conclusion that no chain transfer by Me-EG could take place during the copolymerization, probably due to the steric demands, and that the ester end-groups of the polymer were formed exclusively upon chain initiation. Furthermore, this was, to our knowledge, the first report where the structure of low molecular weight CO/propene copolymer was unambiguously supported by MALDI-TOF measurements.
It is common to expect that under high CO pressure the dicationic palladium complexes will exchange the weakly bound solvent ligands with the better coordinating CO molecules. Upon coordination to Pd, carbon monoxide is activated for the attack of nucleophiles such as alcohols, water, etc. The attack can take place via different routes, e.g. directly from the outer coordination sphere on the carbon atom of coordinated CO or via the coordination of a nucleophile to Pd prior to the attack on CO. For the latter, the ligand substitution in the fourth coordination position of square-planar Pd complexes is expected to be facilitated due to the trans- and cis-influence of the diphosphine and one CO ligand. Additionally, despite the relative bulkiness of the diphosphines applied for the catalysis of the CO/alkene copolymerization with palladium, one apical position of the square-planar palladium center still remains accessible, as demonstrated e.g. by the crystal structure elucidation of the [(dppp)Pd(phen)(CH2NO2)][PF6] (phen = phenanthrene) complex.77 Thus, the nucleophile can coordinate at the apical position above the square-planar palladium complex and attack the CO ligand, or substitute one of the ligands in the coordination plane of Pd followed by attack on CO.
Copolymerization activity of the above described dicationic palladium systems also depends on the solubility of the catalyst and the monomer in the reaction medium. In the copolymerization of pure 1-alkenes e.g. 1-hexene with CO, activation of the [(dppp)Pd(MeCN)2][BF4]2 catalyst by methanol or hexanol was found to be less effective in comparison to diethylene glycol (DEG) which was explained as follows:50 methanol and hexanol form homogenous solutions with hexene, and the solubility of the dicationic palladium catalyst in such media is rather poor, making a homogenous catalysis impossible. In contrast, DEG is immiscible with hexene and they form a two-phase system allowing the catalyst to dissolve in the polar phase, where the activation of the catalyst takes place. Growth of the polymer chain on the catalyst enhances its solubility in the non-polar phase, making further chain propagation possible in pure hexene. Obviously, an intense stirring is necessary to increase the effectiveness of such two-phase copolymerization processes. Interestingly, this approach remains effective for a variety of 1-olefins. The reaction is, however, very sensitive to the temperature and gives the best results at ca. 25 °C, at which the constant activity of the catalyst is maintained over several days. Increase of the temperature leads to catalyst decomposition, as noticed by the formation of black Pd(0).
Decomposition of the palladium catalysts under CO/alkene copolymerization conditions is commonly associated with the intermediate formation of palladium hydrides and their derivatives, which can disproportionate to give metallic palladium as one of the products. The mechanism of Pd–H formation and its drop-out of the catalytic cycle in the presence of protic compounds is considered to be rather complex and ambiguous.78 The relatively easy reduction of palladium(II) catalyst to Pd(0) can, however, be suppressed by the addition of oxidants, among which benzoquinone is the most commonly used. The effect of a moderate excess of benzoquinone in the Pd catalytic system can be seen from the increase of the catalyst productivity and the copolymerization rate in comparison with the benzoquinone-free system.43,73 This effect is attributed to the oxidation of palladium hydride species and their catalytically inactive derivatives by benzoquinone to give Pd(II) before the formation of black palladium(0). Fortunately, the addition of benzoquinone seems to have no strong influence on the catalyst activation, the polymer chain initiation and its growth, since the polymers formed both in the presence of and without benzoquinone do not differ in their molecular weight.43
4. Conclusion and perspective
Summarizing the discussion on activation of late transition metal catalysts for olefin polymerization and copolymerization, we would like to draw attention to the fact that, despite a vast number of reports on the synthesis of polymers, the comprehensive experimental mechanistic studies of these processes are not as numerous as in case of earlier transition metal catalysts. Probably, this is one of the reasons why some aspects of olefin polymerization and especially of their copolymerization with CO still remain very disputable. Among them, for example, are the questions of CO/olefin copolymer chain initiation and transfer, which seem to be specifically dependent on the conditions of copolymerization.By comparison to the explained cases of the activation of late transition metal polymerization catalysts, several analogies can obviously be observed. The α-diimine complexes as well as the different complexes for alkene/CO copolymerizations show clear similarities with metallocene polymerization catalysts concerning the possible activation pathways.
Differences to early transition metal complexes mainly result from the inherent character of the catalytically active metal. The reduced electrophilicity of the cationic Ni(II) or Pd(II) catalysts, even more pronounced in neutral complexes, allows the presence of polar additives or functionalities, be it comonomers or solvents. Reversible coordination of polar groups or electron donors is elegantly exploited by certain activation methods. Compared with each other, the similarities of the metal α-diimine and the diphosphine–Pd complex are even greater, as directly comparable square planar catalyst structures and the similar reactivity show. Reversible polar group addition and chelate formation are found in both systems, and are even the necessary features which allow the strictly alternating polymerization of alkenes and CO.
To find a suitable activation method for a catalyst system, a wide variety of reactions are available, which allow optimization and adaptation to specific polymerization conditions. System inherent factors like steric hindrances, solvents, reaction temperature or the need for in situ investigations of the reaction mechanism are to be considered, as these factors can limit the number of possible reactions drastically. With the general concepts a good basis for modification and new reactions are available.
The very interesting SCC concept is a way to get around the usual problems of in situ activation, at least to a certain extent. In cases of highly reactive complexes, the catalyst is quantitatively active at the very beginning of the polymerization. An increasing number of reported SCC catalysts firstly provides a basic pool of suitable systems and secondly enhances the understanding of the effects of modifications. More detailed investigations of polymerization processes leading to a better comparability and more insight into substitution effects would be desirable in the literature.
References
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1203 CrossRef CAS.
- P. Kuhn, D. Sémeril, D. Matt, M. J. Chetcuti and P. Lutz, Dalton Trans., 2007, 515–528 RSC.
- (a) Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059–2062 CrossRef CAS; (b) Z. Guan, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3680–3692 CrossRef CAS.
- L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479–1493 CrossRef CAS.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS.
- G. Müller and B. Rieger, Prog. Polym. Sci., 2002, 27, 815–851 CrossRef CAS.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS.
- S. Mecking, Angew. Chem., Int. Ed., 2001, 40, 534–540 CrossRef CAS.
- (a) T. C. Chung and D. Rhubright, Macromolecules, 1993, 26, 3019–3025 CrossRef CAS; (b) M. J. Schneider, R. Schafer and R. Mülhaupt, Polymer, 1997, 38, 2455–2459 CrossRef CAS; (c) C. E. Wilen, H. Luttikhedde, T. Hjertberg and J. H. Nasman, Macromolecules, 1996, 28, 8569–8575 CrossRef.
- M. M. Marques, S. G. Correia, J. R. Ascenso, A. F. G. Ribeiro, P. T. Gomes, A. R. Dias, P. Foster, M. D. Rausch and J. C. W. Chien, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2457–2469 CrossRef CAS.
- (a) E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS; (b) J.-N. Pédeutour, K. Radhakrishnan, H. Cramail and A. Deffieux, Macromol. Rapid Commun., 2001, 22, 1095–1123 CrossRef CAS.
- R. Blom and I. M. Dahl, Macromol. Chem. Phys., 1999, 200, 442–449 CrossRef CAS.
- P. C. Möhring and N. J. Coville, J. Org. Chem., 1994, 479, 1–29 CrossRef.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- (a) H. tom Dieck, M. Svoboda and J. Kopf, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1978, 33, 1381–1385; (b) H. tom Dieck, M. Svoboda and T. Greiser, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1981, 36, 823–832; (c) M. Svoboda and H. tom Dieck, J. Organomet. Chem., 1980, 191, 321–328 CrossRef.
-
(a) M. S. Brookhart, L. K. Johnson, C. M. Killian, S. D. Arthur, J. Feldman, E. F. McCord, S. J. McLain, K. A. Kreutzer, A. M. A. Bennett, E. B. Coughlin, S. D. Ittel, A. Parthasarathy, D. J. Tempel, WO Pat., 9623010, 1996 Search PubMed;
(b) For ligand compositions: M. S. Brookhart, L. K. Johnson, S. D. Arthur, J. Feldman, K. A. Kreutzer, A. M. A. Bennett, E. Coughlin, S. D. Ittel, A. Parthasarathy, D. J. Tempel, US Pat., 5886224, 1999 Search PubMed.
- M. Schmid, R. Eberhardt, M. Klinga, M. Leskelä and B. Rieger, Organometallics, 2001, 20, 2321–2330 CrossRef CAS.
- D. Meinhard, M. Wegner, G. Kipiani, A. Hearly, P. Reuter, S. Fischer, O. Marti and B. Rieger, J. Am. Chem. Soc., 2007, 129, 9182–9191 CrossRef CAS.
- S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 10634–10635 CrossRef CAS.
- D. Tempel, L. K. Johnson, R. L. Huff, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6686–6700 CrossRef CAS.
- D. J. Tempel and M. Brookhart, Organometallics, 1998, 17, 2290–2296 CrossRef CAS.
- L. H. Schultz, D. J. Tempel and M. Brookhart, J. Am. Chem. Soc., 2001, 123, 11539–11555 CrossRef CAS.
- (a) L. Deng, T. K. Woo, L. Cavallo, P. M. Margl and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 6177–6186 CrossRef CAS; (b) L. Deng, P. Margl and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 1094–1100 CrossRef CAS; (c) A. Michalak and T. Ziegler, Kinet. Catal., 2006, 47, 310–325 CAS.
- For a reported β-agostic species in a (P⁁P)M(C2H5)+ complex: F. M. Conroy-Lewis, L. Mole, A. D. Redhouse, S. A. Litster and J. L. Spencer, J. Chem. Soc., Chem. Commun., 1991, 1601–1603 Search PubMed.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS.
- S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888–899 CrossRef CAS.
- A. Michalak and T. Ziegler, J. Am. Chem. Soc., 2001, 123, 12266–12278 CrossRef CAS.
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Ittel, D. Tempel, C. M. Killian, L. K. Johnson and M. Brookhart, Macromolecules, 2007, 40, 410–420 CrossRef CAS.
- A. C. Gottfried and M. Brookhart, Macromolecules, 2003, 36, 3085–3100 CrossRef and references therein.
- C. M. Killian, L. K. Johnson and M. Brookhart, Organometallics, 1997, 16, 2005–2007 CrossRef CAS.
- X. Zeng and K. Zetterberg, Macromol. Chem. Phys., 1998, 199, 2677–2681 CrossRef CAS.
- M. Rudler-Chauvin and H. Rudler, J. Organomet. Chem., 1977, 134, 115–119 CrossRef CAS.
- R. F. de Souza, R. S. Mauler, L. C. Simon, F. F. Nunes, D. V. S. Vescia and A. Cavagnolli, Macromol. Rapid Commun., 1997, 18, 795–800 CrossRef CAS.
- D. Pappalardo, M. Mazzeo and C. Pellecchia, Macromol. Rapid Commun., 1997, 18, 1017 CrossRef CAS.
- For the synthesis of HB(Ar′)4: M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 1992, 11, 3920 Search PubMed.
- S. Borkar, H. Yennawar and A. Sen, Organometallics, 2007, 26, 4711–4714 CrossRef CAS.
- W. Li, X. Zhang, A. Meetsma and B. Hessen, J. Am. Chem. Soc., 2004, 126, 12246–12247 CrossRef CAS.
- B. Temme, G. Erker, J. Karl, H. Luftmann, R. Fröhlich and S. Kotila, Angew. Chem., Int. Ed. Engl., 1995, 34, 1755–1757 CrossRef CAS.
- G. Erker, Acc. Chem. Res., 2001, 34, 309–317 CrossRef CAS.
- J. W. Strauch, G. Erker, G. Kehr and R. Fröhlich, Angew. Chem., Int. Ed., 2002, 41, 2543–2546 CrossRef CAS.
- F. Ballauf, O. Bayer, L. Leichmann, Ger. Pat., 863711, 1941 Search PubMed.
- W. Reppe, A. Mangini, US Pat., 2577208, 1951 Search PubMed.
- E. Drent and P. H. M. Budzelaar, Chem. Rev., 1996, 96, 663–682 CrossRef CAS.
- E. Drent, Eur. Pat. Appl., 121965 A2, 1984 Search PubMed.
- R. A. M. Robertson and D. J. Cole-Hamilton, Coord. Chem. Rev., 2002, 225, 67–90 CrossRef CAS.
- C. Bianchini and A. Meli, Coord. Chem. Rev., 2002, 225, 35–66 CrossRef CAS.
- B. Milani, G. Corso, G. Mestroni, C. Carfagna, M. Formica and R. Seraglia, Organometallics, 2000, 19, 3435–3441 CrossRef CAS.
- J. Durand, E. Zangrando, M. Stener, G. Fronzoni, C. Carfagna, B. Binotti, P. C. J. Kamer, Ch. Mueller, M. Caporali, P. W. N. M. van Leeuwen, D. Vogt and B. Milani, Chem.–Eur. J., 2006, 12, 7639–7651 CrossRef CAS.
- A. S. Abu-Surrah and B. Rieger, Top. Catal., 1999, 165–177 CrossRef CAS.
- D. Perez-Foullerat, U. W. Meier, S. Hild and B. Rieger, Macromol. Chem. Phys., 2004, 205, 2292–2302 CrossRef CAS.
- E. Drent, R. van Dijk, R. van Ginkel, B. van Oort and R. I. Pugh, Chem. Commun., 2002, 964–965 RSC.
- A. K. Hearley, R. J. Nowack and B. Rieger, Organometallics, 2005, 24, 2755–2763 CrossRef CAS.
- J.-T. Chen and A. Sen, J. Am. Chem. Soc., 1984, 106, 1506–1507 CrossRef CAS.
- A. Sen, J.-T. Chen, W. M. Vetter and R. R. Whittle, J. Am. Chem. Soc., 1987, 109, 148–156 CrossRef.
- P. Margl and T. Ziegler, J. Am. Chem. Soc., 1996, 118, 7337–7344 CrossRef.
- C. S. Shultz, J. Ledford, J. M. DeSimone and M. Brookhart, J. Am. Chem. Soc., 2000, 122, 6351–6356 CrossRef CAS.
- J. Schwarz, E. Herdtweck, W. A. Herrmann and M. G. Gardiner, Organometallics, 2000, 19, 3154–3160 CrossRef CAS.
- D. Meinhard, F. Hollmann, W. Huhn, U. Thewalt, M. Klinga and B. Rieger, Organometallics, 2004, 23, 5637–5639 CrossRef CAS.
- A. Macchioni, G. Bellachioma, G. Cardachi, M. Travaglia, C. Zuccaccia, B. Milani, G. Corso, E. Zangrando, G. Mestroni, C. Carfagna and M. Formica, Organometallics, 1999, 18, 3061–3069 CrossRef CAS.
- B. Binotti, G. Bellachioma, G. Cardaci, C. Carfagna, C. Zuccaccia and A. Macchioni, Chem.–Eur. J., 2007, 13, 1570–1582 CrossRef CAS.
- J. Chatt, L. M. Vallarino and L. M. Venanzi, J. Chem. Soc., 1957, 3413–3416 RSC.
- R. J. Nowack, A. K. Hearley and B. Rieger, Z. Anorg. Allg. Chem., 2005, 631, 2775–2781 CrossRef CAS.
- M. A. Zuideveld, B. H. G. Swennenhuis, M. D. K. Boele, Y. Guari, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 2002, 2308–2317 RSC.
- A. Scarel, J. Durand, D. Franchi, E. Zangrando, G. Mestroni, B. Milani, S. Gladiali, C. Carfagna, B. Binotti, S. Bronco and T. Gragnoli, J. Organomet. Chem., 2005, 690, 2106–2120 CrossRef CAS.
- O. Daugulis, M. Brookhart and P. S. White, Organometallics, 2002, 21, 5935–5943 CrossRef CAS.
- J. Ledford, C. S. Shultz, D. P. Gates, P. S. White, J. M. DeSimone and M. Brookhart, Organometallics, 2001, 20, 5266–5276 CrossRef CAS.
- F. Ozawa, T. Hayashi, H. Koide and A. Yamamoto, J. Chem. Soc., Chem. Commun., 1991, 1469–1470 RSC.
- M. A. Zuideveld, P. C. J. Kamer, P. W. N. M. van Leeuwen, P. A. A. Klusener, H. A. Stil and C. F. Roobeek, J. Am. Chem. Soc., 1998, 120, 7977–7978 CrossRef.
- K. Nozaki, N. Sato, Y. Tonomura, M. Yasutomi, H. Takaya, T. Hiyama, T. Matsubara and N. Koga, J. Am. Chem. Soc., 1997, 119, 12779–12795 CrossRef CAS.
- G. K. Barlow, J. D. Boyle, N. A. Cooley, T. Ghaffar and D. F. Wass, Organometallics, 2000, 19, 1470–1476 CrossRef CAS.
- W. A. Herrmann, C. Broßmer, T. Priermeier and K. Öfele, J. Organomet. Chem., 1994, 481, 97–108 CrossRef CAS.
- Y. Koide, S. G. Bott and A. R. Barron, Organometallics, 1996, 15, 2213–2226 CrossRef CAS.
- E. Drent, J. A. M. Broekhoven and M. J. Doyle, J. Organomet. Chem., 1991, 417, 235–251 CrossRef CAS.
- P. W. N. M. van Leeuwen, M. A. Zuideveld, B. H. G. Swennenhuis, Z. Freixa, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz and A. L. Spek, J. Am. Chem. Soc., 2003, 125, 5523–5539 CrossRef CAS.
- A. S. Abu-Surrah, R. Wursche, B. Rieger, G. Eckert and W. Pechhold, Macromolecules, 1996, 29, 4806–4807 CrossRef CAS.
- A. Mücke and B. Rieger, Macromolecules, 2002, 35, 2865–2867 CrossRef.
- B. Milani, G. Corso, E. Zangrando, L. Randaccio and G. Mestroni, Eur. J. Inorg. Chem., 1999, 2085–2093 CrossRef CAS.
- C. Bianchini, A. Meli and W. Oberhauser, Dalton Trans., 2003, 2627–2635 RSC.
| This journal is © The Royal Society of Chemistry 2008 |