Hypervalent organobismuth(III) carbonate, chalcogenides and halides with the pendant arm ligands 2-(Me2NCH2)C6H4 and 2,6-(Me2NCH2)2C6H3†‡
Hans J.
Breunig
*a,
Lucia
Königsmann
a,
Enno
Lork
a,
Mihai
Nema
b,
Nicky
Philipp
a,
Cristian
Silvestru
*b,
Albert
Soran
b,
Richard A.
Varga
b and
Roxana
Wagner
a
aInstitut für Anorganische und Physikalische Chemie, Universität Bremen, D-28334, Bremen, Germany. E-mail: breunig@chemie.uni-bremen.de; Fax: +49 421 218 4042; Tel: +49 421 218 2266
bFacultatea de Chimie si Inginerie Chimica, Universitatea Babes-Bolyai, RO-400028, Cluj-Napoca, Romania. E-mail: cristi@chem.ubbcluj.ro; Fax: +40 264 590818; Tel: +40 264 593833
First published on 27th February 2008
Abstract
R2BiOH (1) [R = 2-(Me2NCH2)C6H4] and (R2Bi)2O (2) are formed by hydrolysis of R2BiCl with KOH. Single crystals of 2 were obtained by air oxidation of (R2Bi)2. The reaction of R2BiCl and Na2CO3 leads to (R2Bi)2CO3 (3). 3 is also formed by the absorption of CO2 from the air in solutions of 1 or 2 in diethyl ether or toluene. (R2Bi)2S (4) is obtained from R2BiCl and Na2S or from (R2Bi)2 and S8. Exchange reactions between R2BiCl and KBr or NaI give R2BiX [X = Br (5), I (6)]. The reaction of RBiCl2 (7) with Na2S and [W(CO)5(THF)] gives cyclo-(RBiS)2[W(CO)5]2 (8). cyclo-(R′BiS)2 (9) [R′ = 2,6-(Me2NCH2)2C6H3] is formed by reaction of R′BiCl2 and Na2S. The structures of 1–9 were determined by single-crystal X-ray diffraction.
Introduction
Aryl ligands with one or two pendant arms, i.e. 2-(Me2NCH2)C6H4, 2,6-(Me2NCH2)2C6H3 and other aryl groupsortho-substituted with CH2NMe2 or related pendant arms, received attention in organobismuth(III) chemistry.1,2 Most of the publications were concerned with the synthesis and characterization of organobismuth halides useful as starting materials for further chemistry, e.g. RR′BiCl (R′ = 4-MeOC6H4, 4-MeC6H4, Ph, 4-ClC6H4, 1-naphthyl),3RPhBiBr,3 [2-{Me2N(Me)CH}C6H4]R′BiCl (R′ = Ph, 1-naphthyl),3,4 R2BiCl,5 RBiX2 (X = Cl, I),5R[(Me3Si)2CH]BiCl6 [R = 2-(Me2NCH2)C6H4], [2,6-(Me2NCH2)2C6H3]BiX2 (X = Cl,7,8Br, I8), [2,6-(Me2NCH2)2C6H3]2BiCl and [2,6-(Me2NCH2)2C6H3](Me)BiI.8 Studies on few compounds of other types were also reported, e.g. [{2-(Me2NCH2)C6H4}2Bi]+[PF6]−,9 [2-(Me2NCH2)C6H4]Bi[C6H4{C(CF3)2O}-2],10,11 and RBi[C6H4{C(CF3)2O}-2] [R = 2,6-(Me2NCH2)2-4-R′C6H2, R′ = H, tBu].11 In our recent work12–15 we have shown that 2-(Me2NCH2)C6H4 and 2,6-(Me2NCH2)2C6H3groups provide stabilisation by sterical protection and internal coordination of the amino groups for organobismuth compounds, e.g. low-valent species such as cyclo-[2-(Me2NCH2)C6H4]4Bi4,12 R4Bi2 [R = 2-(Me2NCH2)C6H4,12 2,6-(Me2NCH2)2C6H3,14], the first functionalized organobismuth(III) derivatives of the type [2-(Me2NCH2)C6H4]BiCl[(XPR2)(YPR′2)N] (X, Y = O, S, Se; R, R′ = Me, Ph),13 the first peroxo derivative [{2,6-(Me2NCH2)2C6H3}2Bi]2(O2),14 and [2-(Me2NCH2)C6H4]2BiAu(C6F5)2, a compound which exhibits the first metallophilic AuI⋯BiIII closed-shell interaction.15 Intramolecular N→Bi interactions were observed both in the solid state and in solution in all these compounds which can be described, transcending the octet theory, as hypervalent species.16Organobismuth chalcogenides of the type R2BiEH, (R2Bi)2E, (RBiE)n (E = chalcogen, R = alkyl, aryl) are of interest as potential precursors for semiconducting bismuth chalcogenides, Bi2E3, and because they can display interesting molecular and supramolecular architectures. The first studies on compounds of this type were performed in the 19th century, when alkylbismuth(III) derivatives, i.e.Me2BiOH, (Me2Bi)2S, MeBiO, EtBiO, MeBiS, were reported.17–21 These early results are however questionable.21 Well defined compounds of the type (R2Bi)2E [R = Me, n-Pr, p-Tol, Mes, (Me3Si)2CH] and (RBiE)n [R = 2,4,6-{(Me3Si)2CH}3C6H2, n = 2, E = O; n = 3, E = S] have been synthesised since 1980,22–30 and crystal structures were determined for sterically protected bis(diorganobismuth)chalcogenides, (R2Bi)2E [R = Mes, E = O,24,25 S, Se;24 R = (Me3Si)2CH, E = S, Te26], and the organobismuth oxide, [2,4,6-{(Me3Si)2CH}3C6H2BiO]2.30
Organobismuth carbonates of the types (R2Bi)2CO3 have not yet been described in the literature. The closest related bismuth compounds are diorganobismuth carboxylates, e.g. PhBi(O2CMe)2 or Ph2Bi(O2CMe).31
We report here on the formation and structures of hypervalent organobismuth compounds R2BiOH (1), (R2Bi)2O (2), (R2Bi)2CO3 (3), (R2Bi)2S (4), R2BiBr (5), R2BiI (6), RBiCl2 (7), cyclo-(RBiS)2[W(CO)5]2 (8) [R = 2-(Me2NCH2)C6H4], and cyclo-(R′BiS)2 (9) [R′ = 2,6-(Me2NCH2)2C6H3]. The synthesis of 7 was published before.5 Compounds 8 and 9 were briefly reported in a previous conference report.32 Related antimony compounds e.g. cyclo-(RSbS)2 and cyclo-(RSbS)2[W(CO)5] [R = 2-(Me2NCH2)C6H4] were already described by us.33
Results and discussion
Diorganobismuth compounds
The diarylbismuth hydroxide [2-(Me2NCH2)C6H4]2BiOH (1) as well as the corresponding oxide [{2-(Me2NCH2)C6H4}2Bi]2O (2) are formed by hydrolysis of R2BiCl with KOH in a two-phase system of water and diethyl ether in an inert atmosphere. Single crystals of 1·2H2O precipitated after exposing the dried ether phase to the air for 12 h. Conversion of the hydroxide into the oxide can be achieved by treatment of solution of 1 with anhydrous Na2SO4. Prolonged exposure to the air leads to the absorption of CO2 and formation of the carbonate [{2-(Me2NCH2)C6H4}2Bi]2CO3 (3). Compound 3 is also obtained by reaction of Na2CO3 with R2BiCl in CH2Cl2, or when R2BiCl is reacted with KOH in toluene/water in air. Another way for the synthesis of 2 is the insertion of oxygen into the Bi–Bi bond of the dibismuthane R2Bi–BiR2 [R = 2-(Me2NCH2)C6H4]. Single crystals of 2 were obtained serendipitously after addition of elemental selenium to a solution of R2Bi–BiR2 in petroleum ether, due to the reaction of the dibismuthane with traces of air. The reaction of R2Bi–BiR2 with S8 in petroleum ether gives [{2-(Me2NCH2)C6H4}2Bi]2S (4) in 60% yield. Compound 4 is also formed from R2BiCl and Na2S in water/diethyl ether. Halide exchange reactions between R2BiCl and KBr or NaI in ethanol give [2-(Me2NCH2)C6H4]2BiBr (5) and [2-(Me2NCH2)C6H4]2BiI (6). The reactions leading to 1–6 are summarized in Scheme 1. | ||
| Scheme 1 | ||
Compounds 1, 2 and 3 are colourless crystalline compounds, soluble in organic solvents. 1 and 2 are hygroscopic. The sulfide 4 and the halides 5 and 6 are air-stable yellow solids soluble in organic solvents. MS and NMR data of 1–6 as well as elemental analytical data of 3–6 are consistent with the anticipated formulas. Elemental analyses of samples of 1 and 2 prepared by hydrolysis gave only approximate values, probably due to varying amounts of residual water. The room-temperature NMR spectra of 1–6 exhibit only one set of resonances for the two organic groups attached to a metal centre, thus suggesting their equivalence at the NMR time scale. A fluxional behaviour, similar to that observed previously for the related [2-(Me2NCH2)C6H4]2SbCl,34 is supported by the quite broad 1H resonances observed for the methyl protons and the AB spin systems for the CH2groups. Characteristic ions are present in the mass spectra of 1–6 which all contain [R2Bi+] as the base peak. In the case of 4 and 5 the molecular ion is observed.
Single crystals suitable for X-ray diffraction studies were obtained from diethyl ether (1·2H2O), petroleum ether (2), CH2Cl2–hexane (3) or benzene (3·C6H6), acetone (4) and ethanol (5 and 6). The molecular structures are shown in Fig. 1–4 and selected interatomic distances and angles are listed in Tables 1 and 2.
| 1·2H2O | 2 | 4 | 5 | 6 | |
|---|---|---|---|---|---|
| a E = O for 1 and 2, S for 4, Br for 5, and I for 6. | |||||
| Bi(1)–C(1) | 2.279(8) | 2.268(14) | 2.287(6) | 2.254(6) | 2.267(4) |
| Bi(1)–C(10) | 2.267(8) | 2.249(15) | 2.262(6) | 2.255(6) | 2.256(4) |
| Bi(1)–E(1)a | 2.189(6) | 2.124(9) | 2.5558(17) | 2.8452(7) | 3.0723(6) |
| Bi(1)–N(1) | 2.715(7) | 2.828(12) | 2.900(5) | 2.534(5) | 2.514(4) |
| Bi(1)–N(2) | 3.099(9) | 3.183(10) | 3.043(6) | 3.151(5) | 3.148(3) |
| O(1)–H(1) | 0.840(6) | ||||
| E(1)–Bi(1)–C(1) | 90.8(3) | 88.6(5) | 91.02(17) | 92.89(16) | 94.25(12) |
| E(1)–Bi(1)–C(10) | 85.5(3) | 95.7(4) | 94.60(16) | 88.57(16) | 87.77(12) |
| C(1)–Bi(1)–C(10) | 94.4(3) | 93.9(5) | 95.5(2) | 95.5(2) | 94.90(16) |
| N(1)–Bi(1)–E(1) | 159.6(2) | 157.2(3) | 156.8(1) | 166.19(12) | 168.11(9) |
| N(1)–Bi(1)–C(1) | 70.0(3) | 69.1(4) | 68.8(2) | 73.3(2) | 73.86(15) |
| N(1)–Bi(1)–C(10) | 88.9(3) | 81.5(4) | 76.8(2) | 93.5(2) | 93.02(15) |
| N(1)–Bi(1)–N(2) | 89.6(2) | 113.5(3) | 105.6(1) | 77.63(15) | 78.47(10) |
| N(2)–Bi(1)–E(1) | 105.9(2) | 85.0(2) | 90.1(1) | 115.53(10) | 112.56(7) |
| N(2)–Bi(1)–C(1) | 152.5(3) | 156.7(4) | 162.4(2) | 144.5(2) | 145.7(1) |
| N(2)–Bi(1)–C(10) | 66.1(3) | 64.5(4) | 66.8(2) | 66.1(2) | 66.6(1) |
| Bi(1)–O(1)–H(1) | 109.4(5) | ||||
| Bi(1)–E(1)–Bi(1a) | 109.3(7) | 98.17(8) | |||
| 3 | 3·C6H6 | 3 | 3·C6H6 | ||
|---|---|---|---|---|---|
| a O(X) = O(1) for 3, and O(2) for 3·C6H6. b O(Y) = O(3) for 3, and O(1) for 3·C6H6. c O(Z) = O(2) for 3, and O(3) for 3·C6H6. | |||||
| Bi(1)–C(1) | 2.272(8) | 2.257(7) | Bi(2)–C(19) | 2.274(9) | 2.280(8) |
| Bi(1)–C(10) | 2.269(9) | 2.260(8) | Bi(2)–C(28) | 2.247(8) | 2.270(8) |
| Bi(1)–O(X)a | 2.280(5) | 2.256(5) | Bi(2)–O(Z)c | 2.245(6) | 2.260(5) |
| Bi(1)–O(Y)b | 2.879(6) | 2.924(6) | Bi(2)–O(1) | 2.973(5) | 2.999(5) |
| Bi(1)–N(1) | 2.635(7) | 2.658(7) | Bi(2)–N(3) | 2.722(8) | 2.632(7) |
| Bi(1)–N(2) | 3.411(10) | 3.177(7) | Bi(2)–N(4) | 2.918(10) | 3.251(8) |
| C(37)–O(1) | 1.317(10) | 1.243(10) | |||
| C(37)–O(2) | 1.293(9) | 1.282(9) | |||
| C(37)–O(3) | 1.243(10) | 1.299(9) | |||
| O(X)–Bi(1)–C(1)a | 85.5(3) | 83.4(2) | O(Z)–Bi(2)–C(19)c | 88.1(3) | 83.7(2) |
| O(X)–Bi(1)–C(10)a | 89.7(3) | 92.7(2) | O(Z)–Bi(2)–C(28)c | 86.9(3) | 89.1(2) |
| C(1)–Bi(1)–C(10) | 92.3(3) | 94.2(3) | C(19)–Bi(2)–C(28) | 94.0(3) | 94.0(3) |
| N(1)–Bi(1)–O(X)a | 157.0(2) | 153.7(2) | N(3)–Bi(2)–O(Z)c | 156.9(2) | 154.8(2) |
| N(1)–Bi(1)–C(1) | 71.5(3) | 70.4(3) | N(3)–Bi(2)–C(19) | 70.4(3) | 71.2(3) |
| N(1)–Bi(1)–C(10) | 91.1(3) | 90.5(2) | N(3)–Bi(2)–C(28) | 86.3(3) | 90.9(3) |
| N(1)–Bi(1)–N(2) | 77.0(2) | 84.4(2) | N(3)–Bi(2)–N(4) | 115.3(3) | 79.2(2) |
| N(2)–Bi(1)–O(X)a | 123.2(2) | 120.6(2) | N(4)–Bi(2)–O(Z)c | 82.2(2) | 123.1(2) |
| N(2)–Bi(1)–C(1) | 139.3(3) | 147.9(2) | N(4)–Bi(2)–C(19) | 159.8(3) | 143.5(2) |
| N(2)–Bi(1)–C(10) | 62.7(3) | 65.8(3) | N(4)–Bi(2)–C(28) | 68.0(3) | 65.2(3) |
| O(1)–C(37)–O(2) | 115.9(8) | 121.9(7) | |||
| O(1)–C(37)–O(3) | 122.3(7) | 121.8(7) | |||
| O(2)–C(37)–O(3) | 121.8(9) | 116.3(7) | |||
| Bi(1)–O(X)–C(37)a | 107.0(5) | 110.3(5) | Bi(2)–O(Z)–C(37)c | 115.9(6) | 111.9(5) |
 | ||
| Fig. 1 ORTEP representation at 40% probability and atom numbering scheme for (RN1,RN2)-1 isomer. The water molecules are omitted. | ||
![ORTEP representation at 30% probability and atom numbering scheme for (SN1,RN2)-2 isomer [symmetry equivalent atoms (−x, y, 0.5 −z) are given by “a”]. Hydrogen atoms are omitted.](/image/article/2008/DT/b717127g/b717127g-f2.gif) | ||
| Fig. 2 ORTEP representation at 30% probability and atom numbering scheme for (SN1,RN2)-2 isomer [symmetry equivalent atoms (−x, y, 0.5 −z) are given by “a”]. Hydrogen atoms are omitted. | ||
 | ||
| Fig. 3 ORTEP representation at 30% probability and atom numbering scheme for (a) (SN1,RN2,SN3,SN4) and (b) (SN1,RN2,SN3,RN4) isomers in the crystals of 3 and 3·C6H6 (benzene molecule is not shown), respectively. Hydrogen atoms are omitted. | ||
 | ||
| Fig. 4 ORTEP representation at 30% probability and atom numbering scheme for (RN1,SN2)-6 isomer. | ||
A common feature for all diorganobismuth(III) compounds 1–6 is that a metal centre is coordinated stronger by the nitrogen atom of one amine pendant arm, in trans to the chalcogen or halogen atom [see N(1)–Bi(1)–E(1) angles, E = O, S, Br, I for 1·2H2O, 2 and 4–6 (Table 1) and N(1)–Bi(1)–O(X) and N(3)–Bi(2)–O(Z) angles for 3 and 3·C6H6 (Table 2)], while the nitrogen atom of the other amine group exhibits a weak intramolecular interaction trans to a carbon atom [see N(2)–Bi(1)–C(1) angles for 1·2H2O, 2 and 4–6 (Table 1) and N(2)–Bi(1)–C(1) and N(4)–Bi(2)–C(19) angles for 3 and 3·C6H6 (Table 2)]. The strength of the Bi(1)–N(1) interaction in trans to the halogen atom in the bromide 5 [2.534(5) Å] and the iodide 6 [2.514(4) Å] is similar to that found for the related chloride [2.570(5) Å].5 By contrast, a remarkable difference was found for the Bi(1)–N(1) interaction in trans to the chalcogen atom, e.g. it is considerably stronger in 1·2H2O [Bi(1)–N(1) 2.715(7) Å] than in the oxide 2 [2.828(12) Å] and the sulfide 4 [2.900(5) Å]. In 3·C6H6 the strength of these interactions is even stronger [Bi(1)–N(1) 2.658(7), Bi(2)–N(3) 2.632(7) Å], while for the solvent-free carbonate 3 an asymmetry in the two strong intramolecular N→Bi interactions was observed [Bi(1)–N(1) 2.635(7), Bi(2)–N(3) 2.722(8) Å]. The weaker N→Bi intramolecular interaction is of the same magnitude in compounds 1·2H2O, 2, 4–6 [Bi(1)–N(2) range 3.043(6)–3.183(10) Å] and compares well with the value observed in the corresponding [2-(Me2NCH2)C6H4]2BiCl used as starting material [3.047(5) Å].5 A considerable difference should be noted between the two weaker N→Bi intramolecular interactions in the molecular unit of the carbonate, i.e. Bi(1)–N(2) and Bi(2)–N(4) 3.411(10) and 2.918(10) Å in 3, and 3.177(7) and 3.251(8) Å in 3·C6H6, respectively. If both these intramolecular N→Bi interactions per metal atom are considered [cf. sums of the corresponding covalent, ∑rcov(Bi,N) 2.22 Å, and van der Waals radii, ∑rvdW(Bi,N) 3.94 Å],35 the overall coordination becomes distorted square-pyramidal [(C,N)2BiX cores] and the compounds can be described as hypervalent 12-Bi-5 species.16,36
The bismuth–chalcogen distances in the hydroxide 1·2H2O [Bi(1)–O(1) 2.189(6) Å], the oxide 2 [Bi(1)–O(1) 2.124(9) Å] and the sulfide 4 [Bi(1)–S(1) 2.5558(17) Å] are similar to the values found in the few related compounds described so far, i.e.(Mes2Bi)2O·EtOH [2.10(2), 2.12(2) Å],24(Mes2Bi)2O [2.075(8), 2.064(7) Å],25 (Mes2Bi)2S [2.520(7), 2.545(6) Å],24 and [((Me3Si)2CH)2Bi]2S [2.572(1), 2.557(1) Å].26
The Bi(1)–O(1)–Bi(1a) angle in 2 [109.3(7)°] is more acute than in (Mes2Bi)2O·EtOH [117.1(8)°]24 or (Mes2Bi)2O [124.6(4)°],25 while the Bi(1)–S(1)–Bi(1a) angle in 4 [98.17(8)°] is similar to that found for the related (Mes2Bi)2S [98.7(2)°],24 but considerably larger than in [(Me3Si)2Bi]2S [92.48(4)°].26 The Bi⋯Bi distances are 3.465(1) and 3.863(1) Å in 2 and 4, respectively; these values are shorter than the sum of van der Waals-radii for two Bi atoms [∑rvdW(Bi,Bi) 4.80 Å],35 but considerably longer than the Bi–Bi single bond in [2-(Me2NCH2)C6H4]4Bi2 [3.0657(5) Å].12 Apparently in these µ-chalcogeno species the bridging chalcogen atoms behave as clips pressing together the two “soft” bismuth atoms. Due to the relative longer bonds in the Bi–S–Bi bridge this effect is less expressed in the molecule of 4 than in 2 although the central bond angle is more acute in the sulfur compound. Alternatively, the concept of a 3c–2e bond between the two bismuth atoms and the chalcogen atom might explain the short Bi⋯Bi distance in 2 and 4. The torsion angles ϕ (ϕ = Bi–E–Bi–lp; E = O, S, and lp assumed position of the lone pair of electrons at Bi) are 7.1° in 2 and 10.1° in 4, thus corresponding to a syn-syn-conformation.
In the bis(diorganobismuth) carbonates the covalent bismuth–oxygen bonds established by the planar CO3group [Bi(1)–O(1) 2.280(5), Bi(2)–O(2) 2.245(6) Å in 3, and Bi(1)–O(2) 2.256(5), Bi(2)–O(3) 2.260(5) Å in 3·C6H6] are considerably longer than in the hydroxide 1·2H2O and the oxide 2. Additional short intramolecular Bi⋯O interactions are also present [Bi(1)–O(3) 2.879(6), Bi(2)–O(1) 2.973(5) Å in 3, and Bi(1)–O(1) 2.924(6), Bi(2)–O(1) 2.999(5) Å in 3·C6H6]. Not only the CO3 unit but also the two bismuth atoms and two of the directly bonded carbon atoms lie in a plane [deviations from CO3 plane: Bi(1) 0.059, Bi(2) 0.213, C(1) −0.124 and C(28) 0.881 Å in 3; Bi(1) −0.196, Bi(2) 0.455, C(1) −0.108 and C(19) 0.459 Å in 3·C6H6]. The main difference in the quasiplanar (CBi)2CO3 system of 3 and 3·C6H6 is the orientation of the covalent Bi–O bonds and the trans [Scheme 2, (a)] and cis [Scheme 2, (b)] orientation of the Bi–C bonds, respectively, with respect to the Bi2CO3 fragment. The Bi–C bonds of the other aryl groups attached to each metal atom are in trans positions with respect to the (CBi)2CO3 system.
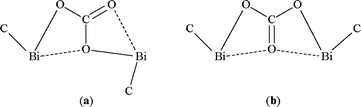 | ||
| Scheme 2 | ||
The structures of the halides 5 and 6 (Fig. 4) are very similar to that established for the corresponding chloride [2-(Me2NCH2)C6H4]2BiCl.5 The bismuth–halogen bond lengths, i.e. Bi(1)–Br(1) 2.8452(7) Å in 5 and Bi(1)–I(1) 3.0723(6) Å in 6, are longer than those observed in Mes2BiBr [Bi–Br 2.696(2) Å]37 or in [Ph2BiI(4-Mepy)] [Bi–I 3.0229(8) Å].38 This elongation is apparently a consequence of the coordination of the amino grouptrans to the halogen atom.
As a result of the intramolecular coordination of the nitrogen atoms to the bismuth two five-membered BiC3N rings are formed. These rings are folded along the Bi(1)⋯Cmethylene axis, with the nitrogen atom lying out of the best plane defined by the residual BiC3 system. This folding induces planar chirality (with the aromatic ring and the nitrogen atom as chiral plane and pilot atom, respectively)39 as described for other related compounds.8,33,34,40–44 Indeed all compounds crystallize as racemates, i.e. 1 : 1 mixtures of (SN1,SN2) and (RN1,RN2) for 1·2H2O, (SN1,RN2) and (RN1,SN2) isomers for 2 and 4–6, (SN1,RN2,SN3,SN4) and (RN1,SN2,RN3,RN4) for 3 and (SN1,RN2,SN3,RN4) and (RN1,SN2,RN3,SN4) for 3·C6H6 (with respect to the two chelate rings at a metal centre).
The crystal of the oxide 2, the carbonate 3 and the sulfide 4 is composed of discrete molecules with no unusual intermolecular contacts. A ribbon-like polymer of (RN1,RN2) and (SN1,SN2) isomers is formed along a axis in the crystal of the hydroxide 1·2H2O based on intermolecular oxygen-hydrogen contacts (see ESI‡). In contrast to the solvent-free carbonate 3, in the crystal of 3·C6H6 pairs of (SN1,RN2,SN3,RN4) and (RN1,SN2,RN3,SN4) isomers are associated into dimer units through weak intermolecular O(3)⋯H(25Ba) (2.43 Å) interactions. Additional relatively weak η6-arene⋯Bi(2) interactions [Bi(2)⋯Phcenter 3.615(1) Å; Bi(2)⋯C distances in the range 3.728–3.993 Å; cf. sums of the corresponding van der Waals radii, ∑rvdW(Bi,C) 4.25 Å35] support this dimer association (Fig. 5). The vector of this interaction is placed trans to a Bi–C bond [C(28)–Bi(2)⋯Phcenter 168.8°]. Similar arene⋯Bi interactions were observed in various organobismuth compounds.2 Dimer association were observed in the crystal of the bromide 5 [Br(1)⋯H(16Aa)methylene 3.10 Å; cf. ∑rvdW(Br,H) 3.15 Å35], but in this case an intermolecular η2-arene⋯Bi interaction is established [η2-Ph⋯Bi(1) 3.705(3) Å; Bi(1)⋯C(11) 3.785(6), Bi(1)⋯C(12) 3.751(7) Å; the rest of the Bi(1)⋯C distances are in the range 4.144–4.548 Å]. Similar dimer units can also be distinguish for the iodide [I(1)⋯H(7Aa)methylene 3.22(1) Å; cf. ∑rvdW(I,H) 3.35 Å;35η2-Ph⋯Bi(1) 3.718(1) Å; Bi(1)⋯C(5a) 3.770(5), Bi(1)⋯C(6a) 3.794(5) Å; the rest of the Bi(1)⋯C distances are in the range 4.127–4.513 Å.] However, in the crystal of 6 these dimers are further associated through iodine-hydrogen contacts [I(1)⋯H(3a′)aryl 3.318(1) Å] into ribbon-like polymers growing along the a axis (Fig. 6). Each polymer is connected to other four related polymers [I(1)⋯H(14b)aryl 3.261(6) Å] thus resulting in a 3D supramolecular architecture (for details, see ESI‡).
![View of dimer association based on intermolecular O⋯H (only hydrogens involved in intermolecular interactions are shown) and η6-arene⋯Bi contacts in the crystal of 3·C6H6 [symmetry equivalent atoms (1 − x, 2 − y, 1 − z) are given by ‘‘a’’].](/image/article/2008/DT/b717127g/b717127g-f5.gif) | ||
| Fig. 5 View of dimer association based on intermolecular O⋯H (only hydrogens involved in intermolecular interactions are shown) and η6-arene⋯Bi contacts in the crystal of 3·C6H6 [symmetry equivalent atoms (1 − x, 2 − y, 1 − z) are given by ‘‘a’’]. | ||
![View of ribbon-like polymer based on intermolecular I⋯H (only hydrogens involved in intermolecular interactions are shown) and η2-arene⋯Bi contacts in the crystal of 6 [symmetry equivalent atoms (1 − x, 1 − y, 1 − z), ( − x, 1 − y, 1 − z) and (1 + x, y, z) are given by ‘‘a’’, “a prime” and “a double prime”, respectively].](/image/article/2008/DT/b717127g/b717127g-f6.gif) | ||
| Fig. 6 View of ribbon-like polymer based on intermolecular I⋯H (only hydrogens involved in intermolecular interactions are shown) and η2-arene⋯Bi contacts in the crystal of 6 [symmetry equivalent atoms (1 − x, 1 − y, 1 − z), ( − x, 1 − y, 1 − z) and (1 + x, y, z) are given by ‘‘a’’, “a prime” and “a double prime”, respectively]. | ||
Monoorganobismuth compounds
The reaction of [2-(Me2NCH2)C6H4]BiCl2 (7)5 with Na2S in liquid ammonia leads to an ill-defined brown solid material, probably containing the heterocycles (RBiS)n as major components. After complexation with [W(CO)5(THF)] in THF the cis isomer of the complex cyclo-(RBiS)2[W(CO)5]2 (8) [R = 2-(Me2NCH2)C6H4] is obtained (Scheme 3). Compound 8 is a red solid which is soluble in organic solvents. | ||
| Scheme 3 | ||
The structures of 7 and 8·0.5C6H6 were established by X-ray diffraction on single crystals obtained at room temperature from water and benzene solutions, respectively. The structure of 7 and the (RN1,RN2)-8 isomer is depicted in Fig. 7 and 8. Relevant interatomic distances and angles are listed in Table 3.
| 7 a | 8·0.5C6H6 | ||||
|---|---|---|---|---|---|
| a Symmetry equivalent atoms (1 − x, −y, 1 − z) are given by ‘‘a’’. | |||||
| Bi(1)–C(1) | 2.225(7) | Bi(1)–C(1) | 2.263(13) | Bi(2)–C(10) | 2.253(11) |
| Bi(1)–Cl(1) | 2.5947(16) | Bi(1)–S(1) | 2.545(3) | Bi(2)–S(1) | 2.731(3) |
| Bi(1)–Cl(2) | 2.8992(16) | Bi(1)–S(2) | 2.745(3) | Bi(2)–S(2) | 2.556(3) |
| Bi(1)–Cl(2a) | 2.8255(16) | Bi(1)–N(1) | 2.568(10) | Bi(2)–N(2) | 2.556(9) |
| Bi(1)–N(1) | 2.458(6) | ||||
| S(1)–W(1) | 2.587(3) | S(2)–W(2) | 2.581(3) | ||
| Cl(1)–Bi(1)–Cl(2) | 174.61(5) | ||||
| N(1)–Bi(1)–Cl(2a) | 158.00(13) | N(1)–Bi(1)–S(2) | 162.8(3) | N(2)–Bi(2)–S(1) | 163.0(3) |
| C(1)–Bi(1)–N(1) | 74.9(2) | C(1)–Bi(1)–S(1) | 95.6(3) | C(10)–Bi(2)–S(2) | 90.6(3) |
| C(1)–Bi(1)–Cl(1) | 88.93(15) | N(1)–Bi(1)–C(1) | 73.4(4) | N(2)–Bi(2)–C(10) | 72.6(4) |
| C(1)–Bi(1)–Cl(2) | 85.90(15) | N(1)–Bi(1)–S(1) | 87.9(3) | N(2)–Bi(2)–S(2) | 86.5(3) |
| C(1)–Bi(1)–Cl(2a) | 86.90(16) | S(2)–Bi(1)–C(1) | 90.0(3) | S(1)–Bi(2)–C(10) | 91.1(3) |
| S(2)–Bi(1)–S(1) | 89.12(9) | S(1)–Bi(2)–S(2) | 89.22(9) | ||
| Cl(1)–Bi(1)–N(1) | 96.13(13) | ||||
| N(1)–Bi(1)–Cl(2) | 84.00(12) | Bi(1)–S(1)–Bi(2) | 91.01(8) | Bi(1)–S(2)–Bi(2) | 90.45(9) |
| Cl(2)–Bi(1)–Cl(2a) | 82.43(5) | ||||
| Cl(2a)–Bi(1)–Cl(1) | 95.78(5) | Bi(1)–S(1)–W(1) | 113.74(11) | Bi(2)–S(1)–W(1) | 102.52(10) |
| Bi(1)–S(2)–W(2) | 106.70(11) | Bi(2)–S(2)–W(2) | 122.17(12) | ||
![ORTEP representation at 50% probability and atom numbering scheme for (R,S)-7 isomer [symmetry equivalent atoms (1 − x, −y, 1 − z) are given by ‘‘a’’].](/image/article/2008/DT/b717127g/b717127g-f7.gif) | ||
| Fig. 7 ORTEP representation at 50% probability and atom numbering scheme for (R,S)-7 isomer [symmetry equivalent atoms (1 − x, −y, 1 − z) are given by ‘‘a’’]. | ||
 | ||
| Fig. 8 ORTEP representation at 20% probability and atom numbering scheme for (RN1,RN2)-8 isomer. Hydrogen atoms and the benzene molecule are omitted. | ||
The crystal of the dichloride 7 consists of centrosymetric dimers involving Cl(2) atoms [Bi(1)–Cl(2) 2.8992(16) Å, Bi(1)–Cl(2a) 2.8255(16) Å] (Fig. 7). The nitrogen of the pendant arm is strong coordinated to the metal [N(1)–Bi(1) 2.458(6) Å] in trans to the bridging halogen atom [N(1)–Bi(1)–Cl(2a) 158.0(1)°]. As expected, the Bi–Cl bonds in the planar Bi2Cl2 fragment are considerably longer than the Bi(1)–Cl(1) bond [2.5947(16) Å]. The geometry at bismuth is described as distorted square-based pyramidal with the aryl group in the apical position and the three chlorides and the nitrogen atom of the amine arm in the basal plane. As described previously for the related diiodide,5 in the crystal of 7 the (R,S) dimers are further associated through weak Bi(1)⋯Cl(1a′) interactions [3.690(2) Å [cf.∑rvdW(Bi,Cl) 4.20 Å35] into a ribbon-like polymer along the a axis (see ESI‡). Similar formation of doubly bridged polymeric chains involving both halogen atoms of a molecular unit were also found for the related PhBiBr2,45 or MeBiCl2.46 If these interactions are considered, the overall Bi coordination geometry in 7 is octahedral.
In the crystal of 7 there are several chloride–hydrogen contacts shorter than the sum of the corresponding van der Waals radii [cf. ∑rvdW(Cl,H) 3.0 Å35]. Thus the ribbon-like polymers are associated into layers through Cl(1a)⋯H(8Cba)methyl contacts [2.840(2) Å] and the parallel layers are connected through further Cl(2)⋯H(4i)phenyl contacts [2.840(2) Å] into a 3D architecture (for details, see ESI‡).
The complex 8·0.5C6H6 crystallizes as a racemate, i.e. 1 : 1 mixture of (SN1,SN2) and (RN1,RN2) isomers (with respect to the two chelate rings corresponding to the two metal centres). The molecule of 8 contains an almost planar Bi2S2 ring [S(1)Bi(1)S(2)/S(1)Bi(2)S(2) dihedral angle of 4.6°; transannular non-bonding distances: Bi(1)⋯Bi(2) 3.766(1) Å; S(1)⋯S(2) 3.715(4) Å] with the aryl substituents on the bismuth atoms in cis configuration with respect to the ring and alternating short and long bismuth–sulfur bonds. Both pendant amino groups are coordinated to bismuth in trans to a Bi-S bond [N(1)–Bi(1)–S(2) 162.8(3)°; N(2)–Bi(2)–S(1) 163.0(3)°]. The strong coordinated nitrogen atoms [Bi(1)–N(1) 2.568(10) Å; Bi(2)–N(2) 2.556(9) Å] are responsible for the asymmetry observed in the length of the bismuth–sulfur bonds within the Bi2S2 ring. The short bismuth–sulfur bonds [Bi(1)–S(1) 2.545(3) Å; Bi(2)–S(2) 2.556(3) Å] are of same magnitude as observed in 3. As expected, the longer Bi–S bonds [Bi(1)–S(2) 2.745(3) Å; Bi(2)–S(1) 2.731(3) Å] are those trans to the nitrogen atoms strongly coordinated to metal centres. The endocyclic angles on the bismuth and sulfur atoms are both close to 90°. The coordination geometry at both bismuth atoms of a molecular unit is thus distorted ψ-trigonal-bipyramidal [(C,N)BiS2 core, hypervalent 10-Bi-4 species],16,36 with nitrogen and sulfur atoms in axial positions. The deviations of the bond angles at the metal centre from the ideal values are mainly due to the constraints imposed by the coordinated amine arm and the Bi2S2 ring (Table 3).
The [RBiS]2 heterocycle is coordinated through the sulfur atoms to W(CO)5 moieties which occupy trans positions relative to the organic ligands. The cis orientation of the metal carbonyl moieties with respect to the Bi2S2 ring is probably a consequence of the steric strain in the molecule, caused by the bulky substituents. The structure of 8 is closely related to the cyclo-[RSbS]2[W(CO)5] where the organic groups are also in cis positions relative to each other and the metal carbonyl unit is coordinated to a sulfur atom.33
The 1H NMR spectrum of 8 in C6D6 exhibits only one set of resonances for the 2-Me2NCH2C6H4groups, thus suggesting that the solid-state structure is preserved in solution too.
In the crystal of 8·0.5C6H6 alternating (SN1,SN2) and (RN1,RN2) isomers are associated into a ribbon-like chain through intermolecular Bi⋯Ocarbonyl interactions [Bi(1)⋯O(10b) 3.126(12) Å; Bi(2)⋯O(2a) 3.438(8) Å; cf. sums of the corresponding covalent, ∑cov(Bi,O) 2.18 Å, and van der Waals radii, ∑vdW(Bi,O) 3.80 Å35] (Fig. 9), which vectors lie approximately trans to a Bi–S bond [S(1)–Bi(1)⋯O(10b) 169.7(2)°; S(2)–Bi(2)⋯O(2a) 155.1(2)°]. If these intermolecular interactions are considered, the overall coordination can be described as distorted square-pyramidal [(C,N)BiS2O core], with the carbon atom in the apical position.
![View of the chain polymeric association in the crystal of 8·0.5C6H6. For clarity, only the five-membered BiC3N rings are shown [symmetry equivalent atoms (1 − x, −y, 1 − z) and (2 − x, 1 − y, 2 − z) are given by “a” and “b”, respectively].](/image/article/2008/DT/b717127g/b717127g-f9.gif) | ||
| Fig. 9 View of the chain polymeric association in the crystal of 8·0.5C6H6. For clarity, only the five-membered BiC3N rings are shown [symmetry equivalent atoms (1 − x, −y, 1 − z) and (2 − x, 1 − y, 2 − z) are given by “a” and “b”, respectively]. | ||
Since the 2-(Me2NCH2)C6H4 ligand was found to prevent the polymerization thus leading to soluble (RBiS)n species we decided to investigate the effect of the use of aryl ligands with two pendant arms able to provide internal coordination to bismuth. The reaction of [2,6-(Me2NCH2)2C6H3]BiCl27,8 with Na2S in CH3CN/water mixture affords the isolation of cyclo-[{2,6-(Me2NCH2)2C6H3}BiS]2 (9) as a yellow solid. Compound 9 is stable for indefinite time in open atmosphere in solid state, but slowly decomposes in solution of chloroform, methylene chloride, benzene or toluene, even under inert atmosphere, to form insoluble black bismuth sulfide and the corresponding triorganobismuth derivative. Decomposition is highly accelerated by heating. The 1H NMR spectrum at room temperature showed broad singlet signals for methyl and methylene protons which suggest a dynamic behaviour in solution. The low solubility and the decomposition of 9 in solution prevents further investigation at variable temperature. The highest ion in the EI-MS corresponds to the molecular fragment of cyclo-[R′BiS]2.
Yellow, single crystals of 9 were grown by diffusion using a chloroform-hexane system. The solid-state structure was established by single-crystal X-ray diffraction. The molecular structure of 9 is depicted in Fig. 10 and relevant interatomic distances and angles are listed in Table 4.
| a Symmetry equivalent atoms (1 − x, 1 − y, −z) are given by “a”. | |||
|---|---|---|---|
| Bi(1)–C(1) | 2.260(5) | ||
| Bi(1)–S(1) | 2.5770(16) | Bi(1)–S(1a) | 2.5784(17) |
| Bi(1)–N(1) | 2.834(6) | Bi(1)–N(2) | 2.851(4) |
| C(1)–Bi(1)–S(1) | 98.13(14) | C(1)–Bi(1)–S(1a) | 99.65(14) |
| S(1)–Bi(1)–S(1a) | 85.01(5) | ||
| N(1)–Bi(1)–C(1) | 67.3(2) | N(2)–Bi(1)–C(1) | 67.5(2) |
| N(1)–Bi(1)–S(1) | 76.9(1) | N(2)–Bi(1)–S(1a) | 76.4(8) |
| N(1)–Bi(1)–S(1a) | 155.5(1) | N(2)–Bi(1)–S(1) | 153.7(1) |
| N(1)–Bi(1)–N(2) | 114.1(1) | ||
| Bi(1)–S(1)–Bi(1a) | 94.99(5) | ||
![ORTEP representation at 40% probability and atom numbering scheme for (RN1,SN2,SN1a,RN2a)-9. Hydrogen atoms are omitted [symmetry equivalent atoms (1 − x, 1 − y, −z) are given by “a”].](/image/article/2008/DT/b717127g/b717127g-f10.gif) | ||
| Fig. 10 ORTEP representation at 40% probability and atom numbering scheme for (RN1,SN2,SN1a,RN2a)-9. Hydrogen atoms are omitted [symmetry equivalent atoms (1 − x, 1 − y, −z) are given by “a”]. | ||
The crystals of 9 consist of dinuclear units of the trans-(RN1,SN2,SN1a,RN2a) isomer (with respect to the two chelate rings at a metal centre) which has a crystallographically imposed inversion symmetry. The aryl substituents occupy trans positions relative to the central, planar four-membered Bi2S2 ring. The transannular Bi(1)⋯Bi(1a) non-bonding distance [3.800(1) Å] in similar to that found in complex 8 [Bi(1)⋯Bi(2) 3.766(1) Å]. However, some remarkable differences should be noted. The size of the endocyclic angles on the bismuth and sulfur atoms is different, i.e. S(1)–Bi(1)–S(1a) 85.01(5)° and Bi(1)–S(1)–Bi(1a) 94.99(5)°. Moreover, the bismuth–sulfur bonds are equivalent [Bi(1)–S(1) 2.5770(16) Å; Bi(1)–S(1a) 2.5784(17) Å]. This is the result of the (N,C,N)-coordination pattern of the organic ligand, with both nitrogen atoms coordinated in a cis arrangement to the bismuth, each almost trans to a sulfur atom [N(1)–Bi(1)–S(1a) 155.5(1)°; N(2)–Bi(1)–S(1) 153.7(1)°]. The two bismuth–nitrogen interactions in 9 are of the same strengths [Bi(1)–N(1) 2.834(6) Å; Bi(1)–N(2) 2.851(4) Å], but weaker than in the related chloride species [2.561(3), 2.570(4) Å] where the nitrogen atoms of the organic ligand are coordinated to bismuth in an almost trans arrangement.8 The resulting geometry at the bismuth centre is distorted square pyramidal [(N,C,N)BiS2 core, hypervalent 12-Bi-5 species],16,36 with the carbon atom in the apical position.
In the crystal of 9 intermolecular Bi⋯Bi contacts [3.917(1) Å; cf. ∑vdW(Bi,Bi) 4.80 Å]34 are observed between dinuclear units, which leads to formation of a chain like polymer (see ESI‡).
Conclusions
New organobismuth(III) compounds containing pendant arm ligands 2-(Me2NCH2)C6H4 and 2,6-(Me2NCH2)2C6H3 were prepared and their hypervalent nature was investigated both in solution and in the solid state. The hydroxide [2-(Me2NCH2)C6H4]2BiOH (1) can be converted easily into the oxide [{2-(Me2NCH2)C6H4}2Bi]2O (2) or the carbonate [{2-(Me2NCH2)C6H4}2Bi]2CO3 (3) and the processes are reversible. In the solid state for all diorganobismuth(III) derivatives reported in this study the metal centre is coordinated more strongly by the nitrogen atom of one amine pendant arm trans to the chalcogen or halogen atom, while the nitrogen atom of the other amine group exhibits a weak intramolecular interaction trans to a carbon atom. If both these intramolecular N→Bi interactions per metal atom are considered the overall coordination becomes distorted square-pyramidal [(C,N)2BiX cores] and the compounds can be described as hypervalent 12-Bi-5 species. The cis isomer of the heterocyclic monoorganobismuth(III) sulfide was isolated as a complex, cyclo-[{2-(Me2NCH2)C6H4}BiS]2[W(CO)5]2 (8), which contains metal carbonyl fragments coordinated to the sulfur atoms. Both bismuth atoms of a molecular unit are in a distorted ψ-trigonal-bipyramidal environment [(C,N)BiS2 core, hypervalent 10-Bi-4 species]. The heterocyclic sulfidecyclo-[{2,6-(Me2NCH2)2C6H3}BiS]2 (9) was obtained as stable solid which contains a distorted square pyramidal (N,C,N)BiS2 core (hypervalent 12-Bi-5 species). The crystal of the oxide 2, the carbonate 3 and the sulfide 4 is composed of discrete molecules, while for the other compounds different degree of association was found in solid state.Experimental
General procedures
Room-temperature NMR spectra were recorded in dried solvents on a BRUKER DPX 200 instrument operating at 200 MHz for 1H and 50 MHz for 13C, respectively. 1H and 13C chemical shifts are reported in δ units (ppm) relative to the residual peak of solvent (ref. CHCl3: 1H 7.26, 13C 77.0 ppm; C6H6: 1H 7.16, 13C 128.06 ppm). Mass spectra were recorded with a FINNIGAN MAT 8200 spectrometer and IR spectra with an FT-IR SPEKTRUM 1000 (Perkin Elmer). Elemental analyses were performed by Mikroanalytisches Laboratorium Beller in Göttingen. All manipulations were carried out under an inert atmosphere of argon using Schlenk techniques. Solvents were dried and freshly distilled under argon prior to use. [2-(Me2NCH2)C6H4]2BiCl,5 [2-(Me2NCH2)C6H4]BiCl2,5 [2-(Me2NCH2)C6H4]4Bi2,12 and [2,6-(Me2NCH2)2C6H3]BiCl2,8 were prepared according to published methods.Method A. A solution of [2-(Me2NCH2)C6H4]2BiCl (2.0 g, 3.9 mmol) in diethylether was reacted with KOH (0.11 g, 1.95 mmol) in 10 mL water as described for the synthesis of 1. When the dried and filtered diethyl ether phase was exposed to the atmosphere for one week the solvent evaporated and crystals of 3 (1.64 g, 83%) were obtained. The compound was recrystallized from benzene as 3·C6H6, mp 200–203 °C.
Method B. A solution of Na2CO3 (0.2 g, 1.9 mmol, 100% excess) in 10 mL distilled water was added to a solution of [2-(Me2NCH2)C6H4]2BiCl (1.0 g, 1.9 mmol) in 25 mL CH2Cl2. The mixture was stirred at room temperature for 24 h and then filtered. The water phase was extracted two times with CH2Cl2 (2 × 20 mL). The organic phase was dried over anhydrous Na2SO4, then filtered. Removal of the solvent in vacuum gave 3 (0.30 g, 59%), mp 157–160 °C. Anal. Calc. for C37H48Bi2N4O3 (1014.77): C, 43.79; H, 4.77. Found: C, 43.95; H, 4.63%. 1H NMR (C6D6): δ 1.91 (24 H, s, CH3), AB spin system with A at 3.01 and B at 3.76 ppm (8 H, CH2, 2JHH 13.0 Hz), 7.21 (12 H, m, H-3,4,5, C6H4), 8.77 (4 H, d, H-6, C6H4, 3JHH 7.0 Hz). 1H NMR (CDCl3): δ 2.23 (24 H, s, CH3), AB spin system with A at 3.40 and B at 3.80 ppm (8 H, CH2, 2JHH 13.0 Hz), 7.33 (12 H, m, H-3,4,5, C6H4), 8.29 (4 H, d, H-6, C6H4, 3JHH 7.0 Hz). 13C NMR (C6D6): δ 45.42 (s, CH3), 67.95 (s, CH2), 129.29 (s, C-4), 129.58, 130.69 (s, C-3,5), 139.91 (s, C-6), 146.42 (s, C-2), 188.85 (s, C-1). MS (EI, 70 eV, 200 °C), m/z (%): 969 (1) [M+-CO2], 836 (3.37) [R3Bi2O+], 493 (1) [R2BiO+], 477 (100) [R2Bi+], 387 (10) [RBiCO2+], 342 (12) [RBi+], 134 (67) [R+] (R = Me2NCH2C6H4). IR: (Nujol) ν(CO) 1283(m), 1520(s) cm−1.
Method A. Elemental sulfur (0.05 g, 1.57 mmol) was added to a solution of [2-(Me2NCH2)C6H4]4Bi2 (1.51 g, 1.57 mmol) in 30 mL petroleum ether at −30 °C and the mixture was stirred for 2.5 h. The colour changed from red to yellow and a yellow powder deposited. The solvent was removed with a syringe and the remaining solid was washed with petroleum ether, then dried under vacuum to give 4 (0.90 g, 60%).
Method B. A solution of Na2S·9H2O (0.24 g, 1.0 mmol) in 10 mL water was added to a suspension of [2-(Me2NCH2)C6H4]2BiCl (1.00 g, 1.95 mmol) in 50 mL toluene. The mixture was stirred for 2 h, the organic layer was separated, dried over anhydrous Na2SO4 and filtered through a glass frit. Removal of the solvent in vacuum gave 4 (0.66 g, 69%), mp 150–152 °C. Anal. Calc. for C36H48Bi2N4S (986.82): C, 43.82; H, 4.90. Found: C, 43.75; H, 4.81%. 1H NMR (C6D6): δ 1.95 (24 H, s, CH3), AB spin system with A at 3.20 and B at 3.49 ppm (8 H, CH2, 2JHH 13.0 Hz), 7.18 (12 H, s, br, H-3,4,5, C6H4), 8.99 (4 H, d, H-6, C6H4, 3JHH 5.8 Hz). 13C NMR (C6D6): δ 45.34 (s, CH3), 67.82 (s, CH2), 127.15 (s, C-4), 129.21, 130.35 (s, C-3,5), 141.10 (s, C-6), 145.19 (s, C-2), 170.39 (s, C-1). MS (EI, 70 eV, 220 °C), m/z (%): 986 (6) [M+], 852 (96) [R3Bi2S+], 477 (100) [R2Bi+], 134 (33) [R+] (R = Me2NCH2C6H4).
Crystal structures
The details of the crystal structure determination and refinement for compounds 1·2H2O, 2–7, 8·0.5C6H6 and 9 are given in Tables 5 and 6. Data were collected on Stoe IPDS (1·2H2O) Siemens P4 (2, 3·C6H6, 4, 6, 7) and Bruker SMART APEX (3, 5, 8·0.5C6H6, 9) diffractometers, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). For this purpose the crystals were attached with Kel-F oil (1·2H2O, 2, 3·C6H6, 4, 6, 7) or epoxy glue (3, 5, 8·0.5C6H6, 9) to a glass fiber. For 1·2H2O, 2–4, 6 and 7 the crystals were cooled under a nitrogen stream at low temperature, while data for 5, 8·0.5C6H6 and 9 were collected at room temperature. The structures were refined with anisotropic thermal parameters. The hydrogen atom attached to oxygen in 1·2H2O was located from the difference map. The other hydrogen atoms were refined with a riding model and a mutual isotropic thermal parameter. For structure solving and refinement the software package SHELX-97 was used.47 The drawings were created with the Diamond program.48| 1·2H2O | 2 | 3 | 3·C6H6 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|---|
| Empirical formula | C18H29BiN2O3 | C36H48Bi2N4O | C37H48Bi2N4O3 | C43H54Bi2N4O3 | C36H48Bi2N4S | C18H24N2BiBr | C18H24N2BiI |
| M | 530.41 | 970.74 | 1014.75 | 1092.86 | 986.80 | 557.28 | 604.27 |
| T/K | 173(2) | 173(2) | 297(2) | 198(2) | 173(2) | 297(2) | 173(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | C2/c | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | P21/n | P21/n |
| a/Å | 9.149(2) | 12.812(3) | 17.063(7) | 13.902(2) | 27.939(3) | 9.5102(9) | 9.548(2) |
| b/Å | 26.827(5) | 14.393(9) | 9.502(4) | 14.011(2) | 7.366(1) | 16.2013(16) | 16.277(4) |
| c/Å | 8.393(2) | 19.969(4) | 24.891(10) | 14.107(4) | 18.947(2) | 12.4073(12) | 12.616(2) |
| α/° | 90.00 | 90.00 | 90.00 | 99.17(1) | 90.00 | 90.00 | 90.00 |
| β/° | 108.61(1) | 98.02 | 106.916(7) | 92.64(2) | 112.43(1) | 90.972(2) | 91.555(16) |
| γ/° | 90.00 | 90.00 | 90.00 | 109.55(1) | 90.00 | 90.00 | 90.00 |
| V/Å3 | 1952.3(7) | 3646(3) | 3861(3) | 2541.6(9) | 3604.3(7) | 1911.4(3) | 1959.9(7) |
| Z | 4 | 4 | 4 | 2 | 4 | 4 | 4 |
| No. reflections collected | 5612 | 4022 | 27069 | 13112 | 9509 | 20240 | 10604 |
| No. independent reflections | 4428 | 3185 | 6802 | 11503 | 4089 | 3894 | 4516 |
| R int | 0.037 | 0.047 | 0.048 | 0.037 | 0.048 | 0.053 | 0.053 |
| Absorption correction | DIFABS49 | DIFABS49 | Multiscan50 | DIFABS49 | DIFABS49 | Multiscan50 | DIFABS49 |
| µ(Mo-Kα)/mm−1 | 9.048 | 9.671 | 9.140 | 6.949 | 9.839 | 11.313 | 10.570 |
| R1 [I > 2σ(I)] | 0.0470 | 0.0596 | 0.0502 | 0.0495 | 0.0357 | 0.0387 | 0.0284 |
| wR2 | 0.1216 | 0.1180 | 0.0970 | 0.1398 | 0.0727 | 0.0763 | 0.0603 |
| GOF on F2 | 1.027 | 1.015 | 1.229 | 1.033 | 0.989 | 1.155 | 1.048 |
| 7 | 8·0.5C6H6 | 9 | |
|---|---|---|---|
| Empirical formula | C18H24N2Bi2Cl4 | C31H24Bi2N2O10S2W2 | C24H38Bi2N4S2 |
| M | 828.16 | 1434.30 | 864.66 |
| T/K | 173(2) | 297(2) | 297(2) |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c |
P |
P21/n |
| a/Å | 9.0380(18) | 11.8825(10) | 6.435(2) |
| b/Å | 8.1540(16) | 12.0273(11) | 11.581(4) |
| c/Å | 16.107(3) | 16.1174(14) | 19.000(6) |
| α/° | 90.00 | 99.254(2) | 90.00 |
| β/° | 100.95(3) | 96.479(2) | 99.094(5) |
| γ/° | 90.00 | 115.567(2) | 90.00 |
| V/Å3 | 1165.4(4) | 2007.4(3) | 1398.2(8) |
| Z | 2 | 2 | 2 |
| No. reflections collected | 13075 | 21482 | 10948 |
| No. independent reflections | 2249 | 8136 | 2847 |
| R int | 0.053 | 0.054 | 0.040 |
| Absorption correction | DIFABS49 | Multiscan50 | Multiscan50 |
| µ(Mo-Kα)/mm−1 | 15.543 | 14.606 | 12.737 |
| R1 [I > 2σ(I)] | 0.0217 | 0.0515 | 0.0282 |
| wR2 | 0.0670 | 0.1187 | 0.0612 |
| GOF on F2 | 1.195 | 1.090 | 1.089 |
CCDC reference numbers 658669 (1·2H2O), 658670 (2), 653119 (3), 658671 (3·C6H6), 658672 (4), 652894 (5), 658673 (6), 658674 (7), 652893 (8·0.5C6H6) and 652892 (9).
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b717127g
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft, the Universität Bremen and the Ministry of Education and Research of Romania (CNCSIS, Research Project No. 709/2007; Excellency Research Program, Research Projects No. 19/2006 and CEx-05-D11-16/2005) for financial support. We also thank the National Center for X-Ray Diffraction (“Babes-Bolyai” University, Cluj-Napoca, Romania) for the support in the solid state structure determinations.References
- Organobismuth Chemistry, ed. H. Suzuki and Y. Matano, Elsevier Science B.V., Amsterdam, 2001 Search PubMed.
- C. Silvestru, H. J. Breunig and H. Althaus, Chem. Rev., 1999, 99, 3277 CrossRef CAS.
- H. Suzuki, T. Murafuji, Y. Matano and N. Azuma, J. Chem. Soc., Perkin Trans. 1, 1993, 2969 RSC.
- T. Murafuji, N. Azuma and H. Suzuki, Organometallics, 1995, 14, 1542 CrossRef CAS.
- C. J. Carmalt, A. H. Cowley, R. D. Culp, R. A. Jones, S. Kamepalli and N. C. Norman, Inorg. Chem., 1997, 36, 2770 CrossRef CAS.
- H. J. Breunig, I. Gheşner, M. E. Gheşner and E. Lork, Inorg. Chem., 2003, 42, 1751 CrossRef CAS.
- D. A. Atwood, A. H. Cowley and J. Ruiz, Inorg. Chim. Acta, 1992, 198–200, 271 CrossRef CAS.
- A. P. Soran, C. Silvestru, H. J. Breunig, G. Balázs and J. C. Green, Organometallics, 2007, 26, 1196 CrossRef CAS.
- C. J. Carmalt, D. Walsh, A. H. Cowley and N. C. Norman, Organometallics, 1997, 16, 3597 CrossRef CAS.
- Y. Yamamoto, X. Chen and K. Akiba, J. Am. Chem. Soc., 1992, 114, 7906 CrossRef CAS.
- Y. Yamamoto, X. Chen, S. Kojima, K. Ohdoi, M. Kitano, Y. Doi and K. Akiba, J. Am. Chem. Soc., 1995, 117, 3922 CrossRef CAS.
- L. Balázs, H. J. Breunig, E. Lork and C. Silvestru, Eur. J. Inorg. Chem., 2003, 1361 CrossRef CAS.
- L. Balázs, O. Stângă, H. J. Breunig and C. Silvestru, Dalton Trans., 2003, 2237 RSC.
- L. Balázs, H. J. Breunig, E. Lork, A. Soran and C. Silvestru, Inorg. Chem., 2006, 45, 2341 CrossRef CAS.
- E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Nema, M. E. Olmos, J. Pérez and C. Silvestru, Chem. Commun., 2007, 571 RSC.
- Chemistry of Hypervalent Compounds, ed. K. Akiba, Wiley-VCH, New York, 1999 Search PubMed.
- F. Dünhaupt, Liebigs Ann. Chem., 1854, 92, 371 CrossRef.
- F. Dünhaupt, J. Prakt. Chem., 1854, 61, 399 Search PubMed.
- A. Marquardt, Ber. Dtsch. Chem. Ges., 1887, 20, 1516 CrossRef.
- A. Marquardt, Ber. Dtsch. Chem. Ges., 1888, 21, 2038.
- M. Wieber, Gmelin Handbuch der Anorganischen Chemie, Vol. 47, Bismut-Organische Verbindungen, Springer Verlag, Berlin, 1977 Search PubMed.
- H. J. Breunig and D. Müller, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1986, 41, 1129.
- M. Wieber and I. Sauer, Z. Naturforsch., B: Chem. Sci., 1987, 42, 695 CAS.
- H. J. Breunig, K. H. Ebert, R. E. Schulz, M. Wieber and I. Sauer, Z. Naturforsch., B: Chem. Sci., 1995, 50, 735 CAS.
- X.-W. Li, J. Lorberth, K. H. Ebert, W. Massa and S. Wocadlo, J. Organomet. Chem., 1998, 560, 211 CrossRef CAS.
- H. J. Breunig, I. Gheşner and E. Lork, J. Organomet. Chem., 2002, 664, 130 CrossRef CAS.
- N. Tokitoh, Y. Arai, R. Okazaki and S. Nagase, Science, 1997, 277, 78 CrossRef CAS.
- N. Tokitoh, Y. Arai, R. Okazaki and S. Nagase, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 124, 371 CrossRef.
- N. Tokitoh, J. Organomet. Chem., 2000, 611, 217 CrossRef CAS.
- T. Sasamori, Y. Arai, N. Takeda, R. Okazaki, Y. Furukawa, M. Kimura, S. Nagase and N. Tokitoh, Bull. Chem. Soc. Jpn., 2002, 75, 661 CrossRef CAS.
- G. B. Deacon, W. R. Jackson and J. M. Pfeiffer, Aust. J. Chem., 1984, 37, 527 CAS.
- H. J. Breunig, L. Balázs, N. Philipp, A. Soran and C. Silvestru, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 853 CrossRef CAS.
- L. M. Opriş, A. Silvestru, C. Silvestru, H. J. Breunig and E. Lork, Dalton Trans., 2004, 3575 RSC.
- L. M. Opriş, A. Silvestru, C. Silvestru, H. J. Breunig and E. Lork, Dalton Trans., 2003, 4367 RSC.
- J. Emsley, Die Elemente, Walter de Gruyter, Berlin, 1994 Search PubMed.
- The N–X–L nomenclature system has been previously described. N valence shell electrons about a central atom X with L ligands: C. W. Perkins, J. C. Martin, A. J. Arduengo III, W. Lau, A. Alegria and K. Kochi, J. Am. Chem. Soc., 1980, 102, 7753 Search PubMed.
- K. H. Ebert, R. E. Schulz, H. J. Breunig, C. Silvestru and I. Haiduc, J. Organomet. Chem., 1994, 470, 93 CrossRef.
- S. C. James, N. C. Norman and A. G. Orpen, J. Chem. Soc., Dalton Trans., 1999, 2837 RSC.
- IUPAC Nomenclature of Organic Chemistry, Pergamon Press, Oxford, 1979 Search PubMed.
- O. Bumbu, C. Silvestru, M. C. Gimeno and A. Laguna, J. Organomet. Chem., 2004, 689, 1172 CrossRef CAS.
- R. A. Varga, C. Silvestru and C. Deleanu, Appl. Organomet. Chem., 2005, 19, 153 CrossRef CAS.
- M. Kulcsar, A. Silvestru, C. Silvestru, J. E. Drake, C. L. B. Macdonald, M. B. Hursthouse and M. E. Light, J. Organomet. Chem., 2005, 690, 3217 CrossRef CAS.
- R. A. Varga, A. Rotar, M. Schürmann, K. Jurkschat and C. Silvestru, Eur. J. Inorg. Chem., 2006, 1475 CrossRef CAS.
- M. Kulcsar, A. Beleaga, C. Silvestru, A. Nicolescu, C. Deleanu, C. Todasca and A. Silvestru, Dalton Trans., 2007, 2187 RSC.
- W. Clegg, M. R. J. Elsegood, R. J. Errington, G. A. Fisher and N. C. Norman, J. Mater. Chem., 1994, 4, 891 RSC.
- H. Althaus, H. J. Breunig and E. Lork, Organometallics, 2001, 20, 586 CrossRef CAS.
- G. M. Sheldrick, SHELX-97, Universität Göttingen, Germany, 1997 Search PubMed.
- DIAMOND—Visual Crystal Structure Information System, CRYSTAL IMPACT, Postfach 1251, D-53002 Bonn, Germany, 2001 Search PubMed.
- N. Walker and D. Stuart, Acta Crystallogr., Sect. A, 1983, 39, 158 CrossRef.
- G. M. Sheldrick, SADABS, Program for area detector adsorption correction, Institute for Inorganic Chemistry, University of Göttingen, Germany, 1996 Search PubMed.
Footnotes |
| † CCDC reference numbers 658669 (1·2H2O), 658670 (2), 653119 (3), 658671 (3·C6H6), 658672 (4), 652894 (5), 658673 (6), 658674 (7), 652893 (8·0.5C6H6) and 652892 (9). For crystallographic data in CIF or other electronic format see DOI: 10.1039/b717127g |
| ‡ Electronic supplementary information (ESI) available: X-Ray crystallographic data in CIF format for 1·2H2O, 2, 3, 3·C6H6 and 4–9; Figures representing the molecular structure of compounds 4 and 5; supramolecular architectures in the crystals of compounds 1·2H2O, 5, 6, 7 and 9. See DOI: 10.1039/b717127g |
| This journal is © The Royal Society of Chemistry 2008 |