Structure of solvated mercury(II) halides in liquid ammonia, triethyl phosphite and tri-n-butylphosphine solution†
Kersti B.
Nilsson
a,
Mikhail
Maliarik
b,
Ingmar
Persson
*a and
Magnus
Sandström
c
aDepartment of Chemistry, Swedish University of Agricultural Sciences, P.O.Box 7015, SE-750 07, Uppsala, Sweden
bIFM-Department of Chemistry, Linköping University, SE-581 83, Linköping, Sweden
cDepartment of Physical, Inorganic and Structural Chemistry, Stockholm University, SE-106 91, Stockholm, Sweden
First published on 4th March 2008
Abstract
Liquid ammonia, trialkyl phosphites, and especially trialkylphosphines, are very powerful electron-pair donor solvents with soft bonding character. The solvent molecules act as strongly coordinating ligands towards mercury(II), interacting strongly enough to displace halide ligands. In liquid ammonia mercury(II) chloride solutions separate into two liquid phases; the upper contains tetraamminemercury(II) complexes, [Hg(NH3)4]2+, and chloride ions in low concentration, while the lower is a dense highly concentrated solution of [Hg(NH3)4]2+ entities, ca. 1.4 mol dm−3, probably ion-paired by hydrogen bonds to the chloride ions. Mercury(II) bromide also dissociates to ionic complexes in liquid ammonia and forms a homogeneous solution for which 199Hg NMR indicates weak bromide association with mercury(II). When dissolving mercury(II) iodide in liquid ammonia and triethyl phosphite solvated molecular complexes form in the solutions. The Raman ν(I–Hg–I) symmetric stretching frequency is 132 cm−1 for the pseudo-tetrahedral [HgI2(NH3)2] complex formed in liquid ammonia, corresponding to DS = 56 on the donor strength scale. For the Hg(ClO4)2/NH4I system in liquid ammonia a 199Hg NMR study showed [HgI4]2− to be the dominating mercury(II) complex for mole ratios n(I−) : n(Hg2+) ≥ 6. A large angle X-ray scattering (LAXS) study of mercury(II) iodide in triethyl phosphite solution showed a [HgI2(P(OC4H9)3)2] complex with the Hg–I and Hg–P bond distances 2.750(3) and 2.457(4) Å, respectively, in near tetrahedral configuration. Trialkylphosphines generally form very strong bonds to mercury(II), dissociating all mercury(II) halides. Mercury(II) chloride and bromide form solid solvated mercury(II) halide salts when treated with tri-n-butylphosphine, because of the low permittivity of the solvent. A LAXS study of a melt of mercury(II) iodide in tri-n-butylphosphine at 330 K resulted in the Hg–I and Hg–P distances 2.851(3) and 2.468(4) Å, respectively. The absence of a distinct I–I distance indicates flexible coordination geometry with weak and non-directional mercury(II) iodide association within the tri-n-butylphosphine solvated complex.
Introduction
This investigation concludes a series where the solvation of mercury(II) halides, HgX2 (X = Cl, Br and I), has been studied in a large number of solvents with widely different electron-pair donor properties, employing structural, thermodynamic and vibrational spectroscopic methods.1–7 The presently studied solvents, liquid ammonia, trialkylphosphines and trialkyl phosphites, are among the strongest electron-pair donor solvents known. The solvent molecules act as ligands in their own right, forming very strong covalent bonds to soft electron acceptors such as mercury(II), and creating solvated complexes with profoundly altered chemical properties.1,2 The relative electron-pair donor abilities, as measured by the donor strength parameter DS, have been determined to be 56 for liquid ammonia (see below), 56 for tri-n-butyl phosphite, and 76 for both triethyl and tri-n-butylphosphine.2Previous Raman vibrational spectroscopic studies of liquid ammonia solutions of mercury(II) chloride and bromide reported complete dissociation to solvated Hg2+ and halide ions, while mercury(II) iodide was proposed to form [HgI(NH3)3]+ complexes and iodide ions.8 There are few structural investigations of mercury(II) halide complexes associated with ammonia in the solid state. Diamminemercury(II) chloride and bromide, Hg(NH3)2X2, described more than a century ago as the “fusible white precipitates”,9 are formed by dissolving the mercury(II) halide in aqueous ammonia in the presence of ammonium halide. No crystal structure has so far been reported for the bromide compound, while the structure of the chloride, [Hg(NH3)2Cl2]∞, shows a pseudo-octahedral configuration around the mercury(II) ion with two short collinear Hg–N bonds, 2.030 Å, and four long double chloro bridges, 2.871 Å.9 The dichlorobis(pyridine)mercury(II) complex, [Hg(NC5H5)2Cl2]∞, displays a similar coordination figure with two relatively long Hg–N distances, 2.266 Å, and four Hg–Cl bond distances at 2.760 Å.10 In the linear diamminemercury(II) complex of [Hg(NH3)2][HgCl3]2 the Hg–N bond distance is 2.074 Å.11 Discrete mercury(II) complexes with pseudo-tetrahedral configurations are formed in [HgI2(NH3)2]S4N4 with the bond distances Hg–N 2.30(2) Å and Hg–I 2.697(1) Å; the I–Hg–I bond angle becomes as large as 120.8° between the strongly bonded iodo ligands.12
The dissociation of mercury(II) halides into solvated mercury(II) and halide ions with trialkylphosphines and trialkyl phosphites by replacing the halide ligands with solvent molecules, causes precipitation of solid [Hg(PR3)n]X2 and [Hg(P(OR)3)n]X2 salts because of the low permittivity of those solvents, and only neutral complexes attain any appreciable solubility. Bis(trialkylphosphine)mercury(II) salts, [Hg(P(CH3)3)2]X2, X = Cl, Br and I, and [Hg(P(C2H5)3)2]Cl2, have been reported to dissociate into [Hg(PR3)2]2+ complexes and halide ions in aqueous solution,13,14 while neutral [Hg(PR3)2X2] complexes form in dichloromethane solution.15,16 Raman and infrared absorption measurements indicated close to tetrahedral coordination geometry for the neutral [HgI2(P(OC2H5)3)2] complex in triethyl phosphite solution, while the stronger Hg–P bonding in the [HgI2(P(C4H9)3)2] complex in tri-n-butylphosphine solution is expected to reduce the I–Hg–I angle.1 For the solid bis(triphenylphosphine)mercury(II) halides, [HgX2(P(C6H5)3)2], the Hg–P bond distances increase and the P–Hg–P bond angles decrease in the order X = Cl, Br and I,17–21 towards nearly regular tetrahedral coordination in the iodide complex.21 Triarylphosphines are weaker electron-pair donors due to the electron withdrawing effect of the aryl group.
199Hg NMR studies on solvated mercury(II) halides in a large number of solvents with different coordinating properties show resonances in a wide chemical shift range, from −1298 ppm of HgI2 in nitromethane to +1112 ppm of HgBr2 in ethylenediamine, relative to 2 mol dm−3mercury(II) perchlorate in 60% aqueous perchloric acid (0 ppm).22 The NMR shifts correlate reasonably well to the electron-pair donor ability of the solvents according to the DS scale, which is suitable for classifying strong electron-pair donor solvents such as those in this study.2 The 199Hg NMR spin–lattice (T1) relaxation times have previously been measured for tetrahedrally and octahedrally coordinated mercury(II) complexes. Both the chemical shift and the relaxation time have been found to depend on the coordination number,23,24 and can aid in characterising the solvation of the mercury(II) halides in the investigated solvents.
Kraus discovered already in 1908 that when adding an alkali metal to liquid ammonia that contains metal ions in low concentration, a two-phase liquid system may form at low temperature and for certain ammonia-to-metal mole ratios (except for caesium).25 More recently, rubidium was examined.26 In such systems a dense bright blue phase appears, coloured by free ammonia-solvated electrons. The lighter, bronze-coloured phase, with delocalised electrons in cavities between ammonia solvated ions, [M(NH3)n]+, obtains the higher metal concentration.27,28 Liquid ammonia solutions can be prepared also of less electropositive metals by cathodic reduction of the metal salt solutions, e.g. of aluminium/aluminium(III) iodide and beryllium/beryllium(II) chloride.28 To our knowledge, liquid/liquid phase separation has so far not been observed for solutions of metal salts in liquid ammonia.
The aims of this study are to determine the structures of the complexes formed when solvating the HgX2 entities (X = Cl, Br, I) of mercury(II) halides with liquid ammonia, triethyl phosphite and tri-n-butylphosphine, and to correlate the structural changes with the coordinating ability of the solvent and with the structure expected from previous vibrational spectroscopic studies.1
Experimental
Sample preparation
Liquid ammonia was prepared by distillation of aqueous ammonia (25%, Merck) as described elsewhere.29Triethyl phosphite and tri-n-butylphosphine (Fluka) were used as purchased. Weighed amounts of the mercury(II) halides (Fluka), dried in desiccators over phosphorus pentoxide before use, were dissolved in liquid ammonia.For the 199Hg NMR studies of [HgIn](2−n)+ complexes in liquid ammonia weighed amounts of ammonium iodide, NH4I (J. T. Baker, Analytical grade) were added to a 4 ml solution of mercury(II) perchlorate trihydrate, Hg(ClO4)2·∼3H2O (G. F. Smith), in liquid ammonia, CHg2+ = 0.6 mol dm−3, in a cooled 10 ml glass vessel with a gastight cap. After dissolution at room temperature, the vessel was cooled again, and the solution transferred to a 5 mm NMR tube (Wilmad®). When dissolving ammonium iodide to high concentration (≥3.2 mol dm−3) together with the mercury(II) perchlorate, the liquid ammonia solution turned yellowish indicating formation of [HgI4]2− complexes.30 The vapour pressure decreased and the viscosity increased as expected from a solution containing dissociated salts. A solution of neutral molecular complexes retains high vapour pressure and low viscosity, as for the solutions of mercury(II) iodide that showed properties similar to pure liquid ammonia.
Weighed amounts of recrystallised and vacuum-dried red mercury(II) iodide were dissolved in triethyl phosphite (Fluka) giving a yellow solution, and by heating in tri-n-butylphosphine (Fluka) giving a colourless melt at 326–330 K. The melt solidifies as a white compound at room temperature and the structural studies were therefore performed at about 330 K. The composition of the studied solutions is given in Table 1. Solid mercury(II) chloride and bromide show visible changes when trialkyl phosphites and -phosphines are the added solvents, but the new phases have low solubility in their respective solvents.
| Solvent | Hg | I | Solvent | μ | ρ |
|---|---|---|---|---|---|
| Triethylphosphite | 1.33 | 2.66 | 4.00 | 45.4 | 1.60 |
| Tri-n-butylphosphine | 1.40 | 2.80 | 3.50 | 47.2 | 1.34 |
Solid ammonia solvated mercury(II) halides were obtained by transferring a cooled liquid ammonia solution (ca. 220 K) of the mercury(II) halide into a 4 ml screw-thread glass vial with a 2 mm PTFE/silicone septum. The solvent slowly evaporated through a micro-hole made in the septum when gradually raising the temperature. In the vial [Hg(NH3)2Cl2]∞ and [Hg(NH3)2Br2]∞ powders formed, as identified by powder diffraction (see below). The solution of mercury(II) iodide left white crystals of a solvated compound, most probably [Hg(NH3)2I2], which rapidly decomposed in air to red mercury(II) iodide.
X-Ray powder diffraction
The white powders of [Hg(NH3)2Cl2]∞, [Hg(NH3)2Br2]∞ and [Hg(NH3)2I2] were measured in transmission mode geometry on a STOE powder diffractometer equipped with a Ge monochromator set to diffract CuKα1 radiation, λ = 1.54060 Å.Large angle X-ray scattering (LAXS)
The X-ray scattering of MoKα-radiation (λ = 0.7107 Å) from the free surface of the triethyl phosphite and tri-n-butylphosphine solutions of mercury(II) iodide were measured on a large angle Θ–Θdiffractometer described earlier.31 The solutions were enclosed in a half-filled cylindrical thin-walled glass container,4 ensuring the in- and out-going X-rays to be perpendicular to the glass-walls at all angles. For the phosphine melt the glass container was placed in a bed of synthetic clay, and a constant current was passed through a Kanthal® wire in the surface of the clay to keep the temperature constant, 330 ± 1 K. The angular dependence of the X-ray absorption in the glass walls has been determined previously.4 The scattered X-ray intensities were measured in the range 4 < Θ <65° at discrete points separated by 0.0335 Å−1 in s, where s = (4π/λ))sinΘ; the scattering angle is 2Θ. The scattering was obscured below Θ ≤ 4° by the upward meniscus in the glass container, and the intensities at low angles were extrapolated. A statistical error of 0.35% was achieved by measuring 40.000 counts twice at each sampling point. The fraction of incoherent scattering contributing to the intensity has been estimated in the usual manner.32Scattering factors, corrections for anomalous dispersion and for incoherent and multiple scattering were applied in the data reduction and correction procedures as described before.4 The experimental intensities were normalized to a stoichiometric unit of volume corresponding to one mercury atom. Spurious peaks below 1.4 Å in the experimental radial distribution function (RDF), which could not be related to interatomic distances within the solvent molecules were removed by a Fourier back-transformation procedure.31 The structure-dependent parts of the intensity functions, i(s), multiplied by the scattering variable s, are displayed in Fig. 2 and 3. The corresponding experimental RDFs, D(r) − 4πr2ρo, were obtained by a Fourier transformation, Fig. 2 and 3. All calculations were carried out by means of the KURVLR33 and STEPLR34 programs.
EXAFS
The standard deviations estimated for the refined parameters in Table 2 are obtained from k3 weighted least squares refinements of the EXAFS function χ(k), and do not include systematic errors of the measurements. These statistical deviations provide a measure of the precision of the results and allow reasonable comparisons, e.g., the significance of relative shifts in the distances. However, the variations in the refined parameters, including the shift in the Eo value (defining k = 0), using different models and data ranges, indicate that the accuracy of the distances given for the separate complexes is within 0.01 to 0.02 Å for well-defined interactions. The “standard deviations” reported in the text have been increased accordingly to include estimated additional effects of systematic errors.
| Sample | Scattering path | N | d | σ 2 | E o | S o 2 |
|---|---|---|---|---|---|---|
| HgCl2 in NH3(l), light phase | ||||||
| [Hg(NH3)4]2+ | Hg–N | 4 | 2.251(8) | 0.014(1) | 12284(1) | 0.98(8) |
| HgBr2 in NH3(l) | ||||||
| [Hg(NH3)4]2+ | Hg–N | 4 | 2.22(2) | 0.014(3) | 12283(2) | 0.83(2) |
| (NH4)2[HgI4] in NH3(l) | ||||||
| HgI42− in NH3(l) | Hg–I | 4 | 2.767(3) | 0.0044(2) | 12290.0 | 0.85(2) |
| Hg–I–I | 12 | 5.06(1) | 0.0035(5) | |||
NMR Measurements
199Hg NMR spectra were recorded at 71.33 MHz (9.4 T) on a Bruker DRX400 spectrometer. Samples containing liquid ammonia were kept in 10 mm NMR tubes with PTFE valves (Wilmad®), while the triethyl phosphite samples were kept in ordinary 10 mm NMR tubes. The spectra were evaluated and the integrals of the signal intensities (I) were obtained using XWinNMR 3.0. The 199Hg chemical shifts were referred to the resonance of 0.1 mol dm−3mercury(II) perchlorate in 0.1 mol dm−3perchloric acid at 293 K.39 The line widths of the signals were obtained by fitting single Lorentzians to each observed signal. Spin–lattice relaxation times of 199Hg in the complexes were obtained at 298 K by the inversion–recovery technique with the standard Bruker T1 program within the XWinNMR 3.0 software package. In all experiments the actual 90° pulse was determined. A relaxation delay greater than 5T1 and at least 12 variable delays (τ) were used in the relaxation time measurements. The intensity of the signals was evaluated by means of the non-linear curve fitting option of the Origin 4.0 program (Microcal Software, Inc.) with three parameters in the equation:40| Iτ = A + Bexp(−τ/T1) | (1) |
For quantitative measurements of the 199Hg signal intensities, 30–60° flip angles were used with the delay between pulses equal to (1–2) × T1. The temperature dependence of the chemical shift was determined in the range 203–323 K for liquid ammonia solutions of mercury(II) chloride and iodide.
Raman Spectroscopy
Raman spectra of the samples were measured by means of an FT-Raman module FRA106/S in combination with a Bruker IFS66/S FT-IR spectrometer. The 1064 nm line from an Nd-YAG-laser was used to irradiate the samples and 10–200 scans were collected at a spectral bandwidth of 2–4 cm−1.Results
Dissolution of mercury(II) halides in liquid ammonia
When adding mercury(II) chloride to liquid ammonia a three-phase (liquid–liquid–solid) system immediately appeared. The solid phase was identified by powder X-ray diffraction as [Hg(NH3)2Cl2]∞. Bubbles, most probably ammonia gas, were also generated due to the heat evolved at the exothermic solvation process; the solvation enthalpies of mercury(II) halides in a wide range of solvents are summarized elsewhere.41 When dissolving mercury(II) bromide or iodide in liquid ammonia, new solid phases and gas formed in the exothermic solvation reactions, but no immiscible liquid–liquid phases were observed. Red HgI2 turned into a white solid, and for HgBr2 powder diffraction showed that the solid phase formed was isomorphous with [Hg(NH3)2Cl2]∞.EXAFS
The EXAFS spectra of the liquid ammonia solutions of the light phase of mercury(II) chloride and of mercury(II) bromide show only light back-scatterers around the mercury(II) ion, consistent with ammonia ligands, see Fig. 1. Model refinements yielded mean Hg–N bond distances of 2.25(2) and 2.22(4) Å, respectively. Furthermore, these EXAFS spectra are similar to previously reported spectra of the tetraamminemercury(II) ion in liquid and aqueous ammonia, and in the solid state.24 The presence of a dominating tetraamminemercury(II) complex strongly suggests complete dissociation of the mercury(II) chloride (light phase) and mercury(II) bromide complexes in liquid ammonia. It was not possible to obtain the EXAFS spectrum of the solution of mercury(II) iodide in liquid ammonia due to its high vapour pressure. However, the EXAFS study of a 0.6 mol dm−3mercury(II) liquid ammonia solution with a large excess of iodide, 3.6 mol dm−3 (Fig. 1), results in the mean Hg–I bond distance 2.77(1) Å, which is consistent with a dominating tetraiodomercurate(II) complex, [HgI4]2−.42![k
3-weighted EXAFS: experimental data (solid line) and theoretical models (dashed) of the analysed mercury(ii) complexes: [Hg(NH3)4]2+ (from HgCl2 in NH3(l), light phase; no offset), [Hg(NH3)4]2+ (from HgBr2 in NH3(l); offset +3) and [HgI4]2− in NH3(l) (offset +8). Corresponding Fourier transforms are shown in Fig. S2.](/image/article/2008/DT/b716134d/b716134d-f1.gif) | ||
| Fig. 1 k 3-weighted EXAFS: experimental data (solid line) and theoretical models (dashed) of the analysed mercury(II) complexes: [Hg(NH3)4]2+ (from HgCl2 in NH3(l), light phase; no offset), [Hg(NH3)4]2+ (from HgBr2 in NH3(l); offset +3) and [HgI4]2− in NH3(l) (offset +8). Corresponding Fourier transforms are shown in Fig. S2.† | ||
The structural parameters obtained from the model refinements of the EXAFS data are summarised in Table 2.
LAXS
The radial distribution function (RDF), D(r) − 4πr2ρo, of a melt of mercury(II) iodide in tri-n-butylphosphine at 330 K (Fig. 2, upper part), shows a peak at 2.85 Å and a marked shoulder at 2.45 Å, which correspond to the Hg–I and Hg–P bond distances, respectively. The broad feature around 1.5 Å corresponds to intramolecular distances in the tri-n-butylphosphine molecule; d(P–C) = 1.81 Å and d(C–C) = 1.54 Å.43 Surprisingly, there is no distinct peak in the RDF corresponding to an I–I distance, as expected for a solvated HgI2 complex. The mean Hg–I and Hg–P bond distances could be refined to 2.851(3) and 2.468(4) Å, respectively, Table 3.![LAXS data: (top) Fourier transforms of model interactions for the “melt” of mercury(ii) iodide in tri-n-butylphosphine at 330 K: separate peak shapes for interactions in the [Hg(P(C4H9)3)2]I2 complex (thick solid line), and within the solvent molecules (solid line). (middle) Experimental D(r) − 4πr2ρo (thick solid line); sum of model interactions (solid line); difference (solid line). (bottom) Structure-dependent LAXS intensity functions, si(s) (solid line); model sicalc(s) (thick solid line).](/image/article/2008/DT/b716134d/b716134d-f2.gif) | ||
| Fig. 2 LAXS data: (top) Fourier transforms of model interactions for the “melt” of mercury(II) iodide in tri-n-butylphosphine at 330 K: separate peak shapes for interactions in the [Hg(P(C4H9)3)2]I2 complex (thick solid line), and within the solvent molecules (solid line). (middle) Experimental D(r) − 4πr2ρo (thick solid line); sum of model interactions (solid line); difference (solid line). (bottom) Structure-dependent LAXS intensity functions, si(s) (solid line); model sicalc(s) (thick solid line). | ||
| Distance | d | σ | n |
|---|---|---|---|
| Triethyl phosphite | |||
| Hg–I | 2.750(3) | 0.059(2) | 2.0 |
| Hg–P | 2.457(4) | 0.068(5) | 2.0 |
| I–I | 4.644(14) | 0.095(8) | 1.0 |
| I–P | 4.09(3) | 0.100(12) | 4.0 |
| Tri-n-butylphosphine | |||
| Hg–I | 2.851(3) | 0.061(2) | 2.0 |
| Hg–P | 2.468(4) | 0.025(5) | 2.0 |
The RDF of the triethyl phosphite solution (Fig. 3, upper part), shows a major sharp peak at 2.75 Å, a fairly broad peak at 4.6 Å, and a shoulder at 4.1 Å, corresponding to the Hg–I, I–I and I–P distances, respectively, within the HgI2(P(OC2H5)3)2 complex. The shoulder at about 1.7 Å corresponds to intramolecular bond distances within the triethyl phosphite molecule, d(P–O) = 1.60 Å, d(O–C) = 1.42 Å and d(C–C) = 1.54 Å.43 The Hg–I, Hg–P, I–I and I–P mean distances, 2.750(2), 2.457(4), 4.644(7) and 4.09(2) Å, respectively, correspond to mean I–Hg–I and I–Hg–P bond angles of 115 and 103°, respectively. The refined parameters of the model for the HgI2(P(OC2H5)3)2 complex are summarized in Table 3.
![LAXS: Fourier transforms of model interactions in the mercury(ii) iodide solution in triethyl phosphite: (top) separate peak shapes for interactions in the [Hg(P(OC2H5)3)2I2] complex (thick solid line), and in the solvent molecules (solid line). (middle) Experimental D(r) − 4πr2ρo (thick solid line); sum of model interactions (solid line); difference (solid line). (bottom) Structure-dependent LAXS intensity functions, si(s) (solid line); model sicalc(s) (thick solid line).](/image/article/2008/DT/b716134d/b716134d-f3.gif) | ||
| Fig. 3 LAXS: Fourier transforms of model interactions in the mercury(II) iodide solution in triethyl phosphite: (top) separate peak shapes for interactions in the [Hg(P(OC2H5)3)2I2] complex (thick solid line), and in the solvent molecules (solid line). (middle) Experimental D(r) − 4πr2ρo (thick solid line); sum of model interactions (solid line); difference (solid line). (bottom) Structure-dependent LAXS intensity functions, si(s) (solid line); model sicalc(s) (thick solid line). | ||
Raman Spectroscopy
The Raman spectrum of HgI2 in triethyl phosphite shows a strong band at 132 cm−1, which can be assigned to the symmetric Hg–I bond stretching, ν(Hg–I). This value corresponds to a value of 56 on the DS scale,2 showing that the electron-pair donor ability of triethyl phosphite is the same as that of tri-n-butyl phosphite, and that the length of the carbon chains of the trialkyl phosphite does not noticeably affect the electron distribution around the phosphorus atom.Raman spectra of liquid ammonia and of the dense liquid phase of the three-phase HgCl2/NH3(l) system are shown in Fig. 4. Apart from the N–H stretching bands attributed to liquid ammonia in the range 3200–3400 cm−1, several new features appeared for the HgCl2/NH3(l) solution in the high wave-number region and also for H–N–H bending (∼1000 cm−1). Curve fitting of the spectrum of the solution revealed new features at ∼3360, 3252 and 3150 cm−1, Fig. S3,† together with high frequency bands, 3375 (νd), 3292 (νs) and 3215 (2δd) cm−1, essentially at the same wave-numbers as in pure liquid ammonia. We attribute the new features in the spectrum to vibrational modes of coordinated ammonia molecules, shifted to lower frequencies by binding to mercury(II) ions.
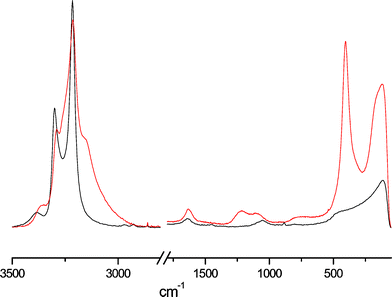 | ||
| Fig. 4 Raman spectra of liquid ammonia (black) and the dense liquid phase of the three-phase HgCl2/NH3(l) system (red). | ||
The strong band at 405 cm−1 is assigned as the symmetric stretching frequency of the Hg–NH3 bonds. The corresponding νs(Hg–N) frequency for the solid [Hg(NH3)4](ClO4)2 compound is 407 cm−1.11,24 The absence of bands, expected at ca. 250 and 180 cm−1 for the Hg–Cl and Hg–Br stretching modes,2 respectively, in the spectra of the liquid ammonia solutions of HgCl2 and HgBr2, supports the assumption that the [Hg(NH3)4]2+ complex is predominating in the dense liquid phase of the three-phase HgCl2/NH3(l) system and in liquid ammonia solutions of HgBr2. The Raman spectra of the light phase of the three-phase HgCl2/NH3(l) system and the solution of HgBr2 resemble the spectrum of pure liquid ammonia, indicating low concentration of the [Hg(NH3)4]2+ complex, see above (Fig. S4†).
The liquid ammonia solution of mercury(II) iodide shows a Hg–N stretching frequency at 356 cm−1 and Hg–I stretching at 132 cm−1, Fig. 5. Previously, Hg–N and Hg–I stretching frequencies of mercury(II) iodide in liquid ammonia were reported at 352 and 123 cm−1, respectively.8 The lower Hg–I frequency could be due to the very high concentration (mole ratio n(HgI2) : n(NH3) = 1 : 7), inducing some association of the mononuclear complexes with weakening of the Hg–I bonds. For the present more dilute solution (mole ratio 1 : 67) the symmetric Hg–I stretching frequency at 132 cm−1 can safely be ascribed to the free distorted pseudotetrahedral [HgI2(NH3)2] complex. Consequently, the DS value of liquid ammonia, which was based on the previously proposed wave-number 123 cm−1,2 should be adjusted from 69 to 56. This revised value indicates that the electron-pair donor ability of liquid ammonia is similar to that of triethyl phosphite.
 | ||
| Fig. 5 Raman spectrum of the solution of the HgI2/NH3(l) system. | ||
The Hg–I stretching frequency at 121 cm−1 obtained for the tetraiodomercurate(II) complex in liquid ammonia solution, see Fig. S5,† is close to the previously reported wave-number of ca. 119 cm−1 for the symmetric stretching of [HgI4]2− in several other solvents.32,44
199Hg NMR
Solutions of the mercury(II) halides in liquid ammonia exhibit a single 199Hg NMR resonance (Fig. 6). The chemical shifts are summarized in Table 4. For the dense liquid phase of the three-phase HgCl2/NH3(l) system the chemical shift at 1188 ppm is close to that of the tetraamminemercury(II) ion in liquid ammonia, 1209 ppm, cf. Table 4.23,24 Quantitative intensity measurements of the signal, after calibration using known concentrations of the hexakis(dimethyl sulfoxide)mercury(II) complex, [Hg(OS(CH3)2)6]2+, allowed the mercury(II) concentration to be estimated to be 1.4 mol dm−3. The 199Hg spin–lattice relaxation time (T1), 5 s, is very similar to that of a Hg(CF3SO3)2 solution in liquid ammonia (Table 4), even though some concentration dependence of T1 can be expected. The relatively long relaxation time indicates high symmetry of the mercury(II) coordination environment,23 and supports a tetrahedral [Hg(NH3)4]2+ complex also in the dense phase of the three-phase HgCl2/NH3(l) system.| Solvate complex/solution | c Hg /mol dm−3 | δ/ppm | Line width/Hz | T 1/s | Temp. coeff./ ppm K−1 | Ref. |
|---|---|---|---|---|---|---|
| a Non-linear temperature dependence of the 199Hg NMR chemical shift. | ||||||
| [Hg(H2O)6]2+/0.1 M HClO4 | 0.3 | −1.4 | 10 | 7.630.47 | −0.40 | 23 |
| [Hg(NH3)4]2+/Hg(CF3SO3)2, NH3(l) | 0.3 | 1209 | 5 | 3.00 ± 0.09 | 22, 23 | |
| [Hg(NH3)4]2+/Hg(ClO4)2·∼3H2O, NH3(l) | 0.4 | 1207 | 5 | 23 | ||
| [Hg(NH3)4]2+/Hg(CF3SO3)2, NH3(l) | 0.3 | 1209 | 5 | 3.000.09 | −1.40 | 22, 23 |
| [Hg(NH3)4]2+/HgCl2, NH3(l) dense phase | 1.4 | 1188 | 5 | 5.11 ± 0.17 | −0.45 | This work |
| [Hg(NH3)4]2+/HgBr2, NH3(l) | >0.1 | 1090–1115 | ∼150 | This work | ||
| [Hg(NH3)2I2]/HgI2, NH3(l) | >0.1 | 351 | 120 | 0.18 ± 0.02 | ∼−6a | This work |
| [Hg(NH3)4]2+/Hg(ClO4)2, NH3(aq) | 0.22 | 1204 | This work | |||
| [Hg(NH3)4]2+/Hg(ClO4)2, NH3(aq) | 0.09 | 1204 | This work | |||
| [Hg(NH3)4]2+/Hg(ClO4)2, NH3(aq) | 0.05 | 1179 | 3.23 ± 0.31 | This work | ||
| [HgCl2(P(OC2H5)3)n]2+/HgCl2, P(OC2H5)3(l) | ∼0.3 | 1740 | This work | |||
| [HgBr2(P(OC2H5)3)n]2+/HgBr2, P(OC2H5)3(l) | ∼0.3 | 1450 | This work | |||
| [HgI2(P(OC2H5)3)n]2+/HgI2, P(OC2H5)3(l) | ∼0.3 | 896 | This work |
 | ||
| Fig. 6 199Hg NMR spectra of the HgX2 (X = CF3SO3, Cl, Br, I) salts in NH3(l). cHg = 0.7 mol dm−3 for Hg(CF3SO3)2 and 1.4 mol dm−3 (Cl) and ≤0.1 mol dm−3 (Br, I). | ||
For mercury(II) bromide solutions in liquid ammonia their 199Hg NMR chemical shift, in the range 1090–1115 ppm, was found to depend slightly on the HgBr2/NH3(l) mole ratio (Table 4). Although the EXAFS results for mercury(II) bromide in liquid ammonia could be interpreted with complete dissociation (see above), the chemical shift of the 199Hg NMR signal differs notably from that of the [Hg(NH3)4]2+ complex in the corresponding mercury(II) chloride and trifluoromethanesulfonate solutions (Fig. 6). This difference, about 100 ppm, together with the concentration dependence and larger line width of the 199Hg NMR signal for the bromide solution (Fig. 6), indicates the presence of minor amounts of mixed [Hg(NH3)4−nBrn](2−n)+ complexes in fast chemical exchange with [Hg(NH3)4]2+.
The solvated mercury(II) iodide molecular complex in liquid ammonia has a very different chemical shift, 351 ppm, and a markedly shorter 199Hg spin–lattice relaxation time, T1 = 0.18 s, than the HgCl2 and Hg(CF3SO3)2 solutions (Table 4 and Fig. 6). The faster 199Hg relaxation can be attributed to the lower symmetry of the [Hg(NH3)2I2] complex, assuming that the chemical shift anisotropy mechanism has an effective contribution to the 199Hg relaxation.23 However, relaxation via scalar coupling to the quadrupolar, fast relaxing 127I nuclei could in this case be an effective mechanism as well.45
The stepwise decrease of the 199Hg chemical shift of the HgX2 species in triethyl phosphite, 1740, 1450 and 896 ppm, in the order X = Cl, Br and I (Table 4), indicates the formation of solvated molecular complexes, [HgX2(P(OC2H5)3)2], rather than ionic dissociation, and corresponds to an increase in the shielding of the mercury nucleus with decreasing electronegativity of the halide. That is an effect common for several mercury halide species, including MeHgX46 and HgX42−,47 and also for the three mercury(II) halides, HgX2 (X = Cl, Br, I), in a wide range of solvents with a large variation of donor strength.22
[HgIn](2−n)+ complexes in liquid ammonia
The formation of [HgIn](2−n)+ (n = 0–4) complexes in liquid ammonia was followed by 199Hg NMR. When changing the n(I−) : n(Hg2+) mole ratio in the Hg(ClO4)2/NH4I/NH3(l) system from 0 to 15 the chemical shift of the single resonance varied enormously covering almost 2500 ppm. The line width also varied strongly with the mole ratio; from ∼20 Hz for n(I−) : n(Hg2+) = 0 and 10, to 2.5 kHz for n(I−) : n(Hg2+) ∼5.5, clearly indicating fast solvent/ligand exchange between several mercury(II) complexes in solution. A detailed study of the exchange dynamics in the Hg(NH3)42+/I−-NH3(l) system is outside the scope of this paper.The 199Hg NMR chemical shift decreases sharply with increasing n(I−) : n(Hg2+) mole ratio, until a constant value of −1205 ppm is reached at the mole ratio 6. This chemical shift, −1205 ppm, can be assigned to the [HgI4]2− complex, because EXAFS and Raman spectroscopy of the sample with n(I−) : n(Hg2+) = 10, shows that [HgI4]2− strongly dominates in a large excess of iodide. The agreement with previously reported δ(199Hg) values, around −1150 ppm for the [HgI4]2- complex in a number of organic solvents, is satisfactory.48
Thus, we conclude that the [Hg(NH3)4]2+ and [HgI4]2− species dominate for mole ratios n(I−) : n(Hg2+) = 0 and ≥6, respectively, while mixed ammine–iodo complexes form at intermediate iodide concentrations. The [Hg(NH3)4]2+ and [HgI4]2− complexes display narrow 199Hg signals, while the fast exchange on the 199Hg NMR time scale for the mixed species drastically broadens the 199Hg resonance. Similar fast ligand exchange and strong dependence of the 199Hg chemical shift on ligand concentration, covering a range of 1100 ppm, was previously reported for the [HgCln](2−n)+ complexes (n = 0–4) in aqueous solution.49
Discussion
Mercury(II) halide complexes in liquid ammonia
The relative volume of the dense lower liquid phase formed when dissolving mercury(II) chloride in liquid ammonia increases when increasing the added amount of solid mercury(II) chloride, and is in equilibrium with the precipitated solid [Hg(NH3)2Cl2]∞ compound. The upper liquid phase, with low total mercury(II) concentration, contains tetraamminemercury(II) and chloride ions. Furthermore, the dense phase most probably consists of tetraamminemercury(II) complexes, hydrogen bonded to chloride ions forming outer-sphere ion pairs. The structure of the tetraamminemercury(II) complex has been described in detail elsewhere.24When dissolving mercury(II) bromide in liquid ammonia only a single liquid phase forms. The 199Hg NMR shift is about 100 ppm lower than that of the tetraamminemercury(II) ion in liquid ammonia, strongly indicating some remaining direct interaction between the mercury(II) and bromide ions. This different behaviour may be an effect of the higher hydrogen bonding ability of chloride than of bromide ions,50 promoting the ion-pair formation with the polarised ammine ligands of the [Hg(NH3)4]2+ complexes in the dense liquid phase of the HgCl2/NH3 system.
The mercury(II)–iodide complexes are sufficiently stable to be retained also in liquid ammonia. The tetraiodomercurate(II) complex that forms in excess of iodide (Fig. 1) shows that mercury(II) binds iodide ions stronger than ammonia molecules; this is also indicated by the higher Hg–I symmetric stretching frequency (132 cm−1) for the ammonia solvated mercury(II) iodide complex [HgI2(NH3)2] than for [HgI4]2− (121 cm−1), see above.
Mercury(II) halide complexes in phosphorus donor solvents
A number of crystal structures of mercury(II) trialkyl- and triarylphosphines and mercury(II) halide trialkyl- and triarylphosphines have been reported; see the summary in Table 5, while no mercury(II) phosphite structures were found. The mean Hg–P bond distances in the solid trialkylphosphine solvated mercury(II) complexes are ca. 2.44, 2.51 and 2.54 Å in linear, trigonal and tetrahedral configuration, respectively, Table 5. For triphenyl- and triarylphosphine [HgX2(PR3)2] complexes the mean Hg–P bond distances are 2.46, 2.54 and 2.56 Å, and the P–Hg–P bond angles 130°, 113° and 109° for X = Cl, Br and I, respectively, Table 5. The decreasing P–Hg–P angles approaching tetrahedral values show that triphenyl and triarylphosphine ligands bind more strongly to mercury(II) than chloride, slightly stronger than bromide, and with almost equal strength as iodide. In such qualitative comparisons of the relative bond strength of ligands in complexes, thiocyanate shows similar behaviour to chloride, while cyanide resembles iodide, as can be expected from comparisons of stability constants of mercury(II) complexes in aqueous solution.68 The short Hg–P bond distances and the large P–Hg–P bond angles in the HgX2(PR3)2 compounds, X = Cl and Br (cf.Table 5) indicate that mercury(II) forms much stronger bonds to trialkylphosphines than to chloride and bromide; structures of Hg(PR3)2I2 compounds have not yet been reported. The structural parameters of the Hg(PR3)2 entities in the perchlorate and acetate salts are similar to those for the Hg(PR3)2X2 compounds, which should be regarded as halide salts of digonal [Hg(PR3)2]2+ complexes. Trialkylphosphines are considerably stronger electron-pair donors than triarylphosphines, cf.Table 5.| Codea | Complex | d(Hg–P)/Å | d(Hg–X)/Å | ∠(P–Hg–P)/° | ∠(X–Hg—X)/° | Ref. |
|---|---|---|---|---|---|---|
| a Codes in ref. 42. | ||||||
| Solids | ||||||
| Triphenylphosphine complexes | ||||||
| BENJIN | Hg(P(C6H5)3)2(NO3)2 | 2.451, 2.451 | (2.508, 2.790) | 131.8 | 51 | |
| BULZAJ | HgCl2(P(C6H5)3)2 | 2.462, 2.478 | 2.545, 2.559 | 134.1 | 110.7 | 17 |
| BULZAJ01 | HgCl2(P(C6H5)3)2 | 2.462, 2.478 | 2.545, 2.559 | 134.1 | 110.7 | 18 |
| NEKNUM | HgCl2(P(C6H5)3)2 | 2.482, 2.482 | 2.523, 2.523 | 123.4 | 103.6 | 19 |
| DOPJAT | HgCl2(P(C4SH3)3)2 | 2.471, 2.513 | 2.518, 2.540 | 128.7 | 107.3 | 52 |
| BULZEN | HgBr2(P(C6H5)3)2 | 2.535, 2.540 | 2.626, 2.633 | 113.0 | 106.9 | 17 |
| BULZEN01 | HgBr2(P(C6H5)3)2 | 2.492, 2.550 | 2.627, 2.637 | 113.0 | 107.1 | 20 |
| DITPHG | HgI2(P(C6H5)3)2 | 2.556, 2.572 | 2.734, 2.763 | 108.9 | 110.5 | 21 |
| BENJEJ | Hg(CN)2(P(C6H5)3)2 | 2.433, 2.588 | 2.189, 2.268 | 108.9 | 107.7 | 53 |
| BENJEJ01 | Hg(CN)2(P(C6H5)3)2 | 54 | ||||
| TCTPHG | Hg(SCN)2(P(C6H5)3)2 | 2.484, 2.494 | 2.566, 2.574 | 118.1 | 96.6 | 55 |
| TCTPHG01 | Hg(SCN)2(P(C6H5)3)2 | 54 | ||||
| SEDGEN | Hg(SCN)2(P(C6H5)3)2 | 2.499, 2.522 | 2.566, 2.571 | 114.1 | 104.7 | 56 |
| Tri-p-methoxyphenylphosphine | ||||||
| GIHYOL | [Hg(P(C21H21O3)3)3](ClO4)2 | 2.500, 2.503, 2.534 | 113.1,115.5, 126.7 | 57 | ||
| Tri(2,4,6-trimethoxyphenyl)phosphine) | ||||||
| PEYKIP | [Hg(P(C6H3(OCH3)3)2][Hg2Cl6] | 2.388, 2.388 | 166.5 | 58 | ||
| PEYKIP01 | [Hg(P(C6H3(OCH3)3)2][Hg2Cl6] | 2.377, 2.377 | 166.3 | 59 | ||
| Dimethylphenylphosphine | ||||||
| DADJOH | [Hg(P(CH3)2C6H5)3)4][Ta2OCl10] | 2.526, 2.526, 2.547, 2.547 | 102.5–111.9 | 60 | ||
| Trialkylphosphine complexes | ||||||
| TCHPHG | [Hg(P(C6H11)3)2](ClO4)2 | 2.444, 2.445 | 170.3 | 61 | ||
| ACHPHG | [Hg(P(C6H11)3)2](CH3OO)2·2H2O | 2.439, 2.440 | 153.0 | 61 | ||
| TAMNIF | [Hg(P(CH2C6H5)3)2](BF4)2 | 2.403, 2.403 | 180.0 | 62 | ||
| TAMNOL | [Hg(P(CH2C6H5)3)2](NO3)2 | 2.416, 2.429 | 2.479, 2.601 | 153.2 | 62 | |
| 2.431, 2.435 | 2.510, 2.523 | 150.9 | ||||
| CLTEHG | HgCl2(P(C2H5)3)2 | 2.393, 2.393 | 2.681, 2.681 | 158.3 | 105.5 | 53 |
| JADDEX | HgCl2(P(CH2CH2CN)3)2 | 2.412, 2.450 | 2.603, 2.668 | 153.9 | 98.1 | 63 |
| 2.452, 2.452 | 2.608, 2.621 | 146.5 | 95.0 | |||
| DUTDIF | [HgCl2(P(C6H11)2(CSNC6H5))2] | 2.452, 2.456 | 2.594, 2.598 | 149.4 | 102.7 | 64 |
| VUDJOT | [HgCl2(P(C9H21N4)2] | 2.453, 2.470 | 2.637, 2.652 | 144.5 | 102.2 | 65 |
| CIZNII | HgBr2(P(CH3)2C2H5)2·(CH3)2CO | 2.44, 2.50 | 2.72, 2.79 | 151.3 | 107.3 | 66 |
| HgBr2(P(CH3)2C2H5)2 | 2.39, 2.48 | 2.79, 2.88 | 150 | 106.9 | 67 | |
| HgBr2(P(C4H9)3)2 | 2.39 | 2.68 | 158.5 | 105.5 | 67 | |
| Solution/melt | ||||||
| HgI2/P(OC4H9)3 | 2.457(4) | 2.750(3) | 115(1) | 103(3) | This work | |
| HgI2/P(C4H9)3 | 2.468(4) | 2.851(3) | This work | |||
The structural parameters determined by LAXS methods for the highly concentrated mercury(II) iodide solution with ca. 2.0 iodide ions and 2.5 tri-n-butylphosphine molecules per mercury(II) atom, Table 1, can similarly be discussed in terms of a melt containing tri-n-butylphosphine solvated mercury(II) complexes [Hg(PR3)2]2+ and iodide ions. The mean Hg–P bond distance, 2.468(4) Å, obtained for the melt is only slightly longer than that of almost linearly coordinated mercury(II) trialkylphosphine complexes in the solid state, 2.44 Å (Table 5). The Hg–I bond distances are quite long, 2.852(3) Å, and the absence of a distinct I–I distance strongly indicates that the Hg–I bonds are weak and non-directional. The slight excess of the strongly coordinating tri-n-butylphosphine ligand can probably to some extent dislocate some of the iodide ions, which for electrostatic reasons are confined to the tri-n-butylphosphine solvated mercury(II) ion, due to the low permittivity of the solvent. The observations described above show that trialkylphosphine is a stronger ligand than iodide to the soft electron-pair acceptor mercury(II). A similar behaviour is found for gold(I), an even softer electron-pair acceptor, where the iodide ion is associated at long distance to almost linear bis(tricyclohexylphosphine)gold(I) complexes.69 In the compound bis(triphenylphosphine)gold(I) iodide the Au–I distance is 0.1 Å shorter,70 consistent with the weaker electron-pair donor properties of triphenylphosphine. The difference in Hg–I bond lengths for the corresponding complexes are very similar (Table 2).
The donor strength parameter DS is 56 for liquid ammonia and tri-n-butyl phosphite, and triethyl phosphite should show similar electron-pair donor properties toward mercury(II).2 However, the phosphites have low permittivity as solvents. The relatively high solubility, as well as the stepwise change in the 199Hg NMR chemical shift of the mercury(II) halides in triethyl phosphite (Table 4), strongly suggest that the molecular HgX2 entities do not dissociate. The coordination number for the triethyl phosphite solvated mercury(II) iodide complex, [HgI2(P(OC2H5)3)2], is therefore expected to be similar to the corresponding [HgI2(NH3)2] complex in liquid ammonia. Vibrational spectroscopy indicated that the coordination around mercury(II) is close to tetrahedral with somewhat stronger bonding to the iodide ion.1,2 The structure observed in solution is consistent with this prediction with the mean Hg–I and Hg–P bond distances 2.750(2) and 2.457(4) Å, respectively. The I–Hg–I bond angle is 115° and the Hg–I bond distance is slightly shorter than that found for the HgI42− complex (2.78 Å), Table 3.
The Hg–P bond is of similar length in the near tetrahedral [HgI2(P(OC2H5)3)2] as in the almost linear bis(trialkylphosphine)mercury(II) complexes, even though the latter Hg–P bonds are much stronger. This indicates that the phosphorus atom has a smaller effective radius in phosphites than in phosphines. This is expected as the oxygen is electron withdrawing while alkyl carbons in phosphines are not. This phenomenon has also been observed for nitrogen donor solvents where metal-to-nitrogen bonds in pyridine are longer than metal-to-nitrogen bonds in acetonitrile, in spite of the significantly stronger bonding in pyridine.71–74
Conclusions
Mercury(II) chloride and bromide dissociate to ions in the strong electron-pair donor solvents liquid ammonia (DS = 56), triethyl phosphite (DS = 56) and tri-n-butylphosphine (DS = 76), while mercury(II) iodide only dissociates in tri-n-butylphosphine. Upon addition of mercury(II) chloride to liquid ammonia a three-phase (liquid–liquid–solid) system immediately appears. The upper light liquid phase contains completely dissociated mercury(II) chloride, present as tetraamminemercury(II) complexes and solvated chloride ions in low concentration, while the dense highly concentrated phase most probably contains tetraamminemercury(II) complexes, hydrogen bonded to chloride ions forming outer-sphere ion pairs. The liquid ammonia solutions of mercury(II) bromide and mercury(II) iodide always form a single liquid phase independent of concentration. The solubility of the mercury(II) halides in the phosphorus donor solvents trialkyl phosphite and trialkylphosphine is low, except for mercury(II) iodide in trialkyl phosphite. Dissociation into ions, combined with low permittivity of these solvents, causes formation of solid [Hg(PR3)n]X2 and [Hg(P(OR)3)n]X2 salts and only the neutral [HgI2(P(OR)3)2] complex has high solubility in trialkyl phosphite.Acknowledgements
We are grateful to Prof. Bo Liedberg, Department of Physics and Measurements, Linköping University, for the use of Raman and infrared spectrometers. The support from the Swedish Research Council is acknowledged. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.References
- I. Persson, M. Sandström and P. L. Goggin, Inorg. Chim. Acta, 1987, 129, 183–197 CrossRef CAS.
- M. Sandström, I. Persson and P. Persson, Acta Chem. Scand., 1990, 44, 653–675 CrossRef CAS.
- M. Sandström, Acta Chem. Scand., Ser. A, 1978, 32, 627–641 Search PubMed.
- I. Persson, M. Sandström, P. L. Goggin and A. Mosset, J. Chem. Soc., Dalton Trans., 1985, 1597–1605 RSC.
- M. Sandström, I. Persson and P. L. Goggin, J. Chem. Soc., Dalton Trans., 1987, 2411–2415 RSC.
- L. Bengtsson, B. Holmberg, I. Persson and Å. Iverfeldt, Inorg. Chim. Acta, 1988, 146, 233–241 CrossRef CAS.
- I. Persson, M. Landgren and A. Marton, Inorg. Chim. Acta, 1986, 116, 135–144 CrossRef CAS.
- D. J. Gardiner, A. H. Haji and B. Straughan, J. Chem. Soc., Dalton Trans., 1978, 705–710 RSC.
- C. H. MacGillavry and J. M. Bijvoet, Z. Kristallogr. Kristallgeom. Kristallphys. Kristallchem., 1936, 94, 231–245 Search PubMed.
- A. J. Canty, C. L. Raston, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1982, 15–18 RSC.
- P. Nockemann and G. Meyer, Z. Anorg. Allg. Chem., 2003, 629, 123–128 CrossRef CAS.
- H. Martan and J. Weiss, Z. Anorg. Allg. Chem., 1984, 515, 225–229 CAS.
- H. Schmidbaur and K.-H. Räthlein, Chem. Ber., 1973, 106, 2491–2507 CAS.
- G. E. Coates and A. Lauder, J. Chem. Soc., 1965, 1857–1864 RSC.
- S. O. Grim, P. J. Luiand and R. L. Keiter, Inorg. Chem., 1974, 13, 342–345 CrossRef CAS.
- V. A. Yamaski and E. Fluck, Z. Anorg. Allg. Chem., 1973, 396, 297–302.
- N. A. Bell, T. D. Dee, M. Goldstein, P. J. McKenna and I. W. Nowell, Inorg. Chim. Acta, 1983, 71, 135–140 CrossRef CAS.
- T. S. Lobana, M. K. Sandhu, M. R. Snow and E. R. T. Tiekink, Acta Crystallogr., Sect. C, 1988, 41, 179–181 CrossRef.
- J. Vicente, R. Clemente and M. C. R. d. Arellano, CCDC code NEKNUM.
- M. Kubicki, S. K. Hadjikakou and M. N. Xanthopoulou, Polyhedron, 2001, 20, 2179–2185 CrossRef CAS.
- L. Fälth, Chem. Scr., 1976, 9, 71 CAS.
- P. Peringer, Inorg. Chim. Acta, 1980, 39, 67–70 CrossRef CAS.
- M. Maliarik and I. Persson, Magn. Reson. Chem., 2005, 43, 835–842 CrossRef CAS.
- K. B. Nilsson, M. Maliarik, I. Persson, A. Fischer, A.-S. Ullström, L. Eriksson and M. Sandström, Inorg. Chem., 2007, 47 DOI:10.1021/ic7013489.
- C. A. Kraus, J. Am. Chem. Soc., 1908, 30, 653–668 CrossRef CAS.
- W. L. Jolly, Modern Inorganic Chemistry, Mcgraw-Hill Book Co., New York, 2nd edn, 1991 Search PubMed.
- W. M. Jolly, Prog. Inorg Chem., 1959, 1, 235 CAS.
- J. C. Wasse, S. Hayama, N. T. Skipper, D. Morrison and D. T. Bowron, J. Phys. Chem. B, 2003, 107, 14452–14456 CrossRef CAS.
- K. B. Nilsson and I. Persson, Dalton Trans., 2004, 1312–1319 RSC.
- (a) T. Griffiths and R. Anderson, J. Chem. Soc., Faraday Trans., 1984, 80, 2361–2374 Search PubMed; (b) R. Peterson, P. Lingane and W. Reynolds, Inorg. Chem., 1970, 9, 680–682 CrossRef CAS.
- H. A. Levy, M. D. Danford, and A. H. Narten, Data Collection and Evaluation with an X-ray Diffraction Designed for the Study of Liquid Structure, Report ORNL 3960, Oak Ridge National Laboratory, Oak Ridge, TN, 1966 Search PubMed.
- G. Johansson and M. Sandström, Acta Chem. Scand., Ser. A, 1977, 31, 132–140.
- G. Johansson and M. Sandström, Chem. Scr., 1973, 4, 195–198 CAS.
- M. Molund and I. Persson, Chem. Scr., 1985, 25, 197–197 CAS.
- A. Thompson, D. Attwood, E. Gullikson, M. Howells, K.-J. Kim, J. Kirz, J. Kortright, I. Lindau, P. Pianatta, A. Robinson, J. Scofield, J. Underwood, D. Vaughan, G. Williams and H. Winick, X-ray Data Booklet, LBNL/PUB-490 Rev. 2, Lawrence Berkeley National Laboratory, Berkeley, CA, 2001 Search PubMed.
- G. N. George and I. J. Pickering, EXAFSPAK-A Suite of Computer Programs for Analysis of X-Ray Absorption Spectra, SSRL, Stanford, CA 1993 Search PubMed.
- (a) FEFF code for ab initio calculations of XAFS: S. I. Zabinsky, J. J. Rehr, A. Ankudinov, R. C. Albers and M. J. Eller, Phys. Rev. B, 1995, 52, 2995–3009 Search PubMed; (b) A. Ankudinov, Ph. D. Thesis, University of Washington, 1996; the FEFF program available from: http://Feff.phys.washington.edu/feff.
- G. A. Jeffrey and M. Vlasse, Inorg. Chem., 1967, 6, 396–399 CrossRef CAS.
- R. J. Goodfellow, in Post-Transition Metals, Copper to Mercury, ed. J. Mason, Plenum Press, New York, 1987 Search PubMed.
- S. Braun, H.-O. Kalinowski, and S. Berger, in 100 and More Basic NMR Experiments, VCH, Weinheim, 1996 Search PubMed.
- I. Persson, M. Landgren and A. Marton, Inorg. Chim. Acta, 1986, 116, 135–144 CrossRef CAS.
- Inorganic Crystal Structure Data Base, National Institute of Standards and Technology, Fachinformationszentrum, Karlsruhe, Release 07/2 and references therein Search PubMed.
- F. H. Allen, S. Bellard, M. D. Brice, B. A. Cartwright, A. Doubleday, H. Higgs, T. Hummelink, C. G. Hummelink-Peters, O. Kennard, W. D. S. Motherwell, J. R. Rodgers and D. G. Watson, The Cambridge Crystallographic Data Centre: computer-based search, retrieval, analysis and display of information, Acta Crystallogr., Sect. B, 1979, 35, 2331–2339 CrossRef and references therein.
- M.-L. Delwaulle, Bull. Soc. Chim. Fr., 1955, 1294 CAS.
- J. W. Akitt and B. E. Mann, NMR and Chemistry, Stanley Thornes, Cheltenham, 2000, 4th edn, pp. 109–117 Search PubMed.
- J. A. Iggo, NMR Spectroscopy in Inorganic Chemistry, Oxford University Press, Oxford, 2002, pp. 35–41 Search PubMed.
- R. Colton and D. Dakternieks, Aust. J. Chem, 1980, 33, 2405–2409 CAS.
- R. J. Goodfellow, in Multinuclear NMR, ed. J. Mason, Plenum Press, New York, 1987, pp. 563–590 Search PubMed.
- G. Klose, F. Volke, G. Peinel and G. Knobloch, Magn. Reson. Chem., 1993, 31, 548–551 CrossRef CAS.
- S. Ahrland, Pure Appl. Chem., 1982, 54, 1451–1468 CAS.
- H. B. Buergi, E. Fischer, R. W. Kunz, M. Parvez and P. S. Pregosin, Inorg. Chem., 1982, 21, 1246–1256 CrossRef CAS.
- D. W. Allen, N. A. Bell, S. T. Fong, L. A. March and I. W. Nowell, Inorg. Chim. Acta, 1985, 99, 157–163 CrossRef CAS.
- N. A. Bell, T. D. Dee, P. L. Goggin, R. Goodfellow, T. Jones, K. Kessler, D. M. McEwan and I. A. Nowell, J. Chem. Res. (S), 1981, 2, 201–202 Search PubMed.
- T. Allman and R. E. Leninski, Inorg. Chem., 1986, 25, 3202–3204 CrossRef CAS.
- R. C. Makhija, A. L. Beauchamp and R. Rivest, J. Chem. Soc., Dalton Trans., 1973, 2447–2450 RSC.
- T. S. Lobana, M. K. Sandhu, D. C. Povey, G. W. Smith and V. Ramdas, J. Chem. Soc., Dalton Trans., 1989, 2339–2341 RSC.
- T. Allman, R. G. Goel and A. L. Beauchamp, Acta Crystallogr., Sect. C, 1988, 44, 1557–1560 CrossRef.
- L.-J. Baker, G. A. Bowmaker, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1993, 3235–3240 RSC.
- M. B. Hursthouse and M. E. Light, unpublished data, code PEYKIP01 in the Cambridge Structure Data Base.
- F. A. Cotton, S. A. Duraj and W. J. Roth, Acta Crystallogr., Sect. C, 1985, 41, 881–883 CrossRef.
- E. C. Alyea, S. A. Dias, G. Ferguson and M. A. Khan, J. Chem. Res. (S), 1979, 360, 4101 Search PubMed.
- G. A. Bowmaker, B. Assadollahzadeh, A. M. Brodie, E. W. Ainscough, G. H. Freeman and G. B. Jameson, Dalton Trans., 2005, 1602–1612 RSC.
- N. A. Bell, L. A. March and I. W. Nowell, Inorg. Chem. Acta, 1989, 156, 195–200 CrossRef CAS.
- S. W. Cowan, D. Daktemieks, R. W. Gable, B. F. Hoskins, C. L. Rolls and E. R. T. Tiekink, Aust. J. Chem., 1986, 39, 547–556 CAS.
- J.-S. Tang, M. A. H. Laramay, V. Young, S. Ringrose, R. A. Jacobson and J. G. Verkade, J. Am. Chem. Soc., 1992, 114, 3129–3131 CrossRef CAS.
- N. A. Bell, M. Goldstein, L. A. March and I. W. Nowell, J. Chem. Soc., Dalton Trans., 1984, 1621–1624 RSC.
- T. Jones, PhD Thesis, Sheffield City Polytechnic, Sheffield, 1979.
- IUPAC Stability Constants Database, Academic Software, Otley, UK, 2000 and references therein Search PubMed.
- G. A. Bowmaker, C. L. Brown, R. D. Hart, P. C. Healy, C. E. F. Rickard and A. H. White, J. Chem. Soc., Dalton Trans., 1999, 881–890 RSC.
- G. A. Bowmaker, J. C. Dyason, P. C. Healy, L. M. Engelhardt, C. Pakawatchai and A. H. White, J. Chem. Soc., Dalton Trans., 1987, 1089–1097 RSC; R. Fränkel, J. Kniczek, W. Ponikwar, H. Nöth, K. Polborn and W. P. Fehlhammer, Inorg. Chim. Acta, 2001, 312, 23–29 CrossRef CAS.
- I. Persson, J. E. Penner-Hahn and K. O. Hodgson, Inorg. Chem., 1993, 32, 2497–2501 CrossRef CAS.
- F. Hultén and I. Persson, Acta Chem. Scand., Ser. A, 1987, 41, 87–92 Search PubMed.
- K. Nilsson and I. Persson, Acta Chem. Scand., Ser. A, 1987, 41, 139–145 Search PubMed.
- S. Ahrland, K. Nilsson, I. Persson, A. Yuchi and J. E. Penner-Hahn, Inorg. Chem., 1989, 28, 1833–1838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XANES, FT data and Raman spectra of the complexes. See DOI: 10.1039/b716134d |
| This journal is © The Royal Society of Chemistry 2008 |