Automated oligosaccharide synthesis†
Peter H.
Seeberger
*
Laboratory for Organic Chemistry, Swiss Federal Institute of Technology (ETH) Zürich, HCI F 315, Wolfgang-Pauli-Str. 10, CH-8093 Zürich, Switzerland. E-mail: seeberger@org.chem.ethz.ch; Fax: 41 44 633 1235; Tel: 41 44 633 2103
First published on 20th August 2007
Abstract
Peptides and oligonucleotides are prepared by automated synthesizers that can be operated by non-specialists. Carbohydrates have been hard to assemble, but the increasing awareness of the biological importance of this class of complex repeating biopolymers has prompted efforts to accelerate their synthesis. This tutorial review defines the state of the art of automated solid phase oligosaccharide synthesis and identifies areas in need of further innovation. Application of the automated synthesis method to prepare a malaria vaccine candidate is briefly highlighted.
 Peter H. Seeberger | Peter H. Seeberger (BS University Erlangen-Nürnberg, PhD University of Colorado) was a postdoctoral fellow with Samuel Danishefsky at Sloan-Kettering Institute in New York before working as an Assistant and Associate Professor at MIT from 1998 to 2003. Since 2003 he has been a Professor at the ETH in Zurich, Switzerland and an Affiliate Professor at the Burnham Institute in La Jolla, CA. The Seeberger group research focuses on the chemistry and biology of carbohydrates and on microreactor chemistry. For this work he has received a number of honors and awards. |
1 Introduction
In the 1960s the syntheses of insulin1 and of the first gene2 were major research projects that occupied synthetic chemistry teams for several years. These total syntheses relied on the assembly of blocks that were then combined into oligomers. Today, peptide and oligonucleotide assembly is routinely carried out using commercially available automated synthesizers that sequentially combine monomeric building blocks. Even non-experts can synthesize most sequences within one day or just order them from service companies that rapidly deliver the desired molecules for all types of biological experiments.3Carbohydrates , the third major class of biopolymers, are less well studied than peptides and oligonucleotides. Access to pure carbohydrates remains challenging and has impeded biological investigations. Isolation of oligosaccharides is tedious, when at all possible and typically yields miniscule amounts of the desired materials. The chemical synthesis of oligosaccharides has been at a stage that is comparable to that of oligopeptide and oligonucleotide synthesis in the 1960s. Each new structure is a major research project that is carried out by specialized laboratories.4
In recent years efforts have been undertaken to create an automated solid-phase oligosaccharide synthesizer that will allow non-specialists to assemble defined oligosaccharides in days rather than years using a defined set of monosaccharide building blocks. The final goal is the development of a general and simple process that is now practised for peptides and oligonucleotides. Although not quite reality yet, we have come a long way toward this goal. At least for a majority of carbohydrate sequences, an automated synthesis scenario is expected to become routine within the next five years. This tutorial review discusses the key challenges and current solutions. To guide those interested in contributing, the remaining open questions are identified. The application potential of automated oligosaccharide synthesis will be highlighted on the example of a carbohydrate -based anti-GPI malaria vaccine candidate.
2 The challenge of oligosaccharide synthesis
Why are carbohydrates more difficult to synthesize than peptides and oligonucleotides? Both peptides and oligonucleotides are strictly linear biopolymers that are assembled from four nucleotide building blocks or 20 proteogenic amino acids respectively. Mammalian carbohydrates are composed of ten different monosaccharides and are typically encountered in nature as glycoconjugates where an oligosaccharide is connected to a lipid or a protein . Even the oligosaccharide portion alone is more complex than either peptides or oligonucleotides.The formation of each glycosidic linkage results in the creation of a new stereogenic center unlike the situation for amide and phosphate diester linkages in peptides and oligonucleotides. Stereochemical control of glycosidic linkage formation is a key challenge. In addition, a host of functional hydroxyl and amine groups around each sugar ring has to be differentiated by the placement of protective groups.
The diversity of biopolymers greatly differs as illustrated for hexamers: A total of 46 (=4096) different hexanucleotide and 206 (=64 million) hexapeptide sequences are possible. In the case of oligosaccharides , based on the ten mammalian monosaccharides , branching and different stereoisomers, it was calculated that 192 billion different structures are theoretically possible.
Until very recently, glycospace diversity made researchers wonder whether an automated approach would ever be possible. A bioinformatics study, based on the most comprehensive currently available databank, revealed that nature actually occupies only a small portion of the vast glycospace. It was calculated that a strictly linear synthesis approach as is practised for oligopeptides and oligonucleotides would require only 36 monosaccharide building blocks to create 75% of mammalian structures. To produce 90% of all structures, a set of 65 building blocks is required. Procurement of such a large number of building blocks, while challenging, is tractable.5
3 Key issues for automated synthesis
The automated synthesis of peptides and oligonucleotides is currently carried out as follows: A set of differentially protected building blocks is used in a strictly linear fashion (Fig. 1). In either case the nucleophile for the formation of the amide or phosphate diester linkage is connected via a linker to a solid support. This linker is only disconnected upon completion of the synthesis. The solid support serves to keep the growing biopolymer chain in a form that can be removed from the reaction mixture by filtration. Such a washing, filtration, washing process lends itself for an automated system that has to control simply the type of solution to be added to the reaction chamber as well as the times for which a given solution remains in contact with the resin. Once the desired linkage is formed, the temporary protecting group has to be removed in order to free the functional group that will serve in the extension of the oligomer. Any unreacted building blocks or side products are simply removed by washing. The purification of the fully assembled molecule is carried out only after the entire oligomer has been cleaved from the support matrix. Purification of the desired product by high pressure liquid chromatography (HPLC) is the norm and had immense impact on the practice of peptide and oligonucleotide synthesis.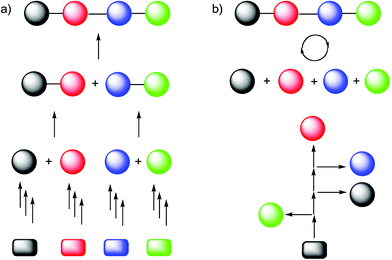 | ||
| Fig. 1 Comparison of the a) total synthesis approach with b) the automated synthesis approach based on a defined set of building blocks. | ||
Given the well-established situation for peptides and oligonucleotides, the challenges for automation of oligosaccharide assembly were clear from the outset:
1) A set of monosaccharide building blocks carrying compatible permanent and temporary protective groups was needed;
2) The first building block would be attached to an insoluble support via a linker that is chemically compatible with all synthetic operations;
3) Coupling and deprotection conditions that are rapid, selective and quantitative had to be established;
4) Capping procedures to minimize deletion sequences need to be incorporated;
5) Real-time monitoring of coupling efficiency was highly desirable;
6) Efficient cleavage of the linker at the end of the synthesis should render the oligosacharide either as the free reducing terminus or in a form that allows for the creation of glycoconjugates;
7) Ready removal of all protective groups; and
8) Purification and quality control of the final product.
The key challenges were clear and most laboratories focused on selected issues. Advances were made not in a chronological order but rather advances in one area resulted in other bottle necks that needed to be addressed. Only recently it has become possible to create an encompassing synthesis approach that addresses all of the above mentioned key challenges. In the following sections the advances will be discussed in detail.
3.1 Synthesis strategy
A solid supported synthesis relies on the attachment of one reaction partner to an insoluble carrier for ready separation by filtration. Either the nucleophile (glycosyl acceptor) or the sugar carrying the anomeric leaving group (glycosyl donor) can in principle be attached to the solid support. Both approaches have been explored.6 It is now widely accepted that the acceptor-bound approach is advantageous. Anchoring of the nucleophile allows for an excess of the reactive donor to be used to drive the reaction to completion. More importantly, side reactions typically occur by decomposition of the reactive species. If the donor is the limiting reagent as for the donor-bound approach, any unproductive side reaction will result in a direct reduction in overall yield. Automated oligosaccharide synthesis relies on the attachment of the nucleophile to the solid support, the acceptor-bound approach.3.2 Solid support resin
The solid phase facilitates the removal of the growing biopolymer chain from all other reagents by filtration. A host of different matrices have been explored over the years, ranging from controlled pore glass (CPG) that is utilized for DNA synthesis to polystyrene resins, the basis for peptide assembly. Loading, swelling, solvent and reagent compatibility, mechanical robustness, price and other factors play a role in selecting the proper support. We found that commercially available chloromethylated polystyrene resins with a loading of 0.3–0.5 mmol g–1 performed best during automated oligosaccharide syntheses.3.3 Linker
The linker, the connection between the solid support and the first monosaccharide , can be viewed as a support-bound protecting group. Consequently, the linker is of utmost importance for the entire synthetic process as its chemical nature determines the reaction conditions that can be used during the assembly, the cleavage conditions required to liberate the final product from the resin and also the form of the oligosaccharide that is being released. Since the linker is now typically used to connect the anomeric position of the reducing end sugar to the support, it is desirable to release either a free reducing end or spacers that enable attachment to a protein carrier or a chip surface.The type of functional group selected as linker has to be orthogonal to all other modes of protection used during the synthesis. We incorporated a double bond that is not found in naturally occurring oligosaccharides . The octenediol linker is linked to the support either via an ether 7 or an ester linkage (Scheme 1).8 The final oligosaccharide product is released by a cross metathesis reaction using Grubbs catalyst in the presence of ethylene or another alkene . With ethylene, the oligosaccharide product is released in form of an n-pentenyl glycoside. This reducing end moiety can either be removed to furnish the free reducing sugar or the double bond can be converted into a aldehyde or thiol group to be linked to carrier proteins and surfaces. Other linkers also have been used6 and alternative anchors should be explored.
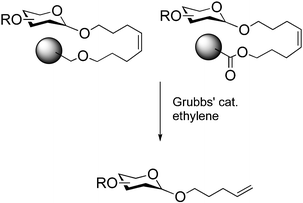 | ||
| Scheme 1 Cleavage of the octene diol linkers by olefin cross metathesis produces n-pentenyl glycosides. | ||
3.4 Monosaccharide building blocks
With an overall synthesis strategy and linker in place, oligosaccharide synthesis relies on the availability of a reliable and versatile set of differentially protected monosaccharide building blocks. After establishing that just 36 building blocks are required to access 75% of mammalian oligosaccharides ,5 we decided on the overall protecting group scheme. Benzyl ethers (Bn), pivaloyl esters (Piv), azides and N-trichloroacetyl (TCA) groups serve as permanent protective groups that will be cleaved only after completion of the synthesis. 9-Fluorenylcarbyloxymethyl (Fmoc), levulinoyl (Lev), benzoyl esters (Bz) and tert-butyldimethylsilyl ethers (TBDMS) have been chosen as temporary modes of protection. These protective groups are placed on the respective hydroxyl and amine groups around each monosaccharide ring during the synthesis of each building block. Alternative protecting group schemes can be envisioned, but for the time being the strategy described here is the first general approach. A leaving group has to be placed on the anomeric carbon to initiate the glycosylation reaction.A host of anomeric leaving groups have been developed over the past 100 years and have served well in the assembly of complex oligosaccharides .9 For the automated synthesis we chose two leaving groups: glycosyl trichloroacetimidates10 and glycosyl phosphates.11 Both anomeric leaving groups are activated by the addition of TMSOTf, that is caustic, but not toxic and result in selective and efficient glycosylations. It has to be emphasized that other anomeric leaving groups may also serve this purpose.
With the protecting and leaving group choices made, a set of monosaccharide building blocks was established (Scheme 2). Access to these building blocks is not trivial and is currently the limiting step for automated oligosaccharide assembly. Novel synthetic routes are needed. Rather than synthesizing each building block separately as has been practised to date, integrated synthetic paths that grant access to several blocks from a common starting material will be needed. Process issues such as compound crystallinity to avoid column chromatography will have to be addressed. With a set of building blocks identified, the development of synthetic pathways has become a key challenge for oligosaccharide synthesis. Soon, a first set of building blocks will become commercially available.
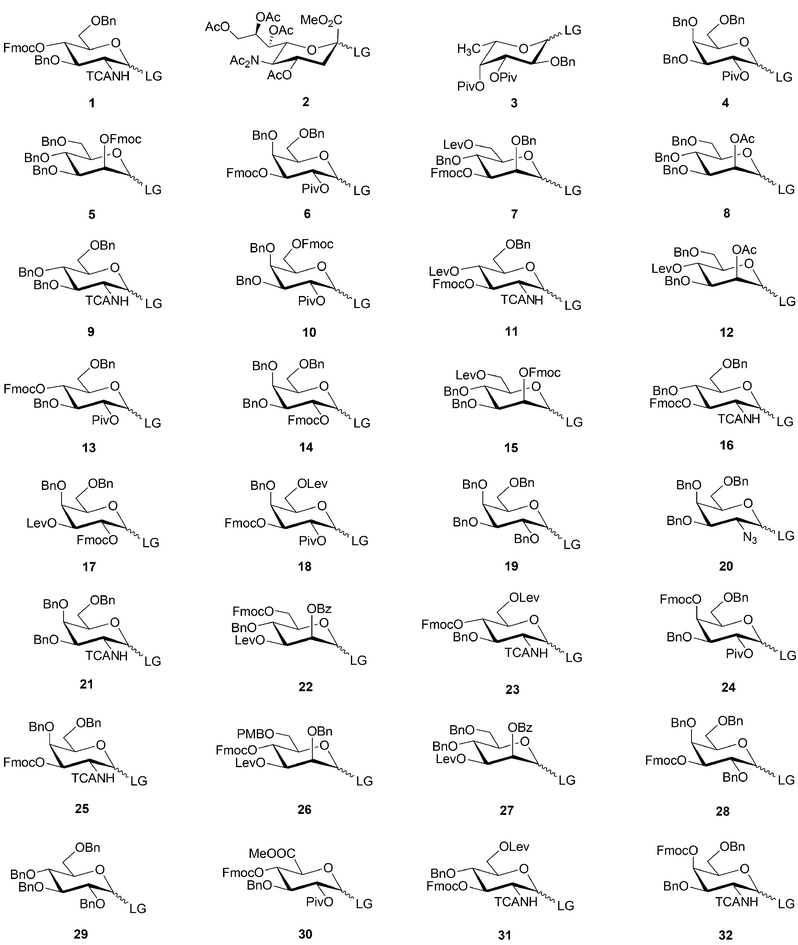 | ||
| Scheme 2 Putative monosaccharide building blocks 1–32 sorted by their relative abundance in mammalian oligosaccharides . Fmoc, Lev and PMB serve as temporary protecting groups, whereas Bn, Ac, Piv and Bz serve as permanent protecting groups. The stereochemistry and the leaving group (LG) at the anomeric center are not defined. Abbreviations: Ac: acetate; Bn: benzyl; Bz: benzoate; Fmoc: 9-fluorenylmethyl carbonyl; Lev: levaloyl; LG: leaving group; Pv: pivaloyl; TCA: trichloroacetate. | ||
3.5 Coupling and deprotection steps
The assembly of the oligosaccharides involves a two-step coupling cycle that is based on the formation of a glycosidic linkage by connecting the solid support-bound nucleophile with the anomeric position of the incoming building block. In the first step of the synthesis the nucleophile is the alcohol of the linker that anchors the growing oligosaccharide chain to the polymeric support. A solution of the building block is added to the reaction chamber where the support polymer resides before delivery of a solution of the coupling agent. A host of protocols has been tested, but currently two standard procedures have evolved.12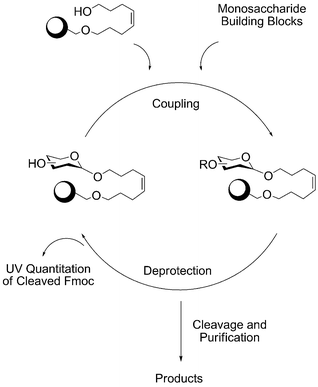 | ||
| Scheme 3 Coupling cycle of automated oligosaccharide synthesis. | ||
When glycosyl phosphate building blocks are used, five equivalents of the building block are delivered followed by the addition of five equivalents of the coupling agent TMSOTf. Following coupling at –15 °C for 15 minutes, the coupling solution is removed from the resin and the process is repeated. Double couplings involving five equivalents of building block each have been shown to result in coupling efficiencies exceeding 98%. It has to be emphasized that a significantly lower excess of building block can often result in similar coupling efficiencies albeit less reliably for different linkages. For some “difficult” couplings triple glycosylations have been employed.
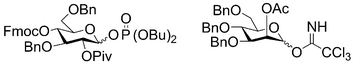 | ||
| Scheme 4 Representative glycosyl phosphate and glycosyl trichloroacetimidate building blocks. | ||
Glycosyl trichloroacetimidates have been a staple for the construction of complex carbohydrates over the past twenty years10 and are now routinely used as building blocks on solid support. As for glycosyl phosphates, five equivalents of building block are used twice, but 0.75 equivalents of the activating agent TMSOTf is sufficient to induce couplings. The temperatures for coupling reactions employing glycosyl trichloroacetimidates vary but are typically in the range of –15 °C up to room temperature.7
The construction of cis-glycosidic linkages such as α-galactosides for example requires lower temperatures in order to achieve higher selectivities. Exact coupling cycles including the number of equivalents of building block to be used as well as temperatures are currently still being optimized and generalized. Standard protocols will be forthcoming in the next year.
| Step | Function | Reagent | Time (min) |
|---|---|---|---|
| 1 | Couple | 5 eq. donor and 5 eq. TMSOTf | 21 |
| 2 | Wash | Dichloromethane | 9 |
| 3 | Couple | 5 eq. donor and 5 eq. TMSOTf | 21 |
| 4 | Wash | N,N-dimethylformamide (DMF) | 9 |
| 5 | Deprotection | 3 × 175 eq. piperidine in DMF or 5 × 10 eq. hydrazine in DMF | 34 |
| 80 | |||
| 6 | Wash | N,N-dimethylformamide (DMF) | 9 |
| 7 | Wash | 0.2 M acetic acid in tetrahydrofuran | 9 |
| 8 | Wash | Tetrahydrofuran | 9 |
| 9 | Wash | Dichloromethane | 9 |
Washing steps follow each coupling to remove all reagents from the solid phase resin. A wash in the solvent used for the coupling, is succeeded by washing steps with other solvents. Solvents that do not swell the polymeric support (e.g.methanol) shrink the resin and expel any unwanted reagents. Prior to deprotection, the resin is swollen in the deprotection solvent.
Selective removal of a specific protective group exposes the hydroxyl group as nucleophile for the next coupling. Fmoc is currently used as standard temporary protecting group for our automated syntheses.8 The Fmoc group is cleaved by treatment with a 20% solution of piperidine in DMF for three times ten minutes. Levulinyl esters have been used for temporary protection since Lev esters can be selectively cleaved in the presence of other esters by the action of hydrazine in DMF. Acetate esters that are cleaved by treatment with sodium methoxide have also been employed.7,13 Silyl ethers as well as several other groups are being pursued actively for temporary protection. This is an area of investigation that still holds many improvements in store.
3.6 Capping and tagging strategies
Most coupling steps are very high yielding, but a portion of hydroxyl groups may fail to be glycosylated during a coupling step. These very hydroxyl groups may react in subsequent steps and thereby produce deletion sequences that are difficult to separate when longer oligosaccharides are prepared. In oligonucleotide synthesis, a capping step to render unreacted nucleophiles mute is routinely employed. Capping is not common for peptide synthesis.3We introduced a capping–tagging strategy to not only cap hydroxyl groups that failed to react, but also to mark them for ready removal following cleavage from the solid support. Ester or silyl ether groups are installed onto unreacted hydroxyl groups. At the same time this procedure introduces an azide or fluorous tag (Scheme 5) that can be used to remove the deletion sequences by reaction with a scavenger resin (A-Tag) or filtration by fluorous chromatography (F-Tag).14
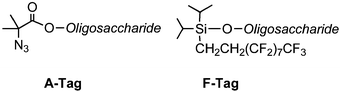 | ||
| Scheme 5 Cap-tags to facilitate removal of deletion sequences. | ||
3.7 Real-time monitoring of coupling efficiency
Rapid assessment of the success of each coupling reaction without having to sacrifice any of the materials on the support is highly desirable. By recognizing a problematic, incomplete coupling instantaneously, the synthesis can be aborted and valuable building blocks can be conserved. When Fmoc groups are cleaved, the piperidine–dibenzofulvene adduct is formed that exhibits a distinctive UV absorption. Measuring the UV absorption of the deprotection solution allows for calculation of the material bound to the polymer support.8 This procedure is routine for automated peptide synthesis and is now routinely incorporated into automated solid phase oligosaccharide synthesis. Other protective groups that carry specific UV labels such as novel silyl ethers and esters are currently under development.3.8 Release and purification of the protected oligosaccharide
Once solid support oligosaccharide synthesis is complete, the fully protected product has to be released from the polymer carrier by cleaving the linker. Scission of the linker has to occur under mild, selective conditions but ideally with complete conversion. The two octene diol linkers (Scheme 1) can be cleaved by olefin cross metathesis.7,8 The oligosaccharides are released as n-pentenyl glycosides when Grubbs' catalyst is added to the resin under an atmosphere of ethylene. The double bond at the reducing terminus can be readily modified15 to install thiol or amine groups for attachment to microarray surfaces and proteins .The solid support resin is transferred from the reaction vessel of the automated synthesizer to a round bottom flask for cleavage. Addition of the catalyst under an atmosphere of ethylene in a balloon initiates the cross metathesis that requires 16 h before the solution containing the liberated product(s) can be filtered away from the resin.
After cleavage from the resin, the result of the synthesis can be assessed by HPLC-mass spectrometry (LC-MS) analysis of a small aliquot of the cleavage reaction (Fig. 2). This analytical method is now routinely used during the development of novel coupling and deprotection chemistries as well as for the optimization of reaction procedures.
 | ||
| Fig. 2 Reverse phase HPLC spectrum of crude, fully protected Globo-H hexasaccharide after automated synthesis and cleavage from solid support (UV-absorbance at 209 nm). | ||
The fully protected oligosaccharide product is separated from any unwanted side products using reverse-phase HPLC. The absorbance of the permanent benzyl ether protective groups at 260 nm greatly facilitates monitoring. Purification of the fully protected oligosaccharides is significantly easier than the removal of closely related side products at the stage of the unprotected carbohydrate ! Thus, guided by the LC-MS results, HPLC purification protocols have been developed to separate even oligosaccharides differing in only one anomeric center as depicted for the example of Globo-H (Fig. 2).16
The separation methods are currently still being improved in order to increase recovery rates since as much as 40–50% of the desired product is lost during HPLC purification. These recovery rates are similar to those obtained for peptide purification but warrant further study.
3.9 Removal of all protective groups
Once pure, fully protected oligosaccharides have been procured, global deprotection is the last step before the final product is obtained. Depending on the protecting groups present in the oligosaccharide , several steps may be required to relieve the oligosaccharides from the masking groups. When N-trichloroacetyl groups are present, those are converted into the corresponding N-acetyl moieties by Zn/Cu and hydrochloric acid.17 At this stage, treatment of the oligosaccharide with sodium in liquid ammonia (Birch reaction), cleaves any benzyl ethers, ester and silyl ether protective groups. This highly efficient process is technically challenging for non-chemists.18Alternatively, a stepwise protocol furnishes the final product. Removal of all temporary protective groups including ester hydrolysis and silyl ether cleavage is followed by palladium catalyzed hydrogenolysis to cleave the benzyl ethers. This multistep procedure is efficient but may require more time than the actual assembly process. The hydrogenation also reduces the n-pentenyl glycoside double bond and precludes attachment of this material to the surface of microarrays or proteins .
3.10 Purification and quality control of the final product
The deprotected oligosaccharide product is purified one last time. Absence of any UV active groups complicates the analysis in stark contrast to oligonucleotides and peptides that contain residues with a specific UV absorbance. Pure oligosaccharide samples are passed through size exclusion chromatography columns and dialyzed to remove the salts accumulated during deprotection.Finally, quality control of the oligosaccharide product remains. Currently, the products are characterized in much the same way as any small organic molecule produced by synthetic means is treated: 1H NMR is of the utmost importance to ascertain anomeric purity by means of the distinct “anomeric region” of the spectrum. 13C NMR and mass spectrometry are also providing insights into structural integrity.
Synthetic peptides and oligonucleotides are now routinely analyzed by mass spectrometry and sequencing in place of an in-depth spectroscopic analysis. Ultimately, such a sitation is desirable for synthetic oligosaccharides as well. Currently, thorough analysis is essential but as the confidence in the reliability of the synthetic process increases quality control will be streamlined.
4 The automated synthesis process
After addressing the different synthetic challenges, the synthetic process was automated.12 All synthetic manipulations were integrated into a synthesis instrument. Process automation is relatively straightforward once the key decisions have been made.A modified Applied Biosystems 433 peptide synthesizer served as the first prototype instrument. This instrument uses argon pressure to drive solutions through teflon lines and selenoid valves are opened and closed under computer control to time the flow. Plastic cartridges containing the building blocks are delivered to a needle that punches a hole into the membrane covering the cartridge top. The building block solution is pushed through the syringe needle into the reaction vessel. This double walled glass vessel contains the solid phase resin on top of a glass frit. Circulation of a cooling fluid around the reactor controls the temperature of the reaction vessel. In the original design of the first prototype the circulating chiller had to be adjusted manually to the desired temperature at the appropriate times, a nuisance during longer syntheses.
 | ||
| Fig. 3 The first automated oligosaccharide synthesizer. | ||
Using a computer program the solutions and solvents are delivered in an orchestrated manner determined by the programmed coupling cycle into the reaction chamber. The entire chamber is vortexed 20% of the time to ensure complete mixing without physically harming the solid support. The solvents and reagents are removed from the resin by argon pressure that moves the solutions through the glass frit. The automated synthesis process is a reiterative execution of few, relatively simple manoeuvres (Scheme 3).
5 Examples of automated oligosaccharide syntheses
In the course of developing different aspects of the automated synthesis process a series of increasingly complex oligosaccharide targets posed as challenge for methodological improvements. Some targets were used merely to explore the formation of specific linkages or connection patterns, while others were selected for their biological relevance. The goal of all syntheses is the development of a general synthetic method to construct any oligosaccharide or oligosaccharide analogue.5.1 Synthesis of a β-glucan dodecasaccharide
The presence of a fungal β-glucan oligosaccharide triggers the soybean plant to release antibiotic phytoalexins. The response initiated by the phytoalexin β-glucans in the host soybean plant is a well-studied defence mechanism. These oligosaccharides had been synthesized previously in solution19 and on the solid support20 and served as a benchmark in our early automation endeavors.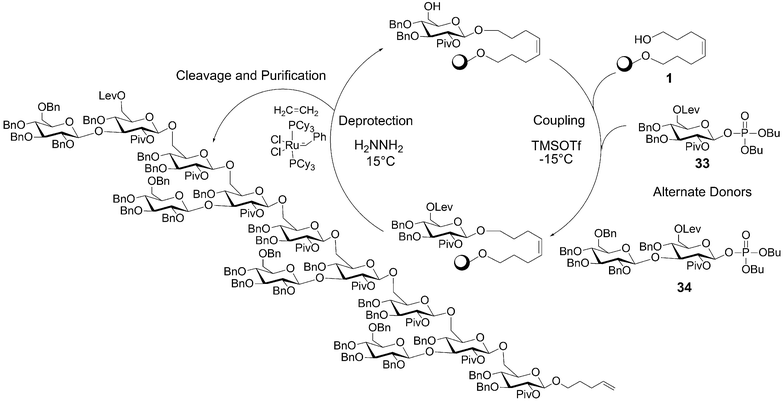 | ||
| Scheme 6 Automated synthesis of a β-glucan dodecamer. Coupling: 25 mmol resin (83 mg, 0.30 mmol g–1 loading); 5 eq. donor 33 or 34 (90 and 170 mg respectively); 5 eq. TMSOTf (1 mL, 0.125 M TMSOTf in CH2Cl2) repeated two times for 15 min each at –15 °C. Deprotection: 4 mL, 0.25 M N2H4 in pyridine–acetic acid (3 : 2) repeated two times for 15 min each at 15 °C. | ||
The synthesis of the branched β-(1 → 3)/β-(1 → 6) glucan structure was accomplished using two glycosyl phosphate building blocks. A levulinoyl ester served as 6-O temporary protecting group and the 2-O-pivaloyl group ensured complete trans-selectivity in the glycosylation reactions. Deprotection of the levulinoyl ester was achieved with a hydrazine solution in pyridine/acetic acid while the phosphate building block was activated with TMSOTf. This linear synthesis used a disaccharide such that alternating elongation with monosaccharide building blocks resulted in a branched structure.
The rapid automated assembly of this complex carbohydrate established the principle of automated synthesis and addressed all of the challenges. Still, a merely linear structure was established containing exclusively trans glycosidic linkages. Real-time monitoring of the synthesis process was not possible at that time.
5.2 Synthesis of blood group determinant and tumor-associated antigens
The Lewis blood group oligosaccharides are a family of ceramide containing glycosphingolipids decorating the exterior of healthy and disease derived cells. The Lewis Y hexasaccharide and dimeric combinations of Lewis antigens, including the Ley–Lex nonasaccharide, are tumor markers that currently are being explored as cancer vaccines.Five monomer building blocks were sufficient for the construction of the three target structures Lewis X, Lewis Y and Ley–Lex. The Fmoc group served for temporary protection of hydroxyl groups and facilitated monitoring of protecting group removal by UV. To account for branching connections via both the C3 and C4 positions of the glucosamine units, an additional levulinoyl ester was employed.
In the course of this synthesis two sets of deprotection conditions were programmed to effect Fmoc and levulinoyl ester removal. Fmoc cleavage was achieved by three exposures to piperidine (20% in DMF). Following each exposure, the solution from the reaction vessel was collected for UV analysis. Removal of the C4 levulinoyl group from the glucosamine was achieved by three exposures to a solution of hydrazine (10% in DMF).
Protected Lewis X pentasaccharide 35 was constructed in just 12 hours and was isolated in 12% yield after HPLC purification. Protected Lewis Y hexasaccharide 36 was completed in 14 hours in an isolated yield of 10%. The solid-phase synthesis of Ley–Lex nonasaccharide 37 was finished after 23 hours; cleavage from the solid support and HPLC purification produced 37 as the major product in 6% isolated yield. Fully protected oligosaccharides were all produced in overall yields comparable or better than previous solution-phase syntheses, but in a fraction of the time previously required.
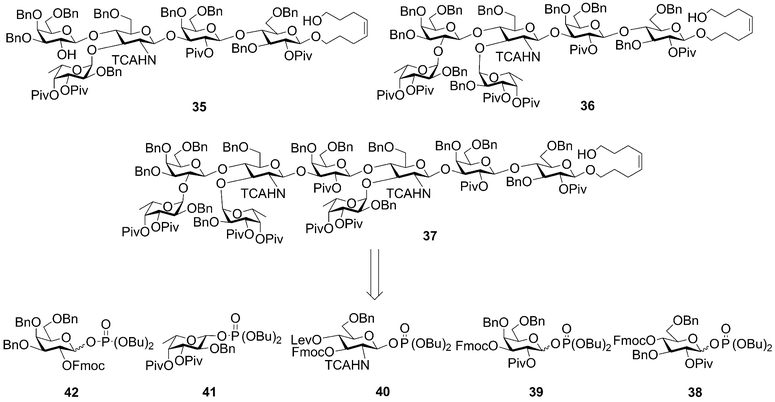 | ||
| Scheme 7 Retrosynthesis of Lewis X pentasaccharide (35), Lewis Y hexasaccharide (36), and Ley–Lex nonasaccharide (37) indicates monosaccharide building blocks 38–42. (Bn, benzyl; Bu, butyl; Piv, pivaloyl; Lev, levulinoyl; Fmoc, 9-fluorenylmethoxycarbonyl; TCA, trichloroacetyl.) | ||
5 Applications of automated oligosaccharide synthesis—an anti-GPI malaria vaccine candidate
Automated oligosaccharide synthesis holds great potential to accelerate the assembly of oligosaccharide antigens to be used in conjugate vaccines. Our laboratory is currently exploring novel vaccine candidates against malaria,21 anthrax,22 leishmaniasis23 and several other viral and bacterial diseases. To illustrate the potential of the method, the impact of automated synthesis on the development of a malaria vaccine candidate is briefly summarized.Malaria infects currently 5% of the world's population, resulting in 100 million clinical cases and 3 million deaths per year. As current treatments are facing increasingly resistant parasites a malaria vaccine would be of great benefit. Much of malaria's mortality is due to an inflammatory cascade initiated by a glycosylphosphatidylinositol (GPI) malarial toxin , released when parasites rupture the host's red blood cells. We demonstrated that anti-GPI vaccination can prevent malarial pathology in an animal model. Mice immunized with chemically synthesized GPI bound to a carrier protein were substantially protected from death caused by malaria parasites. Between 60 and 75% of vaccinated mice survived, compared to a 0 to 9% survival rate for unvaccinated mice.21 While the solution-phase synthesis of 43 allowed for the procurement of much larger amounts of GPI than through isolation of natural GPI, faster access to 43 was important for the further development of anti-GPI malaria vaccine candidates.
The α linkage between inositol and glucosamine presented too great a challenge to a fully automated approach in 2002. Thus, GPI 43 was synthesized via a semi-automated approach. Disaccharide 45 was prepared in solution and tetra-mannosyl fragment 44 was assembled on the automated synthesizer.24 The two fragments were to be joined to fashion a hexasaccharide for further elaboration to vaccine candidate 43. The protein conjugated GPI is currently in preclinical evaluation as a malaria vaccine candidate.
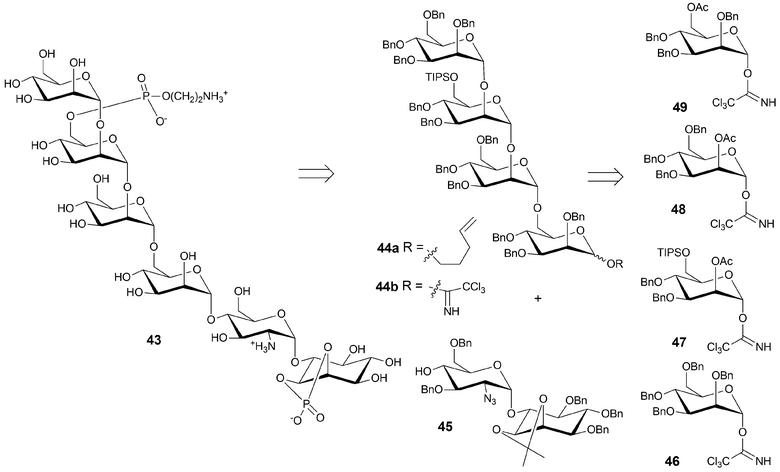 | ||
| Scheme 8 Retrosynthesis of the GPI malaria vaccine candidate. Disaccharide 45 is prepared by solution phase methods, tetrasaccharide 44a by automated synthesis. | ||
6 Conclusions and outlook
The state-of-the-art in automated oligosaccharide synthesis has been summarized. The key challenges en route to a general approach to oligosaccharide synthesis have been identified and initial solutions have been found. Procurement of monosaccharide building blocks, selection of an appropriate solid support, a versatile and robust linker, a quick and effective coupling cycle as well as a synthesis instrument have been achieved. Based on these advances many complex oligosaccharides can be rapidly assembled in good yield. Not all linkages are yet accessible and difficult sequences remain.The feasibility of the general principle is now established and the remaining specific problems will have to be addressed. Building blocks will become commercially available very soon together with a second generation synthesizer. Thus, access to complex carbohydrates will be greatly simplified and enable advances in glycobiology and medical applications of carbohydrates .
Improvements in many aspects of the synthetic process can be envisioned and the importance of automated oligosaccharide synthesis to glycobiology and medicine warrants the attention of synthetic chemists to make carbohydrate synthesis a routine process for non-specialists. Even when automated synthesis will render the majority of syntheses routine in the near future, the multitude of possible carbohydrate sequences will pose challenges for organic chemists for years to come.
References
- J. Meienhofer, E. Schnabel, H. Bremer, O. Brinkhoff, R. Zabel, W. Sroka, H. Klostermeyer, D. Brandenburg, T. Okuda and H. Zahn, Z. Naturforsch., 1963, 18b, 1120 Search PubMed.
- K. L. Agarwal, H. Buchi, M. H. Caruthers, N. Gupta, H. G. Khorana, K. Kleppe, A. Kumar, E. Ohtsuka, U. L. Rajbhandary, J. H. Vandesande, V. Sgaramel, H. Weber and T. Yamada, Nature, 1970, 227, 27.
- (a) R. B. Merrifield, Angew. Chem., Int. Ed. Engl., 1985, 24, 799 CrossRef; (b) M. H. Caruthers, Science, 1985, 230, 281 CrossRef CAS.
- (a) Preparative Carbohydrate Chemistry, ed. S. Hanessian, Marcel Dekker: New York, 1997 Search PubMed; (b) The Organic Chemistry of Sugars, ed. D. E. Levy and P. Fügedi, CRC Press, Boca Raton, 2006 Search PubMed.
- D. B. Werz, R. Ranzinger, S. Herget, A. Adibekian, C.-W. von der Lieth and P. H. Seeberger, submitted for publication.
- P. H. Seeberger and W.-C. Haase, Chem. Rev., 2000, 100, 4349 CrossRef CAS.
- R. B. Andrade, O. J. Plante, L. G. Melean and P. H. Seeberger, Org. Lett., 1999, 1, 1811 CrossRef CAS.
- K. R. Love and P. H. Seeberger, Angew. Chem., Int. Ed., 2004, 43, 602 CrossRef CAS.
- K. Toshima and K. Tatsuta, Chem. Rev., 1993, 93, 1503 CrossRef CAS.
- R. R. Schmidt and W. Kinzy, Adv. Carbohydr. Chem. Biochem., 1994, 50, 21 CAS.
- O. J. Plante, E. R. Palmacci, R. B. Andrade and P. H. Seeberger, J. Am. Chem. Soc., 2001, 123, 9545 CrossRef CAS.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523 CrossRef CAS.
- M. C. Hewitt and P. H. Seeberger, Org. Lett., 2001, 3, 3699 CrossRef CAS.
- E. R. Palmacci, M. C. Hewitt and P. H. Seeberger, Angew. Chem., Int. Ed., 2001, 40, 4433 CrossRef CAS.
- T. Buskas, E. Soderberg, P. Konradsson and B. Fraser-Reid, J. Org. Chem., 2000, 65, 958 CrossRef CAS.
- D. B. Werz, B. Castagner and P. H. Seeberger, J. Am. Chem. Soc., 2007, 129, 2770 CrossRef CAS.
- N. El-Abadla, M. Lampilas, L. Hennig, M. Findeisen, P. Welzel, D. Müller, A. Markus and J. van Heijenoort, Tetrahedron, 1999, 55, 699 CrossRef.
- I. Schon, Chem. Rev., 1984, 84, 287 CrossRef CAS.
- P. Fugedi, W. Birberg, P. J. Garegg and Å. Pilotti, Carbohydr. Res., 1987, 164, 297 CrossRef.
- K. C. Nicolaou, N. Winssinger, J. Pastor and F. DeRoose, J. Am. Chem. Soc., 1997, 119, 449 CrossRef CAS.
- L. Schofield, M. C. Hewitt, K. Evans, M. A. Siomos and P. H. Seeberger, Nature, 2002, 418, 785 CrossRef CAS.
- D. B. Werz and P. H. Seeberger, Angew. Chem., Int. Ed., 2005, 44, 6315 CrossRef CAS.
- X. Liu, S. Siegrist, M. Amacker, R. Zurbriggen, G. Pluschke and P. H. Seeberger, ACS Chem. Biol., 2006, 1, 161 Search PubMed.
- M. C. Hewitt, D. A. Snyder and P. H. Seeberger, J. Am. Chem. Soc., 2002, 124, 13434 CrossRef CAS.
Footnote |
| † The HTML version of this article has been enhanced with colour images. |
| This journal is © The Royal Society of Chemistry 2008 |