Enzyme assays
Jean-Louis
Reymond
*,
Viviana S.
Fluxà
and
Noélie
Maillard
Department of Chemistry and Biochemistry, University of Berne, Freiestrasse 3, Berne, 3012, Switzerland. E-mail: jean-louis.reymond@ioc.unibe.ch; Fax: +41 31 631 80 57; Tel: +41 31 631 43 25
First published on 17th October 2008
Abstract
Enzyme assays are analytical tools to visualize enzyme activities. In recent years a large variety of enzyme assays have been developed to assist the discovery and optimization of industrial enzymes, in particular for “white biotechnology ” where selective enzymes are used with great success for economically viable, mild and environmentally benign production processes. The present article highlights the aspects of fluorogenic and chromogenic substrates, sensors, and enzyme fingerprinting, which are our particular areas of interest.
 Jean Louis Reymond | Jean-Louis Reymond was born in Switzerland in 1963. He graduated from the ETH in Zürich in 1985 and received his PhD from the University of Lausanne in 1989. In 1990 he joined the Scripps Research Institute first as a post-doctoral fellow with R. A. Lerner, then as an assistant professor. In 1997 he moved to the University of Berne, Switzerland, as an associate professor, and became a full professor in 1998. His research touches on three areas of bioorganic chemistry: (1) high-throughput screening assays for biocatalysis; (2) artificial protein design with peptide dendrimers; and (3) small molecule drug discovery. |
 Viviana Fluxá | Viviana Fluxá was born in Santiago de Chile in 1980. She received her MSc in chemistry from Fribourg University in 2006. While at Fribourg, she studied diethylstilbestrol derivatives in the laboratory of Christian Bochet. In March 2006, she joined the Reymond group at the University of Berne in the Department of Chemistry and Biochemistry. Her current research interests include protease profiling with FRET peptides and exploring the biological activity of cyclic peptides libraries. |
 Noélie Maillard | Noélie Maillard, born in Delémont (Switzerland) in 1981, studied chemistry at the University of Neuchâtel, and complete her diploma thesis in inorganic chemistry at the Institute of Chemistry, University of Neuchâtel with Prof. Dr Thomas R. Ward. She started her PhD in 2006 under the supervision of professor Jean-Louis Reymond in the Department of Chemistry and Biochemistry at the University of Bern (Switzerland). Her current research interests include the development of new methods for the screening of peptide dendrimer libraries and its application in different reactions types. |
1. Introduction
Enzyme assays are analytical tools to visualize enzyme activities. In recent years a large variety of enzyme assays have been developed to assist the discovery and optimization of industrial enzymes, in particular for “white biotechnology ” where selective enzymes are used with great success for economically viable, mild and environmentally benign production processes.1,2 In this context the enzyme assays serve to screen collections of enzymes available from strain collections, metagenomic libraries and libraries of mutant enzymes obtained by random or directed mutagenesis from known enzymes. This type of screening must be distinguished from genetic selection experiments, in which a mutation/selection protocol is set up to allow for the survival of microorganisms producing an active enzyme, e.g. by complementation of a biosynthetic pathway in an auxotrophic bacterial or yeast strain. Both high-throughput screening and genetic selection are viable protocols for discovering and improving enzymes.3–12 Important developments of enzyme assays are also constantly occurring in relation to drug discovery efforts, medical diagnostics and in the area of cellular and tissue imaging. Enzyme assays are also used in enzyme model studies.13The majority of enzyme assays are developed to test isolated enzymes or enzyme containing samples such as culture suspensions in 96-well microtiter plates or similar parallel liquid phase systems. The simplest and most practical enzyme assays are based on synthetic substrates that release a colored or fluorescent product upon reaction or induce a directly detectable change in solution such as a precipitation. Many such substrates are commercially available and often serve as reference substrates to determine absolute activities of enzyme samples in Units. Enzyme reactions may also be assayed using indicators which respond indirectly to product formation or substrate consumption. The indicator may be as simple as a pH-indicator and as complex as a functionalized nanoparticle. Many assays are also based on analytical instruments such HPLC, GC, MS, NMR or IR spectrometers. These instruments often allow access to reaction parameters not otherwise accessible such as enantioselectivity and therefore play a critical role in biocatalysis for the discovery and optimization of selective enzymes by directed evolution. Enzyme assays have been the subject of a recent volume providing an overview in the perspective of biocatalysis.14 The present article highlights the aspects of fluorogenic and chromogenic substrates, sensors, and enzyme fingerprinting, which are our particular areas of interest.
2. Chromogenic and fluorogenic substrates
Fluorogenic and chromogenic enzyme substrates form the cornerstone of enzyme assay technology. They incorporate a chromophore whose absorbency or fluorescence properties change as a result of the enzyme reaction. The key advantage of these substrates is that the assay is very simple and the signal produced is directly related to the enzyme-catalyzed reaction. If the colored or fluorescent product is soluble, the assay is well-suited for microtiterplate based assays . On the other hand, if the product is insoluble in the reaction media, the substrate can be used for screening bacterial cultures on agar plates. Almost all examples to date focus on a small family of fluorophores and chromophores, in particular umbelliferones, nitrophenols, fluoresceins , rhodamines and BODIPY dyes, all of which are relatively large aromatic groups which tend to influence both substrate binding (stronger binding), catalytic turnover (reactivity may be lowered or absent compared to non-labeled substrates), and solubility. Encouraging recent reports by Nau and co-workers suggest that new, non-aromatic fluorophores might provide an alternative for FRET type assays .15,16 Alternatively various chemistries can be implemented to separate the enzyme reactive group from the fluorophore in a variety of reaction types, as exemplified with the Clips-O method below.17 Further developments in these directions are still possible and will mark the future of this type of assays .2.1 Phenolate and aniline release
Indican is a chromogenic glycosidase substrate belonging to a family of natural product glycosides found in plants such as Isatis tinctoria and Polygonum tinctorum, the traditional source to produce indigo is by fermentation.18 Cleavage of the glycosidic bond forms an unstable hydroxyindole intermediate, which dimerizes oxidatively at air to form indigo as a blue precipitate. A number of enzyme substrates have been designed following this natural product example. For instance, numerous glycosides of fluorescent or colored phenols are used to test glycosidases, e.g.nitrophenyl β-galactoside (1) for detection of β-galactosidase activity, p-nitrophenyl caproate (2) as a chromogenic lipase substrate, and the nitrophenyl octyl ether 3 to detect cytochrome P450 activity19,20 (Fig. 1). Note that the yellow color of nitrophenolate is only visible above pH 7. Naphthols can be detected indirectly by a secondary reaction with diazonium salts to form azo-dyes, a principle used in cytochemistry to test esterase activities in tissue samples with naphthyl acetate,21 and recently adapted to assay aldolase antibodies in agar plates.22 | ||
| Fig. 1 Chromogenic enzyme substrates releasing nitrophenolate. | ||
The range of phenol release substrates can be extended using indirect release mechanisms, as we first demonstrated with the alcohol dehydrogenase enzyme substrate 5 (Fig. 2). This chiral secondary alcohol is oxidized by the enzyme to form the corresponding ketone6, which is unstable and undergoes a β-elimination reaction catalyzed by bovine serum albumin to produce the blue fluorescent umbelliferone anion 7.23,24 The signal is only visible above pH 7 where the product exists as a fluorescent anion. The same principle allowed substrates for aldolase catalytic antibodies25–27 and proline-type catalysts,28,29transaldolases,30transketolases,31 and lipases.32 A related scheme involving an intermediate hemiacetal provides various lipase and esterase substrates,33–35 as wells as a fluorogenic substrate for Baeyer–Villiger monoxygenases (BVMO) in the form of the 2-aryloxyketone 8,36via the intermediate lactone9 which may be considered as a lactonase-type probe, although it is quite unstable and spontaneously hydrolyzes in the whole cell conditions used to assay BVMO. Substrate 8 and further analogs are currently the only available fluorogenic substrates for BVMO and they are readily obtained by alkylation of various chloroketones with umbelliferone.
 | ||
| Fig. 2 Fluorogenic enzyme substrates with indirect release of umbelliferone. | ||
The ketone or aldehyde leading to β-elimination may also be formed by chemical oxidation of a primary 1,2-diol or 1,2-aminoalcohol reaction product in the so-called Clips-O™ substrates (Fig. 3).17 A typical example is epoxide10, which provides a highly reliable probe of epoxide hydrolase activity for screening in microbial cultures.37 The substrate is hydrolyzed by epoxide hydrolases to form the 1,2-diol 11. In the presence of sodium periodate (NaIO4), the diol is rapidly and quantitatively oxidized to form aldehyde12, which undergoes a β-elimination to form umbelliferone 7 as above. Further examples include thermally stable lipase, amidase and phosphatase substrates,38–41 an HIV-protease substrate,42 and a ceramidase substrate.43
 | ||
| Fig. 3 The Clips-O™ epoxide hydrolase substrate. | ||
Further strategies for indirect phenolate release include intramolecular carbamate or carbonate cyclization. The aldolase antibody substrate 13 features an interesting recent example of this approach (Fig. 4).44 This substrate undergoes sequential retroaldolization, β-elimination and intramolecular carbamate cyclization to release a catechol which can be made visible by formation of the insoluble black precipitate 14 in the presence of Fe(III). Related carbonate and carbamate cyclization are also used in substrate for epoxide hydrolases45 and acylases.46,47
 | ||
| Fig. 4 A chromogenic substrate for aldolase catalytic antibodies. | ||
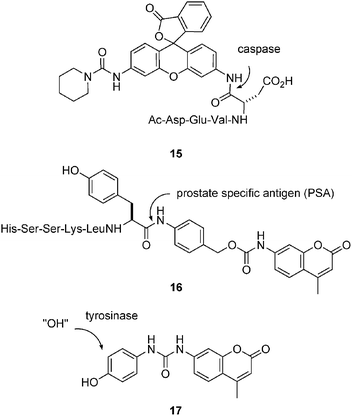
Anilines such as nitrophenyl aniline and rhodamine are frequently converted to amides to form chromogenic or fluorogenic amidase and protease substrates, as in the protease probe 15 designed for caspases (Fig. 5).48 The aniline can also be released indirectly, for example via a “self-immolative” quinone methide mechanism as in the fluorogenic peptide substrate 16 for the prostate specific antigen (PSA),49 or by spontaneous hydrolysis of a urea following oxidation of a phenol to a catechol and subsequently an orthoquinone as in the recently reported tyrosinase substrate 17.50
2.2 FRET
Many enzyme assays are based on FRET (“Förster or Fluorescence Resonance Energy Transfer”) as detection principle, for instance to measure hydrolytic reactions separating fluorophore from quencher in the case of proteases,15,16,51–56cellulases,57 and lipases,58–60 and in the synthetic direction for fucosyl transferases.61Aldehyde18 features an interesting use of FRET, in which addition of a nucleophile to the aldehyde such as an antibody catalyzed aldol addition to form 19 (Fig. 6) removes the intramolecular quenching effect.62–64 Fluorescence of a label can also be modulated by proximity effects such as medium effects. For example, in an aminonitrobenzofurazane-labeled γ-cyclodextrin reported as an α-amylase fluorogenic substrate, cleavage of the cyclodextrin ring by the amylase exposes the fluorophore to water and reduces fluorescence intensity.65 Fluorescent substrates for kinase substrates have been recently reviewed.66 In a recent example, a luminogenic probe was developed for tyrosinephosphorylation based on a short peptide sequence containing an iminodiacetate moiety near the site of phosphorylation. In response to kinase activity, the probe provides a strong luminescence enhancement, resulting from the increased ability of the probe to bind and sensitize Tb3+ and Eu3+ ions upon phosphorylation.67 Fluorescence modulations by aggregation, dilution or phase change are also related to FRET substrates. Thus, lipases can be assayed with 1,3-dioleoyl-2-(4-pyrenylbutanoyl)glycerol in the presence of lipoproteins and albumin .68Ester hydrolysis releases the pyrene carboxylate, which then binds to albumin , resulting in a fluorescence increase. The commercially available FITC-conjugates of casein are used as protease substrates, whereby proteolysis removes the autoquenching and leads to stronger fluorescence of the fluorescein chromophores.69 Recently reported protease-sensitive nanofibers consisting of aggregated β-sheet forming peptides also rely on dilution-induced release of autoquenching upon proteolytic cleavage.70 | ||
| Fig. 6 A FRET probe for following nucleophilic addition reactions to aldehydes. | ||
2.3 Other approaches
The enzyme reaction may also directly modify the chromophore itself as a detection mechanism. For example microbial growth can be monitored by following the activity of nitroreductases that reduce the nitro group of various 7-nitrocoumarins such as 20 to form the corresponding 7-aminocoumarins 21 as a fluorescent products (Fig. 7).71Peroxidases react with a variety of aromatic compounds to form colored products, and indoles are substrates for cytochromes, forming indigo upon secondary oxidation of the hydroxyindole primary product.72Alcohol dehydrogenase and aldolases can be screened using 6-methoxynaphthaldehyde and related substrates.27 The primary amine in substrate 22 is oxidized by monoamine oxidase for form an aldehyde, ultimately forming the fluorescent indole23 by condensation of with the anilineamino group.73 The recent report of an assay for fatty acid dehydrogenases based on chromophore modification by the enzyme is also of interest.74 Luminescent products such as luciferase substrates (bioluminescence)75 or 1,2-dioxetanes (chemiluminescence )76 released from enzyme reactions are also of interest.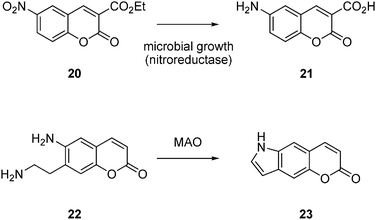 | ||
| Fig. 7 Fluorogenic substrates with direct modification of the chromophore upon reaction. | ||
Substrate dissolution or product precipitation or crystallization may sometimes form the visible signal, without formation of a colored product. Classically, microbial cultures producing active lipases will form a clearing zone on agar plates prepared with tributyrin. Certain polymer degrading enzymes can be screened by recording dissolution of an insoluble substrate, in particular cellulases.77 More sophisticated systems were recently investigated, such as the formation of hydrogels78,79 or dipeptide nanotubes.80 In a recent example, incorporating an ester-containing substrate in a liquid crystal allowed to assay a lipase through its action on the alignment layer.81
2.4 Fluorescence activated cell sorting (FACS)
Fluorescent and/or fluorogenic substrates have been used to directly identify cells expressing active enzymes in liquid culture based on fluorescence-activated cell sorting (FACS), which provides a particularly meaningful application of such substrates in high-throughput screening.82,83 For example mutants of the protease OmpA were displayed on the surface of E. Coli cells and a cell-surface adherent fluorogenic substrate for protease was added to the culture.84,85 Cells expressing an active protease became fluorescent and could be sorted out by FACS, which allowed discovering mutants with a 30-fold improved activity. The same technique has been used for sorting microemulsion droplets where an enzyme gene and expression machinery are compartmentalized together with a fluorogenic substrate system.86–88 Both methods allow to screen very large numbers of variants (>107). Glycosides labeled with Bodipy dyes were used to screen sialyl transferases in living cells, relying on the fact that this substrate but not its sialylated product is cell permeable. Thus, after washing unreacted substrate, cells containing an active enzyme retained the fluorescent product and were separated by fluorescence activated cell sorting.89 A high-throughput screening technique for the identification and isolation of enantioselective enzymes based on FACS has been developed on the idea of labeling each of the two enantiomers with a different fluorescent dye. This method allows to evaluate 108 cells (and number of clones generated) within a few hours.903. Indicator assays
A variety of relatively simple chemosensor systems based on chromogenic or fluorogenic reagents can convert a chemical transformations into a detectable signal, often through a functional group specific reaction or by a separation effect. Biosensors binding to either substrate or product may be used similarly. Such indicator assays can be used to assay reactions of specific, unlabeled substrates, which is necessary whenever a very specific reaction is being optimized. The main drawback of indicator assays is that they are often sensitive to interferences (e.g. other events than catalytic turnover may produce a signal, or turnover may be masked) and may require narrow assay conditions that render them incompatible with certain enzymes. In addition the reporter chemistry may be rate limiting, which prevents their use for kinetic studies. These potential limitations may be overcome for a high-throughput screening application by using proper controls and relying on an endpoint measurement rather than on measuring a reaction rate. As long as they can test authentic substrates, indicator assays will remain one of the best option for enzyme assays in the future.3.1 Enzyme-coupled assays
One of the most straightforward methods to render an enzyme-catalyzed reaction detectable consists in further converting the reaction product by a second enzyme to form a second product and so on, until one of these follow-up reactions produces a detectable signal. The vast majority of enzyme-coupled assays involve an oxido-reductase, usually an alcohol dehydrogease (ADH), using NAD or NADP as cofactors , as exemplified recently in the selection of enantioselective aldolase mutants.91,92 Recent interesting examples include the determination of the enantioselectivity of lipases and esterases using acetate ester substrates on the basis of an acetate detection kit.93 The assay was used recently to select a double mutant of Bacillus Subtilisisesterase with inverted enantioselectivity towards acetylated tertiary alcohols.94 The enantioselectivity of ADH enzymes has also been used to determine the enantioselectivity of alcohols formed by the catalyzed addition of diethyl zinc to aldehydes,95 for a transition metal catalyzed epoxide opening reactions96 and the conversion of benzaldehyde and acetyl cyanide to the corresponding acetylated cyanohydrins.97,98 A pair of enzymes was described that are able to differentiate the 1,2-hexanediol antipodes Lactobacillus kefir alcohol dehydrogenase, highly S selective, and horse liver alcohol dehydrogenase, modestly S selective. This allows one to obtain simultaneous enantioselectivity readouts on two distinct substrates for the Co(III)-salen-mediated hydrolytic kinetic resolution of epoxides.99 It has been shown that only partially enantioselective dehydrogenases are generally sufficient for the ee-determination of chiral alcohols.100The reduction of hydrogen peroxide to water catalyzed by peroxidases occurs with oxidation of various chromogenic dyes, such as ABTS.101Horseradish peroxidase (HRP) and H2O2 as oxidant was used to detect the naphthol product formed by hydroxylation of naphthalene by a P450cam monoxygenase.102,103 Turner and co-workers have developed the reaction to follow the activity of an enantioselective microbial monoamine oxidase (MAO-N) on amines. The MAO-N produces hydrogen peroxide as a byproduct, which is revealed by a peroxidase and 3,3′-diaminobenzidineas a chromogenic reagent.104,105 In a related experiment, the same group has recently reported a novel high-throughput screening method to determine both the rate and the enantioselectivity of asymmetric ketone reduction by ketoreductases (KRED) in the presence of an R-selective alcohol oxidase, and optionally horseradish peroxidase (HRP) and ABTS (Fig. 8).106 An R-selective KRED induces multiple turnovers producing many equivalents of NADP+, or ABTS+ if HRP is added to the assay . By contrast, an S-selective KRED results in only one turnover of NADPH since no R-alcohol is produced as an oxidase substrate.
 | ||
| Fig. 8 Dual-wavelength spectrophotometric tracking of two chromogens (NADP+ and ABTS) for the determination of ketoreductase (KRED) activity and enantioselectivity. | ||
Hydrolytic enzymes such as glycosidases have been used as secondary enzymes to follow the production of chromogenic substrates from non-reactive precursors through a primary enzyme such as a glycosidase, glycosyltransferase, glycosynthases.107–110 Similarly, proteases have been used to track prolylcis–transisomerases,111–113kinases,114,115 and peptide deformylase.116Luciferases produce light by oxidation under consumption of ATP, oxygen and an oxidizable substrate such as luciferin or an aldehyde and reduced flavin. A number of assays have been reported that use a luciferase as secondary enzyme to screen a reaction producing one of the luciferase substrates as a product. The oldest method to quantify ATP relies on firefly luciferase and luciferin.117 Luminescent bacteria have been used to monitor the activity of aldolase catalytic antibodies releasing nonanal.118 The same phenotypic screen was recently used to discover new oxido-reductase enzymes, whereby the substrate spectrum of the bacterial luciferase was explored and extended.119Firefly luciferase was recently used as the substrate undergoing refolding by the chaperone Hsp90.120 An assay for monoamine oxidases A and B was recently reported where the phenol-type substrate for the luciferase is released by β-elimination of a primary aldehyde oxidation product. For inhibitor assay , it should be noted that typically 1–3% of compound libraries inhibit luciferase activities.121
3.2 Functional group selective reagents
Kazlauskas and co-workers have exploited the well-known fact that ester hydrolysis lowers the pH of the reaction medium to develop a colorimetric assay to screen lipases and esterases for enantioselectivity.122,123 pH-indicators have also been used in the context of sol–gel encapsulated enzymes.124 Recently a pH-indicator assay was reported for screening glycosyltransferases based on the acidification induced by glycosyl transfer from UDP-GalNAc,125 and from glycosyl fluorides.126 Functional group selective chromogenic or fluorogenic reagents have been used to detect enzyme activities, including reagents for amines formed by amidases,127,128ammonia from nitrile hydrolysis,129aldehydes from vinyl ester cleavage130,131 and from periodate cleavage of epoxide hydrolysis products,132,133epoxides,134thiols from thiolactones,135,136 phosphorylated peptides from kinase reactions,137–139 UDP from glycosyltransferases,140 dimedone from lipase reactions,141 and amino acids from amidases.142 For example, amino acids can be detected by fluorescence in real-time using the non-fluorescent Cu(II) complex of calcein24. The amino acid displaces Cu(II) from calcein, whereby calcein regains its green fluorescence. This simple assay is suitable to screen acylases, amidases and proteases, as illustrated for the case of aminopeptidase, for which no other fluorescence assay is known (Fig. 9).143,144 | ||
| Fig. 9 A fluorescence assay for aminopeptidase using a calcein sensor. | ||
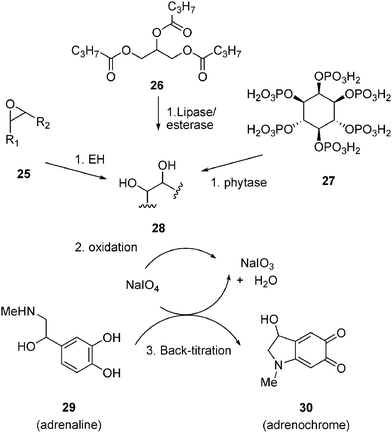
Adrenaline serves as the reporter for detecting for 1,2-diols (28) and 1,2-aminoalcohols by back-titration of sodium periodate (Fig. 10). Adrenaline (29) reacts quantitatively and rapidly with sodium periodate to form the deep red adrenochrome (30), a reaction which is does not take place if a 1,2-diol or 1,2-aminoalcohol has already consumed the periodate oxidizing agent. This provides a practical end-point assay for a variety of hydrolytic enzymes.145,146 The adrenaline assay can be used to screen the enzymatic hydrolysis of epoxides (25) by epoxide hydrolases, triglycerides such as tributyrin26 or various acetate esters147 by lipases and the dephosphorylation of phytic acid (27) by phytases. The adrenaline test for epoxide hydrolase was recently adapted to an automated format for cell culture.148Sodium periodate also decolorizes certain chromophores and the assay was used to screen epoxide hydrolases using fluorescein as periodate reporter.149
 | ||
| Fig. 10 The adrenaline test for enzymes. | ||
In an unusual yet very practical example of indirect sensing of an enzyme reaction, epoxide hydrolase activity on butane-oxide was detected in E. coli cultures on agar-plate using Safranin O.150 Oxidation of the 1,2-diol product by E. Coli modified the membrane potential and lead to accumulation of the red dye in the colonies producing active enzyme, allowing for direct selection.
3.3 Bio- and nano-sensors
While the above example apply simple chemical reactivity principles in the context of enzyme assays , some sensor systems rely on more sophisticated detection schemes with biosensors , vesicles and gold nanoparticles, a type of assay which may be assigned to the “nano” world. The first notable example concerns antibodies for the so-called cat-ELISA assay developed in the context of catalytic antibody research.151–156 Further examples ADP selective aptamers for kinase sensing,157 or lectins for testing glycosyltransferases on microarray displayed substrates.158–161 Vesicles containing synthetic multifunctional pores (SMPs) have been used for enzyme assays .162–166 The SMPs are incorporated in the vesicle membrane and serve as channels for the escape of fluorescein, which results in a fluorescence increase since dilution removes autoquenching. Substrate/product ratios in an enzymatic reaction can be monitored whenever substrate and product differentially modulate the flow of fluorescein through the SMPs. Gold nanoparticles have been used for a variety of enzyme assays . Solutions of Au(III) (HAuCl4) are reduced by NAD(P)H, catechols, or thiols, to form colored suspensions of gold nanoparticles, allowing a colorimetric assay for enzymes such as lactate dehydrogenase,167 acetyl choline esterase168 and tyrosinase.169 Aggregation of gold nanoparticles in suspension induces a color change from red to purple grey, allowing assays for kinases mediated by a biotin–avidin aggregation trigger,170,171 for proteases using a synthetic peptide with a protease-specific sequence flanked by a pair of S-acetyl cysteine residues,172 for alkaline phosphatase173 and ATP sensing174 using charge induced aggregation of nanoparticles, and for endonucleases using two sets of gold nanoparticles coated with complementary single-strand DNA substrates.1754. Fingerprinting
The information content of screening assays can be increased by analyzing multiple substrates simultaneously. The first multi-substrate analysis method was the APIZYM system in the 1960s.176–180 In this method a set of 19 or 32 different enzyme substrates, including chromogenic substrates for lipases and esterases, aminopeptidases, chymotrypsin, trypsin, phosphatases, sulfatases and β-galactosidases, is used in a multi-well format to analyze microbial cultures. The analysis produces a reactivity pattern indicating which enzymes are produced by the microorganism. This information serves to identify the microorganism, and forms the basis of microbial strain identification in hospitals. Enzyme fingerprinting proposes to focus the analysis on a single enzyme using a series of structurally related substrates to characterize its selectivity.181 The data may be used to identify reactive substrates, or for functional classification of the enzymes.40 Generally one analyzes reactivity fingerprints across a series of similar substrates, such as series of peptides , however it should be mentioned that certain enzymes may also catalyze reactions of different functional groups, which is called substrate promiscuity.182,183 Fingerprinting with multiple substrates should be distinguished from the so-called activity-based protein profiling, which is based on labeled suicide enzyme inhibitors to identify reactive enzymes in protein mixtures.184–192 The critical problem of enzyme fingerprinting is to provide a reliable method for producing the data, and most research efforts are currently still focusing on this problem. The true potential of fingerprinting will be realized when reagents will be available for measuring multi-dimensional datasets reporting enzyme selectivities and substrate preferences as readily as what is currently possible with single substrates.4.1 Parallel assays in microtiter plates
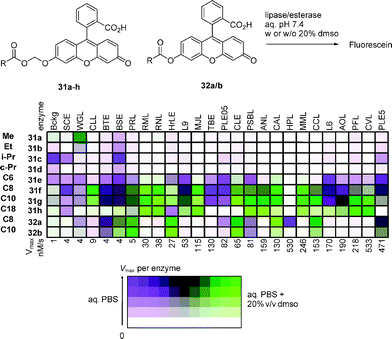
Assays with multiple substrates in microtiterplates have been mostly used for hydrolase profiling.147,193–195 For example, fingerprinting lipases and esterases with 16 periodate-activated fatty acid ester substrates in both enantiomeric forms showed, as expected, that lipases were more active on long-chain substrates, while esterases preferentially cleaved short-chain substrates. More surprisingly, a selectivity for intermediate chain length substrates was apparent as the second principal component of the observed diversity, an information not otherwise accessible (Fig. 11).40 Using a related series of water-soluble esters31a–e and 32a/b derived from fluorescein, fingerprinting showed that lipases and esterases differ from one another by their reactivity in pure aqueous buffer vs. buffer containing 20% dimethyl sulfoxide as co-solvent. Esterases were more active in aqueous environment, while lipases required the cosolvent for highest activity, as shown by the color coded pattern (Fig. 12).35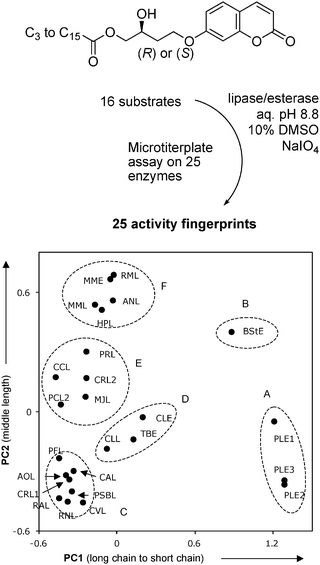
Microtiter-plate based assays have also been used to analyze proteases using multiple peptide substrates to determine the optimal substrate,196–199 in particular with positional scanning libraries of millions of fluorogenic peptidyl coumarinamides as series of 20 or 400 different substrate mixtures.200–208
4.2 On-bead assays
Meldal and co-workers reported the use of synthetic combinatorial libraries of millions of synthetic peptides as FRET substrates to analyze protease reactivity on solid support,209–211 a very practical method still under further development today.212,213 One of the difficulties in the analysis is the necessity to introduce a fluorescent label on the cleaved peptide, which may reduce the reactivity of the protease. We recently reported an assay for on-bead proteolysis of a solid-supported combinatorial library of N-acetylated non-tagged octapeptides AcL (Fig. 13).214 After proteolysis, the free N-termini are simply stained by reductive alkylation with a tagged aldehyde, and the stained beads are analyzed.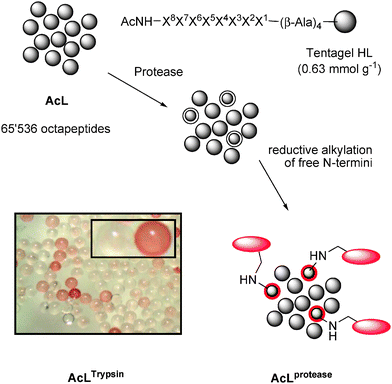 | ||
| Fig. 13 On-bead protease profiling on TentaGel beads. | ||
4.3 Cocktail fingerprinting
Due to its separating power, chromatographic screening offers the possibility to assay several different substrates simultaneously. A single HPLC-analysis returns the activity fingerprint, in which the relative amounts of product formation defining the reactivity profile or fingerprint, can be precisely reproduced if the cocktail composition is controlled. We have demonstrated this principle for a fingerprint analysis of lipases and esterases using a cocktail of 20 monoacyl-glycerol analogs,215 and for proteases using a cocktail of five hexapeptides.216 Such characterization tools may prove useful to identify novel enzymes with unusual selectivities, as well as in the area of diagnostics. Similarly, the classical APIZYM substrate palette for microbial characterization can be formulated as a cocktail reagent, allowing 16 different enzyme reactivities to be determined in a single analysis.217 A cocktail of fluorescent umbelliferyl glycosides was recently used to characterize various glycosidases using an HPLC-based assay .218 Substrate cocktails can also be analyzed by mass spectrometry in a variety of setting including enantioselectivity determination using a mixture of two isotopically labeled pseudo-enantiomers,219–225 for glycosyltransferase reaction using substrate mixtures226–230 or for protease activity determination using a mixture of peptide substrates.2314.4 Microarrays
Miniaturization of fingerprinting (and screening) down to the scale of a few nanoliters per datapoint is possible with microarrays printed on glass slides. A nanospray system was used to homogeneously distribute nanodroplets of a solution containing three fluorogenic protease substrates on a microarray on which spots of enzyme had been previously printed, allowing high-throughput screening of enzyme inhibitors.232 Depositing fluorogenic substrates on poly-lysine-coated glass slides also allows efficient assays of various enzymes in nanodroplets.233 Nanodroplets for enzyme assays can be moved using thermal gradients for mixing.234 Fluorogenic substrates have also been arrayed with covalent attachment to the surface of glass slides to allow activity profiling experiments with proteases and other hydrolytic enzymes using combinatorial series of fluorogenic coumarin-derived substrates35,235,236 and with lipases using substrate of varying acyl chain length 35a–f, relying on chemoselective oxidation of the 1,2-diol product with sodium periodate followed by reaction with rhodamine sulfohydrazide to detect conversion (Fig. 14).237
Microarrays have also been used for an elegant protease profiling method based on combinatorial libraries of PNA-encoded dipeptidyl-rhodamine substrates.238–241 An important improvement in the study of the biology of phosphatases was developed in a microarray. Glass slides having multiple peptide substrates of Ser/Thrphosphatases immobilized on them, can be used to simultaneously determine the preference of the enzymes for the different substrates.242 Protease profiling was also recently reported based on a multiplexed solution phase assay on microarrays.243 Microarrays were also reported for enantioselectivity, for example to estimate the optical purity of amino acids after covalent attachment by reaction with a pseudo-enantiomeric pair of labels bearing two different fluorophores based on Horeau's method.244–246Mass spectrometry was applied for the creation of biochips loaded with label-free oligosaccharide arrays, in order to study glycosyltransferases activities.247
It should be mentioned that it is possible to handle low-volume enzyme assays at the scale of one microliter per assay using silicagel plates pre-impregnated with a fluorogenic substrate as the reaction medium, which provides a practical solution for miniaturization that is much simpler than microarrays.248 A robotic arm is used to dispense the enzyme containing test solutions in a volume of one microliter per assay , which results in a homogeneously dispersed spot on the silicagel surface on which the enzyme reacts evenly with the substrate (Fig. 15).
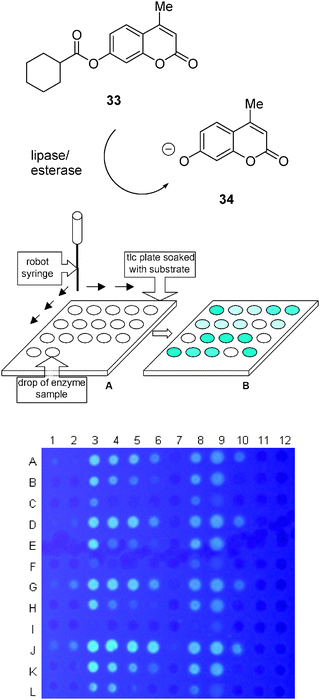 | ||
| Fig. 15 High-throughput screening with microtiter reaction on silicagel plates. | ||
5. Conclusion
In recent years enzyme assays have greatly advanced in their scope and in the diversity of detection principles employed. In the 1990s high-throughput screening of enzyme activity was perceived as a critical bottleneck in enzyme engineering due to the advent of random mutagenesis methods for directed evolution, which multiplied demands for screening by orders of magnitude. Developments of new screening methods based on chemistry, biology and instrumentation have followed to rise to this challenge, in part by reviving and refining older methods.For what concerns future developments, the demand for new enzyme assays remains high in the context of high-throughput screening in enzyme engineering, in particular for sensing regio-, stereo- and enantioselectivity. Remarkably, most of these problems have been in principle solved by instrument-based assays such as NMR, MS, HPLC and MS, not reviewed here, but the methods are often complicated and expensive to implement. Therefore, most application examples in enzyme engineering continue to use fluorogenic and chromogenic substrates and indicator assays as the main screening tool, simply because when available such assays are simple to use and inexpensive. Despite of their apparent drawbacks in terms of possible artefacts, the most useful assays seem to be indicator assays that are compatible with a range of different substrates. In particular enzyme-coupled assays will probably remain high on the list for many years to come, with assays producing a colored precipitate being the most useful for high-throughput screening as they can be applied on agar plates, on paper, or in microtiter plates. Improvement in fingerprinting reagents should also be considered in the future to enable high-throughput screening with a diverse set of substrates simultaneously, which would allow a much more productive use of enzyme libraries.
Acknowledgements
This work was supported by the Swiss National Science Foundation and Protéus SA, Nîmes, France.Notes and references
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS.
- P. Lorenz and H. Zinke, Trends Biotechnol., 2005, 23, 570–574 CrossRef CAS.
- S. Lutz and W. M. Patrick, Curr. Opin. Biotechnol., 2004, 15, 291–297 CrossRef CAS.
- T. W. Johannes and H. Zhao, Curr. Opin. Microbiol., 2006, 9, 261–267 CrossRef CAS.
- K. L. Tee and U. Schwaneberg, Comb. Chem. High Throughput Screening, 2007, 10, 197–217 CrossRef CAS.
- S. B. Rubin-Pitel and H. Zhao, Comb. Chem. High Throughput Screening, 2006, 9, 247–257 CrossRef CAS.
- W. R. Streit and R. A. Schmitz, Curr. Opin. Microbiol., 2004, 7, 492–498 CrossRef CAS.
- S. V. Taylor, P. Kast and D. Hilvert, Angew. Chem., Int. Ed., 2001, 40, 3310–3335 CrossRef.
- G. J. Williams, A. S. Nelson and A. Berry, Cell. Mol. Life Sci., 2004, 61, 3034–3046 CrossRef CAS.
- A. Aharoni, A. D. Griffiths and D. S. Tawfik, Curr. Opin. Chem. Biol., 2005, 9, 210–216 CrossRef CAS.
- M. D. Toscano, K. J. Woycechowsky and D. Hilvert, Angew. Chem., Int. Ed., 2007, 46, 3212–3236 CrossRef CAS.
- H. Lin and V. W. Cornish, Angew. Chem., Int. Ed., 2002, 41, 4402–4425 CrossRef CAS.
- J. Kofoed and J. L. Reymond, Curr. Opin. Chem. Biol., 2005, 9, 656–664 CrossRef CAS.
- J. L. Reymond, Enzyme Assays: High-throughput Screening, Genetic Selection and Fingerprinting, Wiley-VCH, Weiheim, 2005 Search PubMed.
- A. Hennig, D. Roth, T. Enderle and W. M. Nau, ChemBioChem, 2006, 7, 733–737 CrossRef CAS.
- H. Sahoo and W. M. Nau, ChemBioChem, 2007, 8, 567–573 CrossRef CAS.
- F. Badalassi, D. Wahler, G. Klein, P. Crotti and J. L. Reymond, Angew. Chem., Int. Ed., 2000, 39, 4067–4070 CrossRef CAS.
- C. Oberthur, H. Graf and M. Hamburger, Phytochemistry, 2004, 65, 3261–3268 CrossRef.
- E. T. Farinas, U. Schwaneberg, A. Glieder and F. H. Arnold, Adv. Synth. Catal., 2001, 343, 601–606 CrossRef CAS.
- A. Celik, G. A. Roberts, J. H. White, S. K. Chapman, N. J. Turner and S. L. Flitsch, Chem. Commun., 2006, 4492–4494 RSC.
- L. Kass, CRC. Crit. Rev. Clin. Lab. Sci., 1979, 10, 205–223 Search PubMed.
- F. Tanaka, L. Kerwin, D. Kubitz, R. A. Lerner and C. F. Barbas, 3rd, Bioorg. Med. Chem. Lett., 2001, 11, 2983–2986 CrossRef CAS.
- G. Klein and J. L. Reymond, Bioorg. Med. Chem. Lett., 1998, 8, 1113–1116 CrossRef CAS.
- G. Klein and J. L. Reymond, Helv. Chim. Acta, 1999, 82, 400–407 CrossRef CAS.
- N. Jourdain, R. P. Carlon and J. L. Reymond, Tetrahedron Lett., 1998, 39, 9415–9418 CrossRef CAS.
- R. Perez Carlon, N. Jourdain and J. L. Reymond, Chemistry, 2000, 6, 4154–4162 Search PubMed.
- B. List, C. F. Barbas and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15351–15355 CrossRef CAS.
- J. Kofoed, T. Darbre and J. L. Reymond, Chem. Commun., 2006, 1482–1484 RSC.
- J. Kofoed, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2006, 4, 3268–3281 RSC.
- E. Gonzalez-Garcia, V. Helaine, G. Klein, M. Schuermann, G. A. Sprenger, W. D. Fessner and J. L. Reymond, Chem.–Eur. J., 2003, 9, 893–899 CrossRef CAS.
- A. Sevestre, V. Helaine, G. Guyot, C. Martin and L. Hecquet, Tetrahedron Lett., 2003, 44, 827–830 CrossRef CAS.
- E. Leroy, N. Bensel and J. L. Reymond, Adv. Synth. Catal., 2003, 345, 859–865 CrossRef CAS.
- N. Bensel, M. T. Reymond and J. L. Reymond, Chemistry, 2001, 7, 4604–4612 Search PubMed.
- E. Leroy, N. Bensel and J. L. Reymond, Bioorg. Med. Chem. Lett., 2003, 13, 2105–2108 CrossRef CAS.
- Y. Z. Yang, P. Babiak and J. L. Reymond, Helv. Chim. Acta, 2006, 89, 404–415 CrossRef.
- R. Sicard, L. S. Chen, A. J. Marsaioli and J. L. Reymond, Adv. Synth. Catal., 2005, 347, 1041–1050 CrossRef CAS.
- B. Bicalho, L. S. Chen, J. Grognux, J. L. Reymond and A. J. Marsaioli, J. Braz. Chem. Soc., 2004, 15, 911–916 CAS.
- E. Nyfeler, J. Grognux, D. Wahler and J. L. Reymond, Helv. Chim. Acta, 2003, 86, 2919–2927 CrossRef CAS.
- E. M. Gonzalez-Garcia, J. Grognux, D. Wahler and J. L. Reymond, Helv. Chim. Acta, 2003, 86, 2458–2470 CrossRef CAS.
- J. Grognux and J. L. Reymond, ChemBioChem, 2004, 5, 826–831 CrossRef CAS.
- D. Lagarde, H. K. Nguyen, G. Ravot, D. Wahler, J. L. Reymond, G. Hills, T. Veit and F. Lefevre, Org. Process Res. Dev., 2002, 6, 441–445 Search PubMed.
- F. Badalassi, H. K. Nguyen, P. Crotti and J. L. Reymond, Helv. Chim. Acta, 2002, 85, 3090–3098 CrossRef CAS.
- C. Bedia, J. Casas, V. Garcia, T. Levade and G. Fabrias, ChemBioChem, 2007, 8, 642–648 CrossRef CAS.
- M. Shamis, C. F. Barbas and D. Shabat, Bioorg. Med. Chem. Lett., 2007, 17, 1172–1175 CrossRef CAS.
- P. D. Jones, N. M. Wolf, C. Morisseau, P. Whetstone, B. Hock and B. D. Hammock, Anal. Biochem., 2005, 343, 66–75 CAS.
- R. J. Amir and D. Shabat, Chem. Commun., 2004, 1614–1615 RSC.
- Y. Meyer, J. A. Richard, M. Massonneau, P. Y. Renard and A. Romieu, Org. Lett., 2008, 10, 1517–1520 CrossRef CAS.
- Z. Q. Wang, J. F. Liao and Z. J. Diwu, Bioorg. Med. Chem. Lett., 2005, 15, 2335–2338 CrossRef CAS.
- G. B. Jones, C. F. Crasto, J. E. Mathews, L. F. Xie, M. O. Mitchell, A. El-Shafey, A. V. D’Amico and G. J. Bubley, Bioorg. Med. Chem., 2006, 14, 418–425 CrossRef CAS.
- J. P. Germanas, S. Wang, A. Miner, W. Hao and J. M. Ready, Bioorg. Med. Chem. Lett., 2007, 17, 6871–6875 CrossRef CAS.
- E. D. Matayoshi, G. T. Wang, G. A. Krafft and J. Erickson, Science, 1990, 247, 954–958 CrossRef CAS.
- E. K. Kainmuller, E. P. Olle and W. Bannwarth, Chem. Commun., 2005, 5459–5461 RSC.
- E. K. Kainmuller and W. Bannwarth, Helv. Chim. Acta, 2006, 89, 3056–3070 CrossRef.
- R. Warfield, P. Bardelang, H. Saunders, W. C. Chan, C. Penfold, R. James and N. R. Thomas, Org. Biomol. Chem., 2006, 4, 3626–3638 RSC.
- T. Kohl, K. G. Heinze, R. Kuhlemann, A. Koltermann and P. Schwille, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12161–12166 CrossRef CAS.
- J. H. Wosnick, C. M. Mello and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 3400–3405 CrossRef CAS.
- V. Boyer, S. Fort, T. P. Frandsen, M. Schulein, S. Cottaz and H. Driguez, Chem.–Eur. J., 2002, 8, 1389–1394 CrossRef CAS.
- G. Zandonella, L. Haalck, F. Spener, K. Faber, F. Paltauf and A. Hermetter, Chirality, 1996, 8, 481–489 CrossRef CAS.
- M. Duque, M. Graupner, H. Stutz, I. Wicher, R. Zechner, F. Paltauf and A. Hermetter, J. Lipid Res., 1996, 37, 868–876 CAS.
- Y. Z. Yang, P. Babiak and J. L. Reymond, Org. Biomol. Chem., 2006, 4, 1746–1754 RSC.
- T. Maeda and S. Nishimura, Chemistry, 2008, 14, 478–487 Search PubMed.
- F. Tanaka, R. Thayumanavan and C. F. Barbas, J. Am. Chem. Soc., 2003, 125, 8523–8528 CrossRef CAS.
- F. Tanaka, N. Mase and C. F. Barbas, J. Am. Chem. Soc., 2004, 126, 3692–3693 CrossRef CAS.
- H. M. Guo, M. Minakawa and F. Tanaka, J. Org. Chem., 2008, 73, 3964–3966 CrossRef CAS.
- T. Murayama, T. Tanabe, H. Ikeda and A. Ueno, Bioorg. Med. Chem., 2006, 14, 3691–3696 CrossRef CAS.
- D. S. Lawrence and Q. Z. Wang, ChemBioChem, 2007, 8, 373–378 CrossRef CAS.
- M. S. Tremblay, M. Lee and D. Sames, Org. Lett., 2008, 10, 5–8 CrossRef CAS.
- M. Rosseneu, M. J. Taveirne, H. Caster and J. P. Vanbiervliet, Eur. J. Biochem., 1985, 152, 195–198 CrossRef CAS.
- S. S. Twining, Anal. Biochem., 1984, 143, 30–34 CAS.
- B. Law, R. Weissleder and C. H. Tung, Bioconjugate Chem., 2007, 18, 1701–1704 CrossRef CAS.
- A. L. James, J. D. Perry, C. Jay, D. Monget, J. W. Rasburn and F. K. Gould, Lett. Appl. Microbiol., 2001, 33, 403–408 CrossRef CAS.
- A. Celik, R. E. Speight and N. J. Turner, Chem. Commun., 2005, 3652–3654 RSC.
- G. Chen, D. J. Yee, N. G. Gubernator and D. Sames, J. Am. Chem. Soc., 2005, 127, 4544–4545 CrossRef CAS.
- M. K. Froemming and D. Sames, Angew. Chem., Int. Ed., 2006, 45, 637–642 CrossRef CAS.
- W. H. Zhou, M. P. Valley, J. Shultz, E. M. Hawkins, L. Bernad, T. Good, D. Good, T. L. Riss, D. H. Klaubert and K. V. Wood, J. Am. Chem. Soc., 2006, 128, 3122–3123 CrossRef CAS.
- J. A. Richard, L. Jean, A. Romieu, M. Massonneau, P. Noack-Fraissignes and P. Y. Renard, Org. Lett., 2007, 9, 4853–4855 CrossRef CAS.
- B. S. Montenecourt and D. E. Eveleigh, Appl. Environ. Microbiol., 1977, 33, 178–183.
- Z. Yang and B. Xu, Chem. Commun., 2004, 2424–2425 RSC.
- Z. Yang, P. L. Ho, G. Liang, K. H. Chow, Q. Wang, Y. Cao, Z. Guo and B. Xu, J. Am. Chem. Soc., 2007, 129, 266–267 CrossRef CAS.
- L. Adler-Abramovich, R. Perry, A. Sagi, E. Gazit and D. Shabat, ChemBioChem, 2007, 8, 859–862 CrossRef CAS.
- J. Hoogboom, K. Velonia, T. Rasing, A. E. Rowan and R. J. Nolte, Chem. Commun., 2006, 434–435 RSC.
- M. Olsen, B. Iverson and G. Georgiou, Curr. Opin. Biotechnol., 2000, 11, 331–337 CrossRef CAS.
- M. J. Olsen, J. Gam, B. L. Iverson and G. Georgiou, Methods Mol. Biol., 2003, 230, 329–342 Search PubMed.
- M. J. Olsen, D. Stephens, D. Griffiths, P. Daugherty, G. Georgiou and B. L. Iverson, Nat. Biotechnol., 2000, 18, 1071–1074 CrossRef CAS.
- N. Varadarajan, J. Gam, M. J. Olsen, G. Georgiou and B. L. Iverson, Proc. Natl. Acad. Sci. USA, 2005, 102, 6855–6860 CrossRef CAS.
- D. S. Tawfik and A. D. Griffiths, Nat. Biotechnol., 1998, 16, 652–656 CrossRef CAS.
- K. Bernath, M. Hai, E. Mastrobattista, A. D. Griffiths, S. Magdassi and D. S. Tawfik, Anal. Biochem., 2004, 325, 151–157 CrossRef CAS.
- O. J. Miller, K. Bernath, J. J. Agresti, G. Amitai, B. T. Kelly, E. Mastrobattista, V. Taly, S. Magdassi, D. S. Tawfik and A. D. Griffiths, Nat. Methods, 2006, 3, 561–570 CrossRef CAS.
- A. Aharoni, K. Thieme, C. P. Chiu, S. Buchini, L. L. Lairson, H. Chen, N. C. Strynadka, W. W. Wakarchuk and S. G. Withers, Nat. Methods, 2006, 3, 609–614 CrossRef CAS.
- S. Becker, H. Hobenreich, A. Vogel, J. Knorr, S. Wilhelm, F. Rosenau, K. E. Jaeger, M. T. Reetz and H. Kolmar, Angew. Chem., Int. Ed., 2008, 47, 5085–5088 CrossRef CAS.
- G. J. Williams, S. Domann, A. Nelson and A. Berry, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3143–3148 CrossRef CAS.
- T. Woodhall, G. Williams, A. Berry and A. Nelson, Angew. Chem., Int. Ed., 2005, 44, 2109–2112 CrossRef.
- M. Baumann, R. Sturmer and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2001, 40, 4201–4204 CrossRef CAS.
- S. Bartsch, R. Kourist and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2008, 47, 1508–1511 CrossRef.
- P. Abato and C. T. Seto, J. Am. Chem. Soc., 2001, 123, 9206–9207 CrossRef CAS.
- S. Dey, K. R. Karukurichi, W. J. Shen and D. B. Berkowitz, J. Am. Chem. Soc., 2005, 127, 8610–8611 CrossRef CAS.
- A. Hamberg, S. Lundgren, M. Penhoat, C. Moberg and K. Hult, J. Am. Chem. Soc., 2006, 128, 2234–2235 CrossRef CAS.
- A. Hamberg, S. Lundgren, E. Wingstrand, C. Moberg and K. Hult, Chem.–Eur. J., 2007, 13, 4334–4341 CrossRef CAS.
- S. Dey, D. R. Powell, C. Hu and D. B. Berkowitz, Angew. Chem., Int. Ed., 2007, 46, 7010–7014 CrossRef CAS.
- Z. Li, L. Butikofer and B. Witholt, Angew. Chem., Int. Ed., 2004, 43, 1698–1702 CrossRef CAS.
- H. E. Müller, J. Microbiol. Methods, 1984, 2, 101–102 CrossRef.
- H. Joo, A. Arisawa, Z. L. Lin and F. H. Arnold, Chem. Biol., 1999, 6, 699–706 CrossRef CAS.
- H. Joo, Z. L. Lin and F. H. Arnold, Nature, 1999, 399, 670–673 CrossRef CAS.
- M. Alexeeva, A. Enright, M. J. Dawson, M. Mahmoudian and N. J. Turner, Angew. Chem., Int. Ed., 2002, 41, 3177–3180 CrossRef CAS.
- R. Carr, M. Alexeeva, A. Enright, T. S. C. Eve, M. J. Dawson and N. J. Turner, Angew. Chem., Int. Ed., 2003, 42, 4807–4810 CrossRef.
- M. D. Truppo, F. Escalettes and N. J. Turner, Angew. Chem., Int. Ed., 2008, 47, 2639–2641 CrossRef CAS.
- R. A. Dicioccio, C. Piskorz, G. Salamida, J. J. Barlow and K. L. Matta, Anal. Biochem., 1981, 111, 176–183 CAS.
- H. Vankayalapati and G. Singh, Tetrahedron Lett., 1999, 40, 3925–3928 CrossRef CAS.
- D. Indurugalla, J. N. Watson and A. J. Bennet, Org. Biomol. Chem., 2006, 4, 4453–4459 RSC.
- C. Mayer, D. L. Jakeman, M. Mah, G. Karjala, L. Gal, R. A. J. Warren and S. G. Withers, Chem. Biol., 2001, 8, 437–443 CrossRef CAS.
- C. Garciaecheverria, J. L. Kofron, P. Kuzmic, V. Kishore and D. H. Rich, J. Am. Chem. Soc., 1992, 114, 2758–2759 CrossRef CAS.
- J. L. Kofron, P. Kuzmic, V. Kishore, E. Colonbonilla and D. H. Rich, Biochemistry, 1991, 30, 6127–6134 CrossRef CAS.
- J. T. YliKauhaluoma, J. A. Ashley, C. H. L. Lo, J. Coakley, P. Wirsching and K. D. Janda, J. Am. Chem. Soc., 1996, 118, 5496–5497 CrossRef CAS.
- K. Kupcho, R. Somberg, B. Bulleit and S. A. Goueli, Anal. Biochem., 2003, 317, 210–217 CrossRef CAS.
- S. M. Rodems, B. D. Hamman, C. Lin, J. Zhao, S. Shah, D. Heidary, L. Makings, J. H. Stack and B. A. Pollok, Assay Drug Dev. Technol., 2002, 1, 9–19 CrossRef CAS.
- K. T. Nguyen and D. Pei, Methods Mol. Med., 2008, 142, 117–130 Search PubMed.
- O. Molin, L. Nilsson and S. Ansehn, J. Clin. Microbiol., 1983, 18, 521–525 CAS.
- H. Shulman, A. Eberhard, C. Eberhard, S. Ulitzur and E. Keinan, Bioorg. Med. Chem. Lett., 2000, 10, 2353–2356 CrossRef CAS.
- G. Krebs, L. Hugonet and J. D. Sutherland, Angew. Chem., Int. Ed., 2006, 45, 301–305 CrossRef CAS.
- L. Galam, M. K. Hadden, Z. Q. Ma, Q. Z. Ye, B. G. Yun, B. S. J. Blagg and R. L. Matts, Bioorg. Med. Chem., 2007, 15, 1939–1946 CrossRef CAS.
- D. S. Auld, N. T. Southall, A. Jadhav, R. L. Johnson, D. J. Diller, A. Simeonov, C. P. Austin and J. Inglese, J. Med. Chem., 2008, 51, 2372–2386 CrossRef CAS.
- L. E. Janes and R. J. Kazlauskas, J. Org. Chem., 1997, 62, 4560–4561 CrossRef CAS.
- L. E. Janes, A. C. Lowendahl and R. J. Kazlauskas, Chem.–Eur. J., 1998, 4, 2324–2331 CrossRef CAS.
- C. B. Park and D. S. Clark, Biotechnol. Bioeng., 2002, 78, 229–235 CrossRef CAS.
- M. Persson and M. M. Palcic, Anal. Biochem., 2008, 378, 1–7 CrossRef CAS.
- A. Ben-David, G. Shoham and Y. Shoham, Chem. Biol., 2008, 15, 546–551 CrossRef CAS.
- S. J. C. Taylor, R. C. Brown, P. A. Keene and I. N. Taylor, Bioorg. Med. Chem., 1999, 7, 2163–2168 CrossRef CAS.
- E. Henke and U. T. Bornscheuer, Anal. Chem., 2003, 75, 255–260 CrossRef CAS.
- A. Banerjee, R. Sharma and U. C. Banerjee, Biotechnol. Appl. Biochem., 2003, 37, 289–293 CrossRef CAS.
- M. Konarzycka-Bessler and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2003, 42, 1418–1420 CrossRef CAS.
- S. Salahuddin, O. Renaudet and J. L. Reymond, Org. Biomol. Chem., 2004, 2, 1471–1475 RSC.
- C. Mateo, A. Archelas and R. Furstoss, Anal. Biochem., 2003, 314, 135–141 CrossRef CAS.
- K. Doderer, S. Lutz-Wahl, B. Hauer and R. D. Schmid, Anal. Biochem., 2003, 321, 131–134 CrossRef CAS.
- F. Zocher, M. M. Enzelberger, U. T. Bornscheuer, B. Hauer and R. D. Schmid, Anal. Chim. Acta, 1999, 391, 345–351 CrossRef CAS.
- H. Maeda, H. Matsuno, M. Ushida, K. Katayama, K. Saeki and N. Itoh, Angew. Chem., Int. Ed., 2005, 44, 2922–2925 CrossRef CAS.
- O. Khersonsky and D. S. Tawfik, ChemBioChem, 2006, 7, 49–53 CrossRef CAS.
- A. Ojida, Y. Mito-oka, M. Inoue and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 6256–6258 CrossRef CAS.
- S. Yamaguchi, L. Yoshimura, T. Kohira, S. Tamaru and I. Hamachi, J. Am. Chem. Soc., 2005, 127, 11835–11841 CrossRef CAS.
- K. Martin, T. H. Steinberg, L. A. Cooley, K. R. Gee, J. M. Beechem and W. F. Patton, Proteomics, 2003, 3, 1244–1255 CrossRef CAS.
- J. Wongkongkatep, Y. Miyahara, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed., 2006, 45, 665–668 CrossRef CAS.
- C. E. Humphrey, M. A. M. Easson and N. J. Turner, ChemBioChem, 2004, 5, 1144–1148 CrossRef CAS.
- A. L. L. Duchateau, M. G. Hillemans-Crombach, A. van Duijnhoven, R. Reiss and T. Sonke, Anal. Biochem., 2004, 330, 362–364 CrossRef CAS.
- G. Klein, D. Kaufmann, S. Schurch and J. L. Reymond, Chem. Commun., 2001, 561–562 RSC.
- K. E. S. Dean, G. Klein, O. Renaudet and J. L. Reymond, Bioorg. Med. Chem. Lett., 2003, 13, 1653–1656 CrossRef CAS.
- D. Wahler and J. L. Reymond, Angew. Chem., Int. Ed., 2002, 41, 1229–1232 CrossRef CAS.
- J. L. Reymond and V. Fluxa, Nat. Protoc., 2008, 3, 1270 Search PubMed.
- D. Wahler, O. Boujard, F. Lefevre and J. L. Reymond, Tetrahedron, 2004, 60, 703–710 CrossRef CAS.
- D. Kahakeaw and M. T. Reetz, Chem.–Asian J., 2008, 3, 233–238 CrossRef CAS.
- K. Doderer and R. D. Schmid, Biotechnol. Lett., 2004, 26, 835–839 CrossRef CAS.
- B. van Loo, J. H. L. Spelberg, J. Kingma, T. Sonke, M. G. Wubbolts and D. B. Janssen, Chem. Biol., 2004, 11, 981–990 CrossRef CAS.
- D. S. Tawfik, B. S. Green, R. Chap, M. Sela and Z. Eshhar, Proc. Natl. Acad. Sci. USA, 1993, 90, 373–377 CAS.
- G. Macbeath and D. Hilvert, J. Am. Chem. Soc., 1994, 116, 6101–6106 CrossRef CAS.
- F. Benedetti, F. Berti, M. Flego, M. Resmini and E. Bastiani, Anal. Biochem., 1998, 256, 67–73 CrossRef CAS.
- P. Geymayer, N. Bahr and J. L. Reymond, Chem.–Eur. J., 1999, 5, 1006–1012 CrossRef CAS.
- F. Taran, P. Y. Renard, C. Creminon, A. Valleix, Y. Frobert, P. Pradelles, J. Grassi and C. Mioskowski, Tetrahedron Lett., 1999, 40, 1887–1890 CrossRef CAS.
- F. Taran, P. Y. Renard, C. Creminon, A. Valleix, Y. Frobert, P. Pradelles, J. Grassi and C. Mioskowski, Tetrahedron Lett., 1999, 40, 1891–1894 CrossRef CAS.
- R. Nutiu, J. M. Y. Yu and Y. F. Li, ChemBioChem, 2004, 5, 1139–1144 CrossRef CAS.
- P. F. Zatta, K. Nyame, M. J. Cormier, S. A. Mattox, P. A. Prieto, D. F. Smith and R. D. Cummings, Anal. Biochem., 1991, 194, 185–191 CAS.
- L. S. Khraltsova, M. A. Sablina, T. D. Melikhova, D. H. Joziasse, H. Kaltner, H. J. Gabius and N. V. Bovin, Anal. Biochem., 2000, 280, 250–257 CrossRef CAS.
- M. C. Bryan, L. V. Lee and C. H. Wong, Bioorg. Med. Chem. Lett., 2004, 14, 3185–3188 CrossRef CAS.
- M. C. Bryan, O. Plettenburg, P. Sears, D. Rabuka, S. Wacowich-Sgarbi and C. H. Wong, Chem. Biol., 2002, 9, 713–720 CrossRef CAS.
- G. Das, P. Talukdar and S. Matile, Science, 2002, 298, 1600–1602 CrossRef CAS.
- N. Sorde, G. Das and S. Matile, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11964–11969 CrossRef CAS.
- T. Miyatake, M. Nishihara and S. Matile, J. Am. Chem. Soc., 2006, 128, 12420–12421 CrossRef CAS.
- S. M. Butterfield, D. H. Tran, H. Zhang, G. D. Prestwich and S. Matile, J. Am. Chem. Soc., 2008, 130, 3270–3271 CrossRef CAS.
- H. Tanaka and S. Matile, Chirality, 2008, 20, 307–312 CrossRef CAS.
- Y. Xiao, V. Pavlov, S. Levine, T. Niazov, G. Markovitch and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 4519–4522 CrossRef CAS.
- V. Pavlov, Y. Xiao and I. Willner, Nano Lett., 2005, 5, 649–653 CrossRef CAS.
- R. Baron, M. Zayats and I. Willner, Anal. Chem., 2005, 77, 1566–1571 CrossRef CAS.
- Z. Wang, J. Lee, A. R. Cossins and M. Brust, Anal. Chem., 2005, 77, 5770–5774 CrossRef CAS.
- Z. Wang, R. Levy, D. G. Fernig and M. Brust, J. Am. Chem. Soc., 2006, 128, 2214–2215 CrossRef CAS.
- C. Guarise, L. Pasquato, V. De Filippis and P. Scrimin, Proc. Natl. Acad. Sci. USA, 2006, 103, 3978–3982 CrossRef CAS.
- Y. Choi, N. H. Ho and C. H. Tung, Angew. Chem., Int. Ed., 2006, 46, 707–709.
- W. Zhao, W. Chiuman, J. C. Lam, M. A. Brook and Y. Li, Chem. Commun., 2007, 3729–3731 RSC.
- X. Xu, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 3468–3470 CrossRef CAS.
- J. Buissièr, A. Fourcard and L. Colobert, C. R. Hebd. Seances Acad. Sci., Ser. D, 1967, 264, 415–417 Search PubMed.
- P. Nardon, D. Monget, M. L. Didierfichet and G. Dethe, Biomedicine, 1976, 24, 183–190 Search PubMed.
- M. W. Humble, A. King and I. Phillips, J. Clin. Pathol., 1977, 30, 275–277 CrossRef CAS.
- E. Gruner, A. Vongraevenitz and M. Altwegg, J. Microbiol. Methods, 1992, 16, 101–118 CrossRef CAS.
- P. Garcia-Martos, P. Marin, J. M. Hernandez-Molina, L. Garcia-Agudo, S. Aoufi and J. Mira, Mycopathologia, 2001, 150, 1–4 CrossRef CAS.
- J. L. Reymond and D. Wahler, ChemBioChem, 2002, 3, 701–708 CrossRef CAS.
- A. Nath and W. M. Atkins, Biochemistry, 2008, 47, 157–166 CrossRef CAS.
- A. Taglieber, H. Hobenreich, J. D. Carballeira, R. J. Mondiere and M. T. Reetz, Angew. Chem., Int. Ed., 2007, 46, 8597–8600 CrossRef CAS.
- Y. S. Liu, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14694–14699 CrossRef CAS.
- W. Li, J. L. Blankman and B. F. Cravatt, J. Am. Chem. Soc., 2007, 129, 9594–9595 CrossRef CAS.
- D. Greenbaum, K. F. Medzihradszky, A. Burlingame and M. Bogyo, Chem. Biol., 2000, 7, 569–581 CrossRef.
- H. Schmidinger, R. Birner-Gruenberger, G. Riesenhuber, R. Saf, H. Susani-Etzerodt and A. Hermetter, ChemBioChem, 2005, 6, 1776–1781 CrossRef CAS.
- R. Nakai, C. M. Salisbury, H. Rosen and B. F. Cravatt, Bioorg. Med. Chem., 2008 DOI:10.1016/j.bmc.2008.03.018.
- A. Borodovsky, H. Ovaa, W. J. N. Meester, E. S. Venanzi, M. S. Bogyo, B. G. Hekking, H. L. Ploegh, B. M. Kessler and H. S. Overkleeft, ChemBioChem, 2005, 6, 287–291 CrossRef CAS.
- D. C. Greenbaum, W. D. Arnold, F. Lu, L. Hayrapetian, A. Baruch, J. Krumrine, S. Toba, K. Chehade, D. Bromme, I. D. Kuntz and M. Bogyo, Chem. Biol., 2002, 9, 1085–1094 CrossRef CAS.
- M. Bogyo, S. Verhelst, V. Bellingard-Dubouchaud, S. Toba and D. Greenbaum, Chem. Biol., 2000, 7, 27–38 CrossRef CAS.
- R. Srinivasan, X. Huang, S. L. Ng and S. Q. Yao, ChemBioChem, 2006, 7, 32–36 CrossRef.
- A. M. F. Liu, N. A. Somers, R. J. Kazlauskas, T. S. Brush, F. Zocher, M. M. Enzelberger, U. T. Bornscheuer, G. P. Horsman, A. Mezzetti, C. Schmidt-Dannert and R. D. Schmid, Tetrahedron: Asymmetry, 2001, 12, 545–556 CrossRef CAS.
- D. Wahler, F. Badalassi, P. Crotti and J. L. Reymond, Angew. Chem., Int. Ed., 2001, 40, 4457–4460 CrossRef CAS.
- D. Wahler, F. Badalassi, P. Crotti and J. L. Reymond, Chemistry, 2002, 8, 3211–3228 Search PubMed.
- J. C. Powers, J. L. Asgian, O. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS.
- M. Meldal, QSAR Comb. Sci., 2005, 24, 1141–1148 Search PubMed.
- X. S. Puente, L. M. Sanchez, C. M. Overall and C. Lopez-Otin, Nat. Rev. Genet., 2003, 4, 544–558 CrossRef CAS.
- P. L. Richardson, Curr. Pharmaceut. Des., 2002, 8, 2559–2581 Search PubMed.
- D. J. Maly, L. Huang and J. A. Ellman, ChemBioChem, 2002, 3, 17–37.
- N. A. Thornberry, T. A. Rano, E. P. Peterson, D. M. Rasper, T. Timkey, M. Garcia-Calvo, V. M. Houtzager, P. A. Nordstrom, S. Roy, J. P. Vaillancourt, K. T. Chapman and D. W. Nicholson, J. Biol. Chem., 1997, 272, 17907–17911 CrossRef CAS.
- J. L. Harris, B. J. Backes, F. Leonetti, S. Mahrus, J. A. Ellman and C. S. Craik, Proc. Natl. Acad. Sci. USA, 2000, 97, 7754–7759 CrossRef CAS.
- J. L. Harris, P. B. Alper, J. Li, M. Rechsteiner and B. J. Backes, Chem. Biol., 2001, 8, 1131–1141 CrossRef CAS.
- T. A. Rano, T. Timkey, E. P. Peterson, J. Rotonda, D. W. Nicholson, J. W. Becker, K. T. Chapman and N. A. Thornberry, Chem. Biol., 1997, 4, 149–155 CAS.
- C. Pinilla, J. R. Appel, P. Blanc and R. A. Houghten, Biotechniques, 1992, 13, 901–905 CAS.
- A. F. Kisselev, M. Garcia-Calvo, H. S. Overkleeft, E. Peterson, M. W. Pennington, H. L. Ploegh, N. A. Thornberry and A. L. Goldberg, J. Biol. Chem., 2003, 278, 35869–35877 CrossRef CAS.
- Y. Choe, F. Leonetti, D. C. Greenbaum, F. Lecaille, M. Bogyo, D. Bromme, J. A. Ellman and C. S. Craik, J. Biol. Chem., 2006, 281, 12824–12832 CrossRef CAS.
- S. S. Cotrin, L. Puzer, W. A. D. Judice, L. Juliano, A. K. Carmona and M. A. Juliano, Anal. Biochem., 2004, 335, 244–252 CrossRef CAS.
- K. Breddam and M. Meldal, Eur. J. Biochem., 1992, 206, 103–107 CrossRef CAS.
- M. Meldal, I. Svendsen, K. Breddam and F. I. Auzanneau, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3314–3318 CAS.
- P. M. St Hilaire, M. Willert, M. A. Juliano, L. Juliano and M. Meldal, J. Comb. Chem., 1999, 1, 509–523 CrossRef CAS.
- F. M. Alves, I. Y. Hirata, I. E. Gouvea, M. F. M. Alves, M. Meldal, D. Bromme, L. Juliano and M. A. Juliano, J. Comb. Chem., 2007, 9, 627–634 CrossRef CAS.
- O. D. Ekici, A. Karla, M. Paetzel, M. O. Lively, D. H. Pei and R. E. Dalbey, J. Biol. Chem., 2007, 282, 417–425 CAS.
- J. Kofoed and J. L. Reymond, Chem. Commun., 2007, 4453–4455 RSC.
- J. P. Goddard and J. L. Reymond, J. Am. Chem. Soc., 2004, 126, 11116–11117 CrossRef CAS.
- Y. Yongzheng and J. L. Reymond, Mol. Biosyst., 2005, 1, 57–63 RSC.
- R. Sicard, J. P. Goddard, M. Mazel, C. Audiffrin, L. Fourage, G. Ravot, D. Wahler, F. Lefevre and J. L. Reymond, Adv. Synth. Catal., 2005, 347, 987–996 CrossRef CAS.
- S. Park and I. Shin, Org. Lett., 2007, 9, 619–622 CrossRef CAS.
- A. Horeau and A. Nouaille, Tetrahedron Lett., 1990, 31, 2707–2710 CrossRef CAS.
- A. Schoofs and A. Horeau, Tetrahedron Lett., 1977, 3259–3262 CrossRef CAS.
- J. H. Guo, J. Y. Wu, G. Siuzdak and M. G. Finn, Angew. Chem., Int. Ed., 1999, 38, 1755–1758 CrossRef CAS.
- M. T. Reetz, M. H. Becker, H. W. Klein and D. Stockigt, Angew. Chem., Int. Ed., 1999, 38, 1758–1761 CrossRef CAS.
- D. X. Zha, A. Eipper and M. T. Reetz, ChemBioChem, 2003, 4, 34–39 CrossRef CAS.
- M. T. Reetz, C. Torre, A. Eipper, R. Lohmer, M. Hermes, B. Brunner, A. Maichele, M. Bocola, M. Arand, A. Cronin, Y. Genzel, A. Archelas and R. Furstoss, Org. Lett., 2004, 6, 177–180 CrossRef CAS.
- G. DeSantis, K. Wong, B. Farwell, K. Chatman, Z. L. Zhu, G. Tomlinson, H. J. Huang, X. Q. Tan, L. Bibbs, P. Chen, K. Kretz and M. J. Burk, J. Am. Chem. Soc., 2003, 125, 11476–11477 CrossRef CAS.
- C. J. Zea, S. W. MacDonell and N. L. Pohl, J. Am. Chem. Soc., 2003, 125, 13666–13667 CrossRef CAS.
- Y. Yu, K. S. Ko, C. J. Zea and N. L. Pohl, Org. Lett., 2004, 6, 2031–2033 CrossRef CAS.
- M. Yang, M. Brazier, R. Edwards and B. G. Davis, ChemBioChem, 2005, 6, 346–357 CrossRef CAS.
- N. Nagahori and S. I. Nishimura, Chem.–Eur. J., 2006, 12, 6478–6485 CrossRef CAS.
- M. Yang, G. J. Davies and B. G. Davis, Angew. Chem., Int. Ed., 2007, 46, 3885–3888 CrossRef CAS.
- F. Basile, I. Ferrer, E. T. Furlong and K. J. Voorhees, Anal. Chem., 2002, 74, 4290–4293 CrossRef CAS.
- D. N. Gosalia and S. L. Diamond, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8721–8726 CrossRef CAS.
- M. Uttamchandani, X. Huang, G. Y. J. Chen and S. Q. Yao, Bioorg. Med. Chem. Lett., 2005, 15, 2135–2139 CrossRef CAS.
- K. T. Kotz, Y. Gu and G. W. Faris, J. Am. Chem. Soc., 2005, 127, 5736–5737 CrossRef CAS.
- C. M. Salisbury, D. J. Maly and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 14868–14870 CrossRef CAS.
- Q. Zhu, M. Uttamchandani, D. B. Li, M. L. Lesaicherre and S. Q. Yao, Org. Lett., 2003, 5, 1257–1260 CrossRef CAS.
- J. Grognux and J. L. Reymond, Mol. Biosyst., 2006, 2, 492–498 RSC.
- N. Winssinger, R. Damoiseaux, D. C. Tully, B. H. Geierstanger, K. Burdick and J. L. Harris, Chem. Biol., 2004, 11, 1351–1360 CrossRef CAS.
- J. L. Harris and N. Winssinger, Chem.–Eur. J., 2005, 11, 6792–6801 CrossRef CAS.
- F. Debaene, L. Mejias, J. L. Harris and N. Winssinger, Tetrahedron, 2004, 60, 8677–8690 CrossRef CAS.
- J. J. Diaz-Mochon, L. Bialy and M. Bradley, Chem. Commun., 2006, 3984–3986 RSC.
- H. Sun, C. H. Lu, M. Uttamchandani, Y. Xia, Y. C. Liou and S. Q. Yao, Angew. Chem., Int. Ed., 2008, 47, 1698–1702 CrossRef CAS.
- I. A. Kozlov, P. C. Melnyk, J. P. Hachmann, A. Srinivasan, M. Shults, C. F. Zhao, J. Musmacker, N. Nelson, D. L. Barker and M. Lebl, Comb. Chem. High Throughput Screening, 2008, 11, 24–35 CrossRef CAS.
- G. A. Korbel, G. Lalic and M. D. Shair, J. Am. Chem. Soc., 2001, 123, 361–362 CrossRef CAS.
- A. Horeau, Tetrahedron Lett., 1961, 506–512 CrossRef.
- A. Horeau, Tetrahedron Lett., 1962, 965–969 CrossRef CAS.
- L. Ban and M. Mrksich, Angew. Chem., Int. Ed., 2008, 47, 3396–3399 CrossRef CAS.
- P. Babiak and J. L. Reymond, Anal. Chem., 2005, 77, 373–377 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |