Boron-based rotaxanes by multicomponent self-assembly†
Nicolas
Christinat
,
Rosario
Scopelliti
and
Kay
Severin
*
Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland. E-mail: kay.severin@epfl.ch; Fax: +41 (0)21 6939305; Tel: +41 (0)21 6939302
First published on 11th June 2008
Abstract
The multicomponent reaction of 1,2-di(4-pyridyl)ethylene, catechol, 3,5-bis(trifluoromethyl)phenylboronic acid and 1,5-dinaphtho-38-crown-10 or bis-para-phenylene-34-crown-10, respectively, resulted in the formation of rotaxanes, which were characterized by X-ray crystallography.
Over recent years, the perception of rotaxanes has changed substantially. Whereas initially they represented “exotic molecules”1 and an “intriguing synthetic challenge”,2 they are now recognized as key components of molecular nanotechnology.3,4 The basis for this development was the discovery of efficient synthetic routes to construct such mechanically interlocked species.4 The most common strategies rely on preorganized building blocks, which are locked by covalent capture under kinetic control. An interesting alternative is the formation of rotaxanes under thermodynamic control using reversible covalent5 or non-covalent bonds.6 A key advantage of the latter approach is its efficiency, which allows complex structures to be generated, such as an [11]rotaxane7 or rotaxane-based dendrimers, for example.8 To further advance this field, new methods and building blocks for the synthesis of rotaxanes and polyrotaxanes under thermodynamic control are desirable. Below, we describe molecular assembles that—to best of our knowledge—represent the first examples of boron-based rotaxanes. They are obtained in good yield by the assembly of four chemically-distinct components.9
In a continuation of our studies concerning the utilization of boron compounds in supramolecular chemistry,10 we recently observed that bis(dioxaboroles) can be polymerized by the addition of 4,4′-dipyridine or 1,2-di(4-pyridyl)ethylene (DPE).11 Due to coordination to the Lewis acidic boronic esters, the dipyridyl linkers become electron deficient, and strong intrastrand charge-transfer transitions from the electron-rich dioxaborole moieties to the dipyridyl groups are observed.11 This finding was the basis for our investigations concerning boron-based rotaxanes, which are detailed below.
As building blocks for the multicomponent synthesis of a rotaxane, we used DPE, catechol, 3,5-bis(trifluoromethyl)phenylboronic acid and 1,5-dinaphtho-38-crown-1012 (Scheme 1). These compounds were dissolved in a 1 : 2 : 2 : 1 ratio in toluene, and then heated in a Dean–Stark trap. Under these conditions, the catechol was expected to condense with the boronic acid to give a boronic ester. The latter should coordinate to DPEvia dative B–N bonds13 to give an electron-deficient axle. Since the B–N bond formation is reversible, the crown ether can slip onto the axle to form a rotaxane. Spectroscopic and crystallographic analyses showed that this strategy was indeed successful. After cooling to room temperature, a yellow precipitate could be isolated (yield: 67%). NMR spectroscopic analyses of this product showed characteristic signals derived from all four building blocks in a 1 : 2 : 2 : 1 ratio (see ESI†). Clear evidence for the formation of rotaxane 1 was obtained by X-ray crystallography.‡ The structure of 1 is comprised of a dipyridyl axle, two boronic ester stoppers and the crown ether, which is wrapped around the axle (Scheme 1). The molecule has crystallographic C2 symmetry about an axis passing through the center of the ethylenic double bond and the dioxonaphthalene rings. The lengths of the B–O (1.455(14) and 1.489(13) Å) and B–N (1.641(14) Å) bonds are within expected ranges.13 The DPE linker is sandwiched between the two coplanar dioxonaphthalene groups of the crown ether, which are 7.71 Å apart from each other. The plane defined by the pyridyl groups of the axle is slightly twisted with respect to the plane defined by the dioxonaphthalene rings (twist angle: 29.9°). In addition to π-stacking interactions, there are C–H⋯O hydrogen bonds between the α-CH groups of the pyridyl rings and an O-atom of the crown ether.‡
 | ||
| Scheme 1 Synthesis of rotaxane 1. The graphic representation of 1 is based on a crystallographic analysis. | ||
To test whether rotaxanes are also formed with other crown ethers, we performed a multicomponent reaction with bis-para-phenylene-34-crown-1014 instead of 1,5-dinaphtho-38-crown-10. As before, we were able to obtain a yellow precipitate (yield: 63%), which turned out to be rotaxane 2, as evidenced by a crystallographic analysis (see ESI†). The overall structure of 2 is similar to that of 1: the crown ether wraps around the dipyridyl axle, which is connected to two boronic esters via dative B–N bonds (Fig. 1).‡ The unit cell contains two halves of independent rotaxanes with C2 symmetry, which display comparable bond lengths (B–Oav = 1.46 Å; B–Nav = 1.64 Å). Contrary to what was observed for rotaxane 1, the planes defined by the phenylene rings of 2 are nearly coplanar with that defined by the DPE axle. Consequently, the aromatic rings of the crown ether in 2 are closer to each other (7.00 and 7.06 Å). The phenylene rings are aligned with the ethylenic double bond of the axle (Fig. 2, bottom). Several C–H⋯O contacts between the α-CH groups of the pyridyl rings and an O-atom of the crown ether are observed.‡
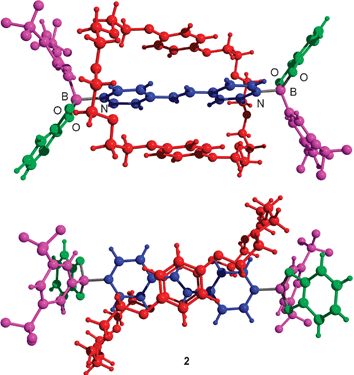 | ||
| Fig. 1 Molecular structure of rotaxane 2 in the crystal. Bottom: view along the C2 symmetry axis; top: view from the side. Only one of the two crystallographically-independent rotaxanes is shown. The co-crystallized toluene molecules are also not shown for clarity. | ||
The multicomponent reaction of DPE, catechol and 3,5-bis(trifluoromethyl)phenylboronic acid with a third crown ether, dibenzo-30-crown-10, also resulted in the formation of a yellow precipitate. NMR spectroscopic analysis of this precipitate revealed the presence of signals derived from all four building blocks. Unfortunately, we were not able to obtain single crystals for a crystallographic analysis. In view of the known tendency of ortho-phenylene-based crown ethers, such as dibenzo-30-crown-10, to bind to cationic guest molecules in a clip-like fashion15 (instead of wrapping around them as required for a rotaxane), we tried to obtain structural information for a closely related analogue. Suitable single crystals were obtained for an aggregate, 3, containing 4,4′-dipyridine as the axle instead of DPE. Compound 3 was formed in 60% yield by the multicomponent reaction of 3,5-bis(trifluoromethyl)phenylboronic acid, 4,4′-dipyridine, catechol and dibenzo-30-crown-10 (see ESI†). Its crystal structure is depicted in Fig. 2. As observed for related guests, such as diquat,15b the crown ether acts as a “molecular clip”16 rather than forming a rotaxane. The phenylene rings of the crown ether show π–π interactions with the pyridyl rings of the axle. In addition, there are a number of weak hydrogen bonds between the pyridyl H-atoms and the O-atoms of the crown ether. The lengths of the B–N (1.636(5) and 1.662(5) Å) and B–O bonds (B–Oav = 1.47 Å) are similar to those observed for 1 and 2.‡
 | ||
| Fig. 2 Molecular structure of host–guest complex 3 in the crystal. The co-crystallized toluene molecule is not shown for clarity. | ||
To obtain further information about the stability of the aggregates in solution, we performed NMR titration experiments with boronic ester 4 and DPE. Ester 4 was rapidly formed when 3,5-bis(trifluoromethyl)phenylboronic acid was heated with an equal amount of catechol (see ESI†). It thus represents a likely intermediate in the assembly of 1 and 2.
The addition of increasing amounts of DPE to 4 resulted in gradual changes in the 1H NMR spectra, indicating that pyridyl adduct formation was fast on the NMR time scale. Fitting of the titration isotherm (see ESI†) to a 2 : 1 binding model gave a first binding constant of K1 = 1.3 (±0.9) 104 M−1 and a second binding constant of K2 = 3.2 (±0.5) 102 M−1. Rotaxane formation can be rationalized by assuming that the crown ether slips onto the mono adduct 5, followed by reversible addition of the second boronic ester stopper to give the adduct 6 (Scheme 2).§ A similar analysis was performed for the interaction of boronic ester 4 with the 4,4′-dipyridyl axle, resulting in slightly lower binding constants of K1 = 1.2 (±0.9) 104 M−1 and K2 = 1.8 (±0.2) 102 M−1.
 | ||
| Scheme 2 Step-wise assembly of adduct 6. | ||
In summary, we have described rotaxanes that are formed in a multicomponent assembly process from a dipyridyl linker, an arylboronic acid, catechol and crown ethers. A unique structural feature of these rotaxanes is that boronic esters are an integral part of the assembly. The boronic esters have a two-fold function: (a) they act as Lewis acids, which renders the coordinated dipyridyl ligands electron-deficient and thus increasing their affinity to electron-rich crown ethers; (b) they act as stoppers. A disadvantage for potential applications in molecular devices is the fact that the highly dynamic B–N bond cannot easily be fixed (e.g. by changing the temperature or solvent). However, the dynamic nature of the B–N bond could be advantageous for the construction of boron-based polyrotaxanes17 or covalent organic rotaxane frameworks, in analogy to the coordination polyrotaxanes and metal–organic rotaxane frameworks described by the groups of Kim and Loeb.18 Current efforts in this direction are being pursued in our laboratory.
This work was supported by the EPFL and the Swiss National Science Foundation. We are grateful to Dr. E. Solari for help with the crystallographic investigations.
Notes and references
- H. W. Franke, Bild Wiss., 1974, 11, 90 Search PubMed.
- D. B. Amabilino, F. M. Rayno and J. F. Stoddart, in Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. B. MacNicol and F. Vögtle, Elsevier Science Ltd., Oxford, 1996, vol. 9, pp. 85 Search PubMed.
- (a) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS; (b) M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028 CrossRef CAS; (c) B. Champin, P. Mobian and J.-P. Sauvage, Chem. Soc. Rev., 2007, 36, 358 RSC; (d) S. Saha and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 77 RSC; (e) R. Beckman, K. Beverly, A. Boukai, Y. Bunimovich, J. W. Choi, E. DeIonno, J. Green, E. Johnston-Halperin, Y. Luo, B. Sheriff, J. F. Stoddart and J. R. Heath, Faraday Discuss., 2006, 131, 9 RSC.
- (a) S. J. Loeb, Chem. Soc. Rev., 2007, 36, 226 RSC; (b) M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211 RSC; (c) F. Aricó, J. D. Badjic, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203 CAS; (d) C. A. Schalley, T. Weiland, J. Brüggemann and F. Vögtle, Top. Curr. Chem., 2005, 248, 141 CAS; (e) K. Kim, Chem. Soc. Rev., 2002, 31, 96 RSC.
- For selected examples, see: (a) F. Aricó, T. Chang, S. J. Cantrill, S. I. Khan and J. F. Stoddart, Chem.–Eur. J., 2005, 11, 4655 CrossRef; (b) M. Horn, J. Ihringer, P. T. Glink and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 4046 CrossRef CAS; (c) Y. Furusho, T. Oku, T. Hasegawa, A. Tsuboi, N. Kihara and T. Tarata, Chem.–Eur. J., 2003, 9, 2895 CrossRef CAS; (d) A. F. M. Kilbinger, S. J. Cantrill, A. W. Waltman, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 3281 CrossRef CAS; (e) P. T. Glink, A. I. Oliva, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 1870 CrossRef CAS.
- For selected examples, see: (a) A. G. Cheetham, T. D. W. Claridge and H. L. Anderson, Org. Biomol. Chem., 2007, 5, 457 RSC; (b) C. A. Hunter, C. M. R. Low, M. J. Packer, S. E. Spey, J. G. Vinter, M. O. Vysotsky and C. Zonta, Angew. Chem., Int. Ed., 2001, 40, 2678 CrossRef CAS; (c) M. J. Gunter, N. Bampos, K. D. Johnstone and J. K. M. Sanders, New J. Chem., 2001, 25, 166 RSC; (d) K.-S. Jeong, J. S. Choi, S.-Y. Chang and H.-Y. Chang, Angew. Chem., Int. Ed., 2000, 39, 1692 CrossRef CAS.
- J. Wu, K. C.-F. Leung and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17266 CrossRef CAS.
- K. C.-F. Leung, F. Aricó, S. J. Cantrill and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 5808 CrossRef CAS.
- For a recent report about the assembly of a catenane using four different building blocks, see: Y. Liu, A. Bruneau, J. He and Z. Abliz, Org. Lett., 2008, 10, 765 Search PubMed.
- (a) N. Christinat, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2008, 47, 1848 CrossRef CAS; (b) N. Christinat, R. Scopelliti and K. Severin, J. Org. Chem., 2007, 72, 2192 CrossRef CAS; (c) N. Christinat, R. Scopelliti and K. Severin, Chem. Commun., 2004, 1158 RSC.
- N. Christinat, E. Croisier, R. Scopelliti, M. Cascella, U. Röthlisberger and K. Severin, Eur. J. Inorg. Chem., 2007, 5177 CrossRef CAS.
- D. B. Amabilino, P. R. Ashton, V. Balzani, S. E. Boyd, A. Credi, J. Y. Lee, S. Menzer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 4295 CrossRef CAS.
- H. Höpfl, J. Organomet. Chem., 1999, 581, 129 CrossRef CAS.
- P. L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. E. Kaifer, D. Philp, M. Pietraszkiewicz, L. Prodi, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 193 CrossRef CAS.
- (a) B. L. Allwood, H. M. Colquhoun, S. M. Doughty, F. H. Kohnke, A. M. Z. Slawin, J. F. Stoddart, D. J. Williams and R. Zarzycki, J. Chem. Soc., Chem. Commun., 1987, 1054 RSC; (b) H. M. Colquhoun, E. P. Goodings, J. M. Maud, J. F. Stoddart, J. B. Wolstenholme and D. J. Williams, J. Chem. Soc., Perkin Trans. 2, 1985, 607 RSC; (c) H. M. Colquhoun, J. F. Stoddart, D. J. Williams, J. B. Wolstenholme and R. Zarzycki, Angew. Chem., Int. Ed. Engl., 1981, 20, 1051 CrossRef.
- F.-G. Klärner and B. Kahler, Acc. Chem. Res., 2003, 36, 919 CrossRef.
- For a review about polyrotaxanes, see: T. Takata, Polym. J., 2006, 38, 1 Search PubMed.
- (a) D. J. Hoffart and S. J. Loeb, Angew. Chem., Int. Ed., 2005, 44, 901 CrossRef CAS; (b) G. J. E. Davidson and S. J. Loeb, Angew. Chem., Int. Ed., 2003, 42, 74 CrossRef CAS; (c) K.-M. Park, D. Whang, E. Lee, J. Heo and K. Kim, Chem.–Eur. J., 2002, 8, 498 CrossRef CAS; (d) E. Lee, J. Kim, J. Heo, D. Whang and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 399 CrossRef CAS; (e) E. Lee, J. Heo and K. Kim, Angew. Chem., Int. Ed., 2000, 39, 2699 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b805437a |
| ‡ Unfortunately, the quality of the structures of rotaxanes 1 and 2 are very low. The axles are well defined, but strong disorder is observed for the ethylene glycol part of the crown ethers and the CF3groups. This disorder, combined with the weakness of the observed diffraction and twinning problems for 1, prevented us from obtaining better results. The distances of the C–H⋯O bonds are therefore not discussed in detail. Data for 1: C76H68B2F12N2O14, Mr = 1482.94, monoclinic, space groupP21/c, a = 11.7382(12), b = 16.517(3), c = 17.832(4) Å, β = 98.670(13)°, V = 3417.7(10) Å3, Z = 2, T = 100(2) K, 40239 reflections collected, 5990 independent reflections, Rint = 0.0786, R1 [I > 2σ(I)] = 0.1633, wR2 (all data) = 0.5002. CCDC 683419. Data for 2: C89H88B2F12N2O14, Mr = 1659.23, triclinic, space groupP-1, a = 13.4013(9), b = 14.5562(10), c = 22.3107(17) Å, α = 96.487(6), β = 107.062(6), γ = 92.798(6)°, V = 4118.6(5) Å3, Z = 2, T = 140(2) K, 26573 reflections collected, 11389 independent reflections, Rint = 0.1597, R1 [I > 2σ(I)] = 0.1543, wR2 (all data) = 0.4318. CCDC 683420. Data for 3: C73H70B2F12N2O14, Mr = 1448.93, monoclinic, space groupP21/c, a = 14.0418(4), b = 22.0219(6), c = 22.2997(6) Å, β = 96.386(3)°, V = 6852.9(3) Å3, Z = 4, T = 140(2) K, 45866 reflections collected, 12085 independent reflections, Rint = 0.0413, R1 [I > 2σ(I)] = 0.0696, wR2 (all data) = 0.2075. CCDC 683421. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b805437a |
| § In view of the numerous species involved, we have not attempted to a derive binding constant for the interaction of 5 with crown ethers. |
| This journal is © The Royal Society of Chemistry 2008 |