Cu-catalyzed stereoselective conjugate addition of arylboronic acids to alkynoates†
Yoshihiko
Yamamoto
*,
Naohiro
Kirai
and
Yu
Harada
Department of Applied Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: omyy@apc.titech.ac.jp; Fax: 81 3 5734 3339; Tel: 81 3 5734 3339
First published on 25th March 2008
Abstract
The CuOAc-catalyzed reaction of internal alkynoates with arylboronic acids proceeded under mild conditions to yield trisubstituted cinnamates stereoselectively.
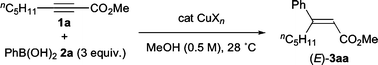
The conjugate addition of organometallic reagents to alkynoates is one of the most efficient methods to obtain synthetically valuable multiply substituted acrylates. For this purpose, organocopper reagents have been typically used,1 since the first report of Corey’s group2a and others.2b,c Organocopper-based methods, however, have several limitations: (a) they usually require stoichiometric amounts of the copper source, (b) the stereochemistry of the resulting alkene depends on both the reaction conditions and nature of the organocopper reagents,1 and (c) they are incompatible with highly reactive functional groups such as aldehydes. Therefore, other methods using catalytic amounts of transition-metal complexes as promoters were developed. Hayashi and co-workers have reported the rhodium-catalyzed hydroarylation of alkynes.3 In their study, they obtained conjugate addition products from two internal alkynoates with phenylboronic acid in high yields. Later, Oh and co-workers also carried out the palladium-catalyzed conjugate addition of arylboronic acids to alkynoates with a good substrate scope.4 Although these catalytic protocols enable the use of commercially available bench-top stable boronic acids and yield trisubstituted α,β-unsaturated esters with moderate to excellent regio- and stereoselectivity, the development of a new method using an inexpensive non-precious metal catalyst would be beneficial. In this regard, the copper-catalyzed addition of Grignard reagents to alkynoates has been reported by Jennings and co-workers.5 It should be noted that they have succeeded in obtaining both syn- and anti-hydroarylation products with moderate to good stereoselectivities by selecting appropriate trialkylsilyl mediators and quenching methods. However, substrate scope in terms of both the organometallic reagent and alkynoate has not been addressed in this copper-catalyzed method. In this communication, we report the copper-catalyzed conjugate addition of differently functionalized arylboronic acids to alkynoates that proceeds under mild reaction conditions to yield syn-hydroarylation products in good yields.
Initially, various transition-metal (TM) salts bearing oxygen ligands were screened as catalyst precursors, because they possibly facilitate the transmetalation with organoboron compounds by donating their ligand to the highly oxophilic boron center.6 Among the TM salts examined [Fe(OAc)2, Fe(acac)3, Co(OAc)2·4H2O, Ni(OAc)2·4H2O, and AgOAc], copper acetates proved to be effective. Thus, the exploratory reaction of methyl 2-octynoate (1a) and 3 equiv. of phenylboronic acid (2a) was carried out with 10 mol% Cu(OAc)2 in MeOH (0.5 M) at a temperature of 28 °C (Scheme 1 and Table 1). TLC analysis indicated that 1a was consumed within 2.5 h, and the chromatographic purification afforded the desired conjugate-addition product 3aa in 96% yield (run 1). It should be noted that 3aa was exclusively obtained as an E-isomer as confirmed from a comparison of its 1H NMR data with those for known compounds (see ESI† ). The methanol solvent is essential for efficient catalytic turnover. The use of ethanol or THF resulted in much lower conversion of 1a, giving 3aa in less than 20% yields. No product was detected when the reaction was carried out in 1,2-dichloroethane for 24 h. When a lower concentration of the substrate (0.25 M) was used, the reaction was complete in 10 h and the yield of 3aa decreased to 84%. Moreover, a trace amount of methyl 2,3-diphenyl-2-octenoate (4) was also detected in the 1H NMR spectra of the crude reaction mixture. The decrease in the amount of 2a (2 equiv.) also decreased the yield (71%, 10 h). A similar high yield was achieved with a lower catalyst loading of 1 mol%; however, the reaction was only complete after a long time (run 2). The reaction with CuOAc proceeded faster to give 3aa in a similar yield (run 3). In this case, a trace amount of 4 was observed in the crude reaction mixture. A similar yield was achieved with even lower loadings of the catalyst (1 mol%) and 2a (1.5 equiv.) (runs 4 and 5). Encouraged by these results, we examined other copper salts, but none of them were superior to the acetates. CuCl and CuBr gave the desired product in comparable yields (runs 6 and 7), although their reactions were only complete after a long time. In contrast, CuI, CuCl2, and CuBr2 hardly exhibited any catalytic activity. Electron-donating ligands such as 2,2′-bipyridine (bipy) and N-heterocyclic carbenes [1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene: SIiPr; or 1,3-dimesitylimidazol-2-ylidene: IMes]7 did not show any positive effect (runs 8–10). Accordingly, CuOAc was employed for further study.
| Run | Cu salt, mol% | Time/h | Yield (%) |
|---|---|---|---|
| a Trace amounts of 4 were detected in 1H NMR spectra of crude samples. b 1.5 equiv. of 2a was used. | |||
| 1 | Cu(OAc)2, 10 mol% | 2.5 | 96 |
| 2 | Cu(OAc)2, 1 mol% | 8 | 95 |
| 3 | CuOAc, 10 mol% | 1 | 95a |
| 4 | CuOAc, 1 mol% | 5 | 94a |
| 5 | CuOAc, 1 mol%b | 6 | 95a |
| 6 | CuCl, 10 mol% | 14 | 93 |
| 7 | CuBr, 10 mol% | 9 | 92a |
| 8 | Cu(OAc)2(bipy), 5 mol% | 2 | 95a |
| 9 | CuCl(SIiPr), 5 mol% | 24 | 80a |
| 10 | CuCl(IMes), 5 mol% | 24 | 61a |
The scope and limitations of the arylboronic acid are summarised in Table 2.8 To ensure complete consumption of the alkynoates, 3 equiv. of arylboronic acid were employed. In the presence of 1–3 mol% CuOAc, o-, m-, and p-tolylboronic acids, 3,5-xylylboronic acid, and 2-naphthylboronic acid gave the corresponding adducts 3ab–3af in high yields (runs 1–5). Similarly, p-halophenylboronic acids 2g–i can be used without the loss of the reactive C(sp2)–halogen bonds that are useful synthetic handles for carrying out further functionalization (runs 6–8). Moreover, carbonyl groups reactive towards Grignard or organolithium reagents are compatible with the present Cu-catalyzed conjugate addition of arylboronic acids (runs 9–11). Consequently, (E)-cinnamates3aj–3al, whose phenyl rings have a formyl, acetyl, or ethoxycarbonyl group, were obtained in 82–93% yields. Although a catalyst loading of 10 mol% was required, m-nitro derivative 3am was also obtained in 94% yield (run 12). However, the use of electron-rich p-methoxyphenylboronic acid2n was problematic, leading to an inseparable mixture of the desired compound 3an, 4,4′-dimethoxybiphenyl, and 1,4-dimethoxybenzene (run 13). The latter side products were formed probably by the Cu-catalyzed homo coupling of 2n and its Ullmann-type coupling with the methanol solvent. In contrast to phenylboronic acids, heteroaryl-, ferrocenyl-, 2-(p-chlorophenyl)ethenyl-, and n-pentylboronic acids did not undergo conjugate addition to alkynoate 1a under the optimal reaction conditions (Fig. 1).
 | ||
| Fig. 1 Reagents which failed to give conjugate addition products. | ||
| Run | 2, Ar | Cu (mol%) | Time/h | 3, yield (%) |
|---|---|---|---|---|
| a Methyl 2-octynoate (1a) was reacted with 3 equiv. of the arylboronic acid in MeOH (0.5 M) at 28 °C. b Yield determined by 1H NMR analysis of a mixture with 4,4′-dimethoxybiphenyl and 1,4-dimethoxybenzene. | ||||
| 1 | 2b, p-MeC6H4 | 1 | 4 | 3ab, 94 |
| 2 | 2c, m-MeC6H4 | 1 | 10 | 3ac, 94 |
| 3 | 2d, o-MeC6H4 | 3 | 6 | 3ad, 91 |
| 4 | 2e, 3,5-Me2C6H3 | 3 | 2 | 3ae, 88 |
| 5 | 2f, 2-naphthyl | 1 | 24 | 3af, 90 |
| 6 | 2g, p-ClC6H4 | 3 | 4 | 3ag, 94 |
| 7 | 2h, p-BrC6H4 | 5 | 24 | 3ah, 94 |
| 8 | 2i, p-IC6H4 | 5 | 3 | 3ai, 91 |
| 9 | 2j, p-OHCC6H4 | 3 | 10 | 3aj, 88 |
| 10 | 2k, p-AcC6H4 | 3 | 5 | 3ak, 93 |
| 11 | 2l, p-EtO2CC6H4 | 3 | 24 | 3al, 82 |
| 12 | 2m, m-O2NC6H4 | 10 | 5 | 3am, 94 |
| 13 | 2n, p-MeOC6H4 | 1 | 24 | 3an, 60b |
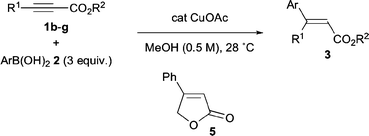
Then, we examined the catalytic conjugate addition of arylboronic acids to various alkynoates 1b–1g (Scheme 2 and Table 3). Alkynoates 1b,c whose alkyl chains have methoxy or chloro substituents were allowed to react with phenylboronic acid (2a), affording the corresponding cinnamates3ba and 3ca in excellent yields (runs 1 and 2). It is noteworthy that protection-free propargyl alcohol derivative 1d can be directly used in our Cu-catalyzed conjugate addition. In this case, the conjugate addition of 2a was followed by in situ lactonization to yield the known 3-phenylbutenolide5, albeit in a moderate yield of 61% (run 3). We then focused on the synthesis of 3,3-diarylacrylates that are difficult to prepare with precise control of stereochemistry via the conventional Horner–Wittig and Wadsworth–Emmons methods. In the presence of 1–3 mol% CuOAc, phenylboronic acids bearing methyl, chloro, and methoxy substituents at the para-position were allowed to react with ethyl phenylpropiolate 1e, affording the corresponding diarylacrylates 3eb, 3eg, and 3en in high yields as single stereoisomers (runs 4–6). It is noteworthy that electron-rich 2n reacted with 1e uneventfully to yield (E)-3en in 91% yield. On the other hand, its stereoisomer (Z)-3fa was selectively obtained by the addition of phenylboronic acid to (p-methoxyphenyl)propiolate1f (run 7). The present method enabled the stereoselective preparation of 3gn possessing o- and p-methoxyphenyl groups (run 8).
 | ||
| Scheme 2 Reaction of various alkynoates 1b–1g. | ||
| Run | 1, R1/R2 | 2, Ar | Conditionsa | 3, yield (%) |
|---|---|---|---|---|
| a All reactions were carried out with CuOAc in 0.5 M MeOH solution at 28 °C. | ||||
| 1 | 1b, MeO(CH2)3/Me | 2a, Ph | 2 mol%, 3 h | 3ba, 92 |
| 2 | 1c, Cl(CH2)3/Me | 2a, Ph | 1 mol%, 2 h | 3ca, 97 |
| 3 | 1d, HOCH2/Me | 2a, Ph | 2 mol%, 2 h | 5, 61 |
| 4 | 1e, Ph/Et | 2b, p-MeC6H4 | 1 mol%, 4 h | 3eb, 90 |
| 5 | 1e, Ph/Et | 2g, p-ClC6H4 | 3 mol%, 12 h | 3eg, 89 |
| 6 | 1e, Ph/Et | 2n, p-MeOC6H4 | 1 mol%, 4 h | 3en (E), 91 |
| 7 | 1f, p-MeOC6H4/Et | 2a, C6H5 | 1 mol%, 24 h | 3fa (Z), 97 |
| 8 | 1g, o-MeOC6H4/Me | 2n, p-MeOC6H4 | 3 mol%, 1 h | 3gn, 88 |
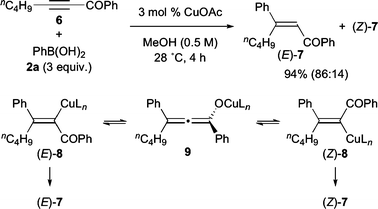
Similar to other alkynyl ester substrates, methyl propiolate and its trimethylsilyl and bromo analogues were also tested, but none of them gave the desired product (Fig. 1). This result is in contrast to that of alkynyl ketone 6 that underwent conjugate addition to yield the corresponding adduct 7 in a high yield with a moderate stereoselectivity of E : Z = 86 : 14 (Scheme 3). The minor stereoisomer (Z)-7 was considered to be formed due to the isomerization of the initially formed vinylcopper species (E)-8 to (Z)-8via transient allenolate 9, as previously reported for the related conjugate additions of organocopper reagents to alkynoates.1,5,9
To obtain further insight into the reaction mechanism, we carried out the reaction of 1a and 2a (1.5 equiv.) in MeOD (Scheme 4). As a result, mono-deuterated (E)-3aa-d was obtained with 74% D content, indicating that the hydroxy group of methanol behaved as a proton donor. Insufficient deuteration might be attributed to the H–D exchange between MeOD and 2a or direct proton transfer from 2a to an alkenylcopper intermediate (see below). Thus, the use of 2-phenyl-5,5-dimethyl-1,3,2-dioxaborinane10 instead of 2a gave (E)-3aa-d with a D content of more than 98%.
 | ||
| Scheme 4 Reactions in MeOD. | ||
Scheme 5 outlines a plausible mechanism of the Cu-catalyzed conjugate addition of arylboronic acids to alkynoates. Transmetallation from the arylboronic acids to copper methoxide (or acetate) proceeds via a four-centered transition state to yield reactive arylcopper species.6 Subsequent carbocupration of the alkynoates produces vinylcopper intermediates, which then undergo protonolysis by methanol to yield the final cinnamates with the concomitant restoration of copper methoxide. In previous cuprate-based methods, a low reaction temperature was required to prevent the isomerization of the initially formed syn-carbocupration adducts to anti-isomers via allenolate intermediates.1,5,9 Because our protocol employs methanol as a solvent, the vinylcopper intermediates undergo facile protonolysis before isomerization, resulting in the stereoselective formation of the syn-hydroarylation products even at ambient temperature.
 | ||
| Scheme 5 Proposed mechanism. | ||
In conclusion, we succeeded in carrying out the catalytic conjugate addition of arylboronates to alkynoates.10 The reaction proceeded in MeOH under mild conditions to yield trisubstituted cinnamates with precise syn-selectivity. This protocol is compatible with phenylboronic acids bearing carbon–halogen bonds as well as carbonyl functional groups.
This research was partially supported by the Ministry of Education, Science, Sports and Culture, Grant-in-Aid for Young Scientists (A), 17685008, and the Global COE program (Tokyo Instituted of Technology). We thank Prof. H. Suzuki (Tokyo Institute of Technology) for use of the GC mass spectrometer, and Prof. K. Tomooka (Kyushu University) for use of the NMR spectrometer.
Notes and references
- K. Nilsson, T. Andersson, C. Ullenius, A. Gerold and N. Krause, Chem.–Eur. J., 1998, 4, 2051 CrossRef CAS , and references cited; G. Li, H.-X. Wei, B. R. Whittlesey and N. N. Batrice, J. Org. Chem., 1999, 64, 1061 Search PubMed; M. C. P. Yeh and P. Knochel, Tetrahedron Lett., 1989, 30, 4799 CrossRef CAS; Y. Yamamoto, H. Yatagai and K. Maruyama, J. Org. Chem., 1979, 44, 1744 CrossRef CAS.
- (a) E. J. Corey and J. A. Katzenellenbogen, J. Am. Chem. Soc., 1969, 91, 1851 CrossRef CAS; (b) J. B. Siddall, M. Biskup and J. H. Fried, J. Am. Chem. Soc., 1969, 91, 1853 CrossRef CAS; (c) J. Klein and R. M. Turkel, J. Am. Chem. Soc., 1969, 91, 6186 CrossRef CAS.
- T. Hayashi, K. Inoue, N. Taniguchi and M. Ogasawara, J. Am. Chem. Soc., 2001, 123, 9918 CrossRef CAS.
- C. H. Oh and J. H. Ryu, Bull. Korean Chem. Soc., 2003, 24, 1563 CAS; C. H. Oh, H. H. Jung, K. S. Kim and N. Kim, Angew. Chem., Int. Ed., 2003, 42, 805 CrossRef CAS; A. K. Gupta, K. S. Kim and C. H. Oh, Synlett, 2005, 457 CAS.
- A. J. Mueller and M. P. Jennings, Org. Lett., 2007, 9, 5327 CrossRef CAS; M. P. Jennings and K. B. Sawant, Eur. J. Org. Chem., 2004, 3201 CrossRef CAS.
- T. Hayashi, M. Takahashi, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 2002, 124, 5052 CrossRef CAS; T. Nishikawa, Y. Yamamoto and N. Miyaura, Organometallics, 2004, 23, 4317 CrossRef CAS.
- S. Okamoto, S. Tominaga, N. Saino, K. Kase and K. Shimoda, J. Organomet. Chem., 2005, 690, 6001 CrossRef CAS; V. Lillo, M. R. Fructos, J. Ramirez, A. A. C. Braga, F. Maseras, M. M. Diaz-Requejo, P. J. Pérez and E. Fernández, Chem.–Eur. J., 2007, 13, 2614 CrossRef CAS.
- Most of the arylboronic acids were purchased and used as received. These boronic acids usually contain varying amounts of the corresponding anhydrides, including triarylboroxins. We hence examined the reaction of alkynoate 1a with triphenylboroxin (1 equivalent) otherwise under the same conditions. The reaction reached completion within 10 h to furnish (E)-3aa in 94% yield. This result shows that the amount of anhydride might influence only the reaction rate.
- J. Klein and R. Levene, J. Chem. Soc., Perkin Trans. 2, 1973, 1971 RSC; J. P. Marino and R. J. Linderman, J. Org. Chem., 1983, 48, 4621 CrossRef CAS; M. Ahlquist, T. E. Nielsen, S. Le Quement, D. Tanner and P.-O. Norrby, Chem.–Eur. J., 2006, 12, 2866 CrossRef CAS.
- During the preparation of this manuscript, the Cu-catalyzed syn-selective conjugate addition of diboron reagents to alkynoates was reported, see: J.-E. Lee, J. Kwon and J. Yun, Chem. Commun., 2008, 733 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/b802231c |
| This journal is © The Royal Society of Chemistry 2008 |