RhI-catalyzed aldol-type reaction of organonitriles under mild conditions†
Akihiro
Goto
a,
Kohei
Endo
ac,
Yu
Ukai
a,
Stephan
Irle
b and
Susumu
Saito
*ab
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Chikusa, Nagoya, 464-8602, Japan. E-mail: susumu@chem.nagoya-u.ac.jp; Fax: +81 52 789 5945; Tel: +81 52 789 5945
bInstitute for Advanced Research, Nagoya University, Chikusa, Nagoya, 464-8601, Japan. E-mail: susumu@chem.nagoya-u.ac.jp; Fax: +81 52 788 6140; Tel: +81 52 788 6140
cDepartment of Chemistry and Biochemistry, School of Advanced Science and Engineering, Waseda University, Ohkubo, Shinjuku, Tokyo, 169-8555, Japan
First published on 26th March 2008
Abstract
An aldol-type reaction of organonitriles with aldehydes was catalyzed by a RhI(OR) species under ambient conditions, and the reaction displayed a broad substrate scope with respect to both organonitrile and aldehyde components.
The nitrile functionality is synthetically versatile for many functional group conversions.1 Incorporation of a nitrile fragment into a carbon framework provides rapid access to a number of useful chemicals such as α-amino and α-hydroxy carboxylic acid derivatives as well as nitrile-branching polymers. Concerned with this goal, cyanation strategies2 have been most widely employed so far. In contrast, aldol-type reactions of alkylnitriles are those in which nitriles serve as enolate equivalents incorporating aldehyde directly. This alternative strategy for introducing the nitrile functionality is an attractive process, providing β-hydroxynitriles, which are potential precursors for pharmaceutically important substances.2 Although pioneering achievements related to this end have been reported recently by Murahashi,3 Verkade,4 Knochel,5 Shibasaki and Kanai6 and others,7 some drawbacks resulting from concomitant dehydration,3 high catalyst loading (∼0.05 molar equiv.) and alkylnitrile loading (ca. 20 molar equiv.)4–6 need improvement in order to increase catalytic efficiency and atom economy. We report here on the first example of the RhI-catalyzed aldol-type reaction of unactivated alkylnitriles under mild conditions, employing lower catalyst and substrate loading, thereby expanding substrate scope with respect to both aldehyde and organonitrile components (Scheme 1).
 | ||
| Scheme 1 General scheme of the Rh-catalyzed nitrile aldol reaction. | ||
The Rh catalyst was prepared by treatment of [Rh(OH)(cod)]2 (0.01 molar equiv.) with Cy3P (0.04 molar equiv.) in toluene at 25 °C for 0.5 h under argon (Cy = c-hexyl). After the solvent and cod were removed by evaporation in vacuum, MeCN (2a: 19 molar equiv.) and benzaldehyde (1a: 1 molar equiv.) were added sequentially to the resulting yellowish viscous oil containing the Rh complex at 25 °C. The resulting suspension was kept at 50 °C with stirring for 24 h, during which time the mixture became a clear solution, giving the β-hydroxynitrile 3aa in an isolated yield of 61% after column chromatography on silica gel. The use of Cy3P alone (0.04 molar equiv.) resulted in complete recovery of 1a. In addition, when [RhCl(cod)]2 was used in place of [Rh(OH)(cod)]2, no reaction was observed, suggesting that the hydroxyl group of Rh worked as a base. In contrast, a slightly better yield of 3aa was obtained with [Rh(OMe)(cod)]2 (70%). Two molar equivalents of Cy3P per Rh ([Rh(OMe)(cod)]2 : Cy3P = 1 : 4) was most appropriate, as equal amounts decreased the yield considerably (40%) and a three-fold excess gave a comparable result (66%). For reference, less basic phosphines (Ph3P, (o-Tol)3P and n-Bu3P) and a phosphite ((PhO)3P) were tested but shown to be far less effective than Cy3P under otherwise identical conditions (3aa: <16% with n-Bu3P, and ∼1% with the others). The use of bidentate phosphines such as dppe, dppp, dppb, and (R)-BINAP resulted consistently in lower conversions (<23%). The solvent screening with [Rh(OMe)(cod)]2–4Cy3P (Rh: 0.01 M; 50 °C, 24 h) suggested that aprotic polar solvents including DMSO, DMF, DMA, NMP and DMI were more promising (3aa: 77–84%). In t-BuOH the reaction proceeded more sluggishly (61%), but better than in MeOH (31%).
Finally, [Rh(OMe)(cod)]2 was tested further, and after an additional set of experiments this was proven to be the best surrogate for [Rh(OH)(cod)]2, affording the highest productivity (99% of 3aa with [Rh(OMe)(cod)]2 (0.01 molar equiv.) and Cy3P (0.04 molar equiv.); Rh: 0.01 M in DMSO, 25 °C, 6 h). We chose DMSO as a representative solvent and further screened additional phosphine ligands (Fig. 1). Among the ones tested we found that R3P ligands or the 2-(1,1′-biphenyl)PR2 ligands 5a (R = Cy) and 5b (R = i-Pr) were the most potent, affording the highest conversion of 2a (>95%). Surprisingly, t-Bu3P or other ArCy2P- and Ar(t-Bu)2P-based phosphines5c–e were totally unsatisfactory (3aa: <5%), suggesting that the reaction is strongly structure-demanding and sensitive to the steric bulk around the outer sphere of Rh.
 | ||
| Fig. 1 Biphenyl-based phosphines tested in the nitrile aldol reaction. | ||
Given the above optimal conditions,‡ the substrate scope was then investigated (Tables 1 and 2). Aromatic, heteroaromatic, α,β-unsaturated and aliphatic aldehydes1a–u were all suitable substrates (Table 1), although a somewhat lower yield was obtained with linear aldehyde1p due to accompanying self-condensation (entry 16). 1,2-Addition predominated with the α,β-unsaturated aldehyde1n (entry 14). The 0.005 and 0.02 molar equivalents of [Rh(OMe)(cod)]2 and Cy3P used (respectively) were enough to achieve a reasonable yield (3aa: 90%) (entry 1). t-BuOH was a better solvent in several cases (entries 16, 18, 19 and 21) in terms of isolated yields of 3. An attempt to reduce the molar equivalents of MeCN from 19 to 2–3 was also successful (Table 2). Even under these nitrile-saving conditions, near-maximum product yields were obtained in several cases (entries 1–4 and 6), albeit with a prolonged reaction time (24 h). Although low diastereoselectivities were obtained from 2b–2e (entries 2–5), this modification reduced the amount of time spent separating and processing the reaction mixture to remove excess alkylnitriles, especially those of a higher molecular weight, from the products (Fig. 2).
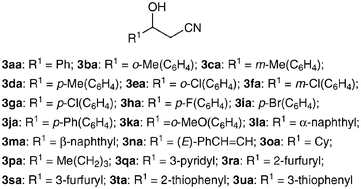 | ||
| Fig. 2 Aldol products 3aa–3ua. | ||
| Entry | Aldehyde | Product | Yield (%)b |
|---|---|---|---|
| a Reagents and conditions: [Rh(OMe)(cod)]2 : Cy3P : 2a : aldehyde = 0.01 : 0.04 : 19 : 1; 25 °C, 6 h in anhydrous DMSO (Rh: 0.01 M). b Yield of isolated, purified products 3aa–3ua (Fig. 2). c 0.005 molar equiv. of [Rh(OMe)(cod)]2 and 0.02 molar equiv. of Cy3P were used. d Reaction time: 18–24 h. e [Rh(OMe)(cod)]2 : Cy3P : 2a = 0.02 : 0.08 : 38; 24 h; Rh: 0.01 M in t-BuOH. f [Rh(OMe)(cod)]2 : Cy3P : 2a = 0.02 : 0.08 :77 ; 24 h; Rh: 0.0066 M in t-BuOH. g [Rh(OMe)(cod)]2 : Cy3P : 2a = 0.02 : 0.08 : 77; 48 h; Rh: 0.005 M in t-BuOH. | |||
| 1 | C6H5CHO (1a) | 3aa | 99 (90)c,d |
| 2 | o-CH3(C6H4)CHO (1b) | 3ba | 97 |
| 3 | m-CH3(C6H4)CHO (1c) | 3ca | 98 |
| 4 | p-CH3(C6H4)CHO (1d) | 3da | 88 |
| 5 | o-Cl(C6H4)CHO (1e) | 3ea | 98 |
| 6 | m-Cl(C6H4)CHO (1f) | 3fa | 99 |
| 7 | p-Cl(C6H4)CHO (1g) | 3ga | 99 |
| 8 | p-F(C6H4)CHO (1h) | 3ha | 99 |
| 9 | p-Br(C6H4)CHO (1i) | 3ia | 99 |
| 10 | p-Ph(C6H4)CHO (1j) | 3ja | 95 |
| 11 | o-CH3O(C6H4)CHO (1k) | 3ka | 93 |
| 12 | (α-naphthyl)CHO (1l) | 3la | 98 |
| 13 | (β-naphthyl)CHO (1m) | 3ma | 95 |
| 14 | (E)-PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHO (1n) CHCHO (1n) |
3na | 89 |
| 15 | CyCHO (1o) | 3oa | 99d |
| 16 | CH3(CH2)3CHO (1p) | 3pa | 76d (83)e |
| 17 |
 (1q) (1q) |
3qa | 99 |
| 18 |
 (1r) (1r) |
3ra | 99f |
| 19 |
 (1s) (1s) |
3sa | 73g |
| 20 |
 (1t) (1t) |
3ta | 56 |
| 21 |
 (1u) (1u) |
3ua | 68e |
| Entry | Nitrile | Product | Yield %b |
|---|---|---|---|
| a Reagents and conditions: [Rh(OMe)(cod)]2 : Cy3P : nitrile : aldehyde = 0.01 : 0.04 : 3 : 1; 25 °C, 24 h in anhydrous DMSO (Rh: 0.03–0.04 M). b Yield of isolated, purified products. c 2 molar equiv. of 2a were used. d Diastereomeric ratio = ca. 1 : 1.2. e [Rh(OMe)(cod)]2 : Cy3P = 0.02 : 0.08; 24 h; Rh: 0.04 M in DMSO. | |||
| 1 | CH3CN (2a) | PhCH(OH)CH2CN (3aa) | 98 (88)c |
| 2 | CH3CH2CN (2b) | PhCH(OH)CH(CH3)CN (4ab) | 98d |
| 3 | CH3(CH2)4CN (2c) | PhCH(OH)CH[(CH2)3CH3]CN (4ac) | 81d,e |
| 4 | (C6H5)CH2CN (2d) | PhCH(OH)CH(C6H5)CN (4ad) | 99d |
| 5 | CH3OCH2CN (2e) | PhCH(OH)CH(OCH3)CN (4ae) | 56d,e |
| 6 | (CH3)2CHCN (2f) | PhCH(OH)C(CH3)2CN (4af) | 95 |
In summary, we have demonstrated that the Rh(OMe)-catalyzed aldol-type reaction of organonitriles with aldehydes is a useful method for obtaining high yields of the corresponding β-hydroxynitriles. The reaction is chemoselective and promising, with catalyst precursor loading (as low as 0.005 molar equiv.) and nitrile loading (as low as 2 molar equiv.) lower than those reported previously.3–6 Investigations into the mechanistic aspects, including ab initio calculations ,§ are now underway in our laboratories.
This work was partially supported by a Grant-in-Aid for Young Scientists (A) and Scientific Research on Priority Areas “Advanced Molecular Transformations of Carbon Resource” from the Ministry of Education, Culture, Sports, Science and Technology, Japan, as well as Asahi Glass and Sumitomo Foundation. SS also greatly appreciates Professor R. Noyori (Nagoya University & RIKEN) for his valuable suggestions and fruitful discussions.
Notes and references
- Chemistry of the Cyano Group, ed. Z. Pappoport and S. Patai, John Wiley & Sons, London, 1970 Search PubMed; A. J. Fatiadi, in The Chemistry of Functional Groups, Supplement C: The Chemistry of Triple-bonded Functional Groups, Part 2, ed. Z. Pappoport and S. Patai, John Wiley & Sons, London, 1983, pp. 1057–1303 Search PubMed; F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035–2077 Search PubMed.
- G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779–794 CrossRef CAS; R. J. H. Gregory, Chem. Rev., 1999, 99, 3649–3682 CrossRef CAS.
- S.-I. Murahashi, T. Naota, H. Taki, M. Mizuno, H. Takaya, S. Komiya, Y. Mizuho, N. Oyasato, M. Hiraoka, M. Hirano and A. Fukuoka, J. Am. Chem. Soc., 1995, 117, 12436–12451 CrossRef CAS; S.-I. Murahashi and T. Naota, Bull. Chem. Soc. Jpn., 1996, 69, 1805–1824 CAS.
- P. Kisanga, D. Mcleod, B. D’Sa and J. Verkade, J. Org. Chem., 1999, 64, 3090–3094 CrossRef CAS.
- A. L. Rodriguez, T. Bunlaksananusorn and P. Knochel, Org. Lett., 2000, 2, 3285–3287 CrossRef CAS; T. Bunlaksananusorn, A. L. Rodriguez and P. Knochel, Chem. Commun., 2001, 745–746 RSC.
- Y. Suto, N. Kumagai, S. Matsunaga, M. Kanai and M. Shibasaki, Org. Lett., 2003, 5, 3147–3150 CrossRef CAS; N. Kumagai, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 13632–13633 CrossRef CAS; Y. Suto, R. Tsuji, M. Kanai and M. Shibasaki, Org. Lett., 2005, 7, 3757–3760 CrossRef CAS; N. Kumagai, S. Matsunaga and M. Shibasaki, Chem. Commun., 2005, 3600–3602 RSC.
- S. Paganelli, A. Schionato and C. Botteghi, Tetrahedron Lett., 1991, 32, 2807–2810 CrossRef CAS; Y. Lin, X. Zhu and M. Xiang, J. Organomet. Chem., 1993, 448, 215–218 CrossRef CAS; R. Kuwano, H. Miyazaki and Y. Ito, Chem. Commun., 1998, 71–72 RSC.
- T. Itoh, K. Mitsukura, W. Kanphai, Y. Takagi, H. Kihara and H. Tsukube, J. Org. Chem., 1997, 62, 9165–9172 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental information and spectral data of new compounds. See DOI: 10.1039/b800634b |
| ‡ General procedure: To a degassed and argon-saturated solution of [Rh(OMe)(cod)]2 (4.8 mg, 0.01 mmol) in anhydrous toluene (0.5 mL) was quickly added a 1.0 M toluene solution of Cy3P (40 µL, 0.04 mmol; commercially available from Aldrich) at 25 °C and the mixture was stirred at this temperature for 0.5 h. After evaporation of any volatile compounds in vacuo (1–3 Torr), degassed and argon-saturated DMSO (1.0 mL) was added to the resulting slurry, followed by sequential addition of CH3CN (2a) (1.0 mL, 19.1 mmol) and PhCHO (1a) (103 µL, 1 mmol). The reaction mixture was stirred at 25 °C for 6 h and was diluted with Et2O to dissolve all the precipitate. The entire mixture was filtered through a short pad of silica gel, transferred into a 100 mL round-bottomed flask with Et2O, evaporated and concentrated. The residue was purified by column chromatography on silica gel (EtOAc–n-hexane = 2 : 1) to give β-hydroxynitrile 3aa8 (146 mg, 99% yield). |
§ Preliminary calculations (B3LYP/LANL2DZ level) revealed that the RhI(OH), complexed with one or two Cy3P, changed their oxidation state upon MeCN addition, giving cationic (Cy3P)nRhIII(OH)(N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CMe) species in both cases. Full accounts on this and further theoretical investigations will appear elsewhere. CMe) species in both cases. Full accounts on this and further theoretical investigations will appear elsewhere. |
| This journal is © The Royal Society of Chemistry 2008 |