Changes in interfacial properties of α-synuclein preceding its aggregation
Emil
Paleček
*a,
Veronika
Ostatná
a,
Michal
Masařík
a,
Carlos W.
Bertoncini†
b and
Thomas M.
Jovin
b
aInstitute of Biophysics, Academy of Sciences of the Czech Republic v.v.i., Kralovopolska 135, 612 65 Brno, Czech Republic. Fax: +420 5 415 17 249; Tel: +420 5 492 46 241E-mail: palecek@ibp.cz
bDepartment of Molecular Biology, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, D-37077, Göttingen, Germany
First published on 15th October 2007
Abstract
Parkinson's disease (PD) is associated with the formation and deposition of amyloid fibrils of the protein α-synuclein (AS). It has been proposed that oligomeric intermediates on the pathway to fibrilization rather than the fibrils themselves are the pathogenic agents of PD, but efficient methods for their detection are lacking. We have studied the interfacial properties of wild-type AS and the course of its aggregation in vitro using electrochemical analysis and dynamic light scattering. The oxidation signals of tyrosine residues of AS at carbon electrodes and the ability of fibrils to adsorb and catalyze hydrogen evolution at hanging mercury drop electrodes (HMDEs) decreased during incubation. HMDEs were particularly sensitive to pre-aggregation changes in AS. Already after 1 h of a standard aggregation assay in vitro (stirring at 37 °C), the electrocatalytic peak H increased greatly and shifted to less negative potentials. Between 3 and 9 h of incubation, an interval during which dynamic light scattering indicated AS oligomerization , peak H diminished and shifted to more negative potentials, and AS adsorbability decreased. We tentatively attribute the very early changes in the interfacial behavior of the protein after the first few hours of incubation to protein destabilization with disruption of long-range interactions. The subsequent changes can be related to the onset of oligomerization. Our results demonstrate the utility of electrochemical methods as new and simple tools for the investigation of amyloid formation.
Introduction
A number of human diseases are associated with protein misfolding resulting in malfunctioning of the cellular machinery.1,2 Particular attention has been focused on a group of diseases in which proteins convert from their normally soluble forms into insoluble fibrils.3–6 The final forms of such aggregates often have a well-defined fibrillar nature known as amyloids. The group of about 20 diseases includes the major neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease (PD) and Creutzfeldt–Jakob disease. PD is the second most common neurodegenerative disease.Recent studies indicate that α-synuclein (AS) is a key player in the pathogenesis of PD and some other neurodegenerative disorders.7 AS was first described in 1988 as a neuron-specific protein localized to the nucleus.8 The primary sequence of the human 14 kDa AS comprises 140 amino acids (aa) and can be sub-divided into three regions: (i) residues 1–60 constitute the N-terminal region containing four 11-aa imperfect repeats with the hexamer motif KTKEGV; (ii) the central region includes the highly amyloidogenic NAC sequence (residues 61–95) containing two additional KTKEGV sequences; and (iii) the C-terminal region comprises amino acids 96–140, rich in acidic residues and prolines and including three highly conserved tyrosines,9 and is presumably disordered under most conditions.
AS is natively unfolded but undergoes aggregation leading to fibrilar structures,7 a process induced in vitro by a variety of means and promoted in vivo by a variety of conditions that generate oxidative stress as well as by numerous point mutations in the AS gene.10–12 Aggregated AS is highly polymorphic, exhibiting a number of different forms depending on the time of incubation and solution conditions, with mature amyloid fibrils constituting the predominant structure in fully aggregated solutions in physiological buffers.7,13–18 AS aggregation in vitro is commonly studied by several methods such as circular dichroism spectroscopy, fluorescence measurements [e.g.thioflavin T (ThioT) binding], electron microscopy and atomic force microscopy.16,17,19–21 Although the central role of AS fibrilization in the pathogenesis of PD is well established,22–24 recent evidence indicates, that the fibril itself may not be the primary pathogenic species.25–29 Unfortunately, the direct detection of defined prefibrilar intermediates (protofibrils), currently presumed to be highly cytotoxic, has proven extremely difficult. The oligomeric species of AS are relatively unstable, transient and present at very low steady-state concentrations.29,30 The incorporation of unusual amino acids into AS and the systematic generation of mutants are just two of the strategies adopted for characterizing intermediates during amyloid formation.31,32
Until our first publication on the subject,33 methods of electrochemical analysis had not been applied in studies of AS. At first sight this protein seems unsuitable for these methods, lacking a redox active center for reversible electrochemistry and cystine or cysteine residues capable of producing signals at mercury electrodes.34 In addition, tryptophan is absent from AS, although there are four tyrosines that can undergo oxidation at carbon electrodes .34–36
Recently we have shown that peptides and proteins , at mercury electrodes, produce a chronopotentiometric peak at highly negative potentials (peak H), which is due to the catalytic hydrogen evolution.34,37,38 This peak differs from the previously described polarographic and voltammetric electrocatalytic signals of proteins 34 (i) by its ability to detect peptides and proteins down to nanomolar and sub-nanomolar concentrations, and (ii) by its remarkable sensitivity to local and global changes in protein structures.34,39 We have previously applied chronopotentiometric and voltammetric methods to investigate (i) the ability of AS to catalyze hydrogen evolution at mercury electrodes, and (ii) the oxidizability of tyrosine residues at carbon electrodes .33,34 We tested various methods of electrochemical stripping analysis in combination with the hanging mercury drop electrode (HMDE). The best results were obtained by the constant current chronopotentiometric analysis (CPSA) that produced a well-developed peak H at low concentrations of the protein .33 Much higher AS concentrations were required to obtain an oxidation peak Y of tyrosine residues at the carbon paste electrodes at +0.8 V by square wave voltammetric (SWV) stripping analysis. Both peak H and Y decreased as a result of AS aggregation in vitro but the changes in peak H were much larger than those of peak Y. In a separate study, carbon electrodes were used in combination with voltammetry to study the aggregation of the Aβ-amyloid peptides involved in Alzheimer's disease.40 A decrease in oxidation peak of tyrosine in the course of the peptide aggregation was observed but pre-aggregation changes were not reported.
In this paper we describe the interfacial behavior of native wild-type AS and investigate its aggregation in vitro by means of peak H and by a.c. impedance measurements. We attribute characteristic changes in both peak H and the adsorption/desorption behavior of AS in the early stage of AS incubation to pre-aggregation, i.e. to an incipient stage of amyloid formation.
Results and discussion
Adsorption/desorption behavior of native AS
The differential capacity C of the electrode double layer is a sensitive indicator of adsorption.41 When a protein is adsorbed at the electrode surface, it removes from the surface ions and molecules of the solvent and at the same time lowers the value of C due to the generally much higher dielectric permittivity of solvents compared to that of protein solutions. We measured the dependence on the polarization potential E of the specific differential capacity (Cs) of the electrode double layer of AS-modified HMDE immersed in a blank background electrolyte. The adsorptive transfer stripping technique was used.33 AS was adsorbed from 50 mM Na phosphate, pH 7.0, at the accumulation time tA of 60 s. Native AS significantly decreased the differential capacity Cs over a wide potential range of about –0.1 to –1.2 V [Fig. 1(a)]. ΔCs at –0.65 V [Fig. 1(c)] increased with increasing AS concentration and saturated between 1 and 2 µM concentration of the protein [ΔCs = 7.37 µF cm–2; Fig. 1(a,c)], suggesting that the full coverage of the electrode was reached. Between 2 and 10 µM AS, practically no changes in ΔCs were observed.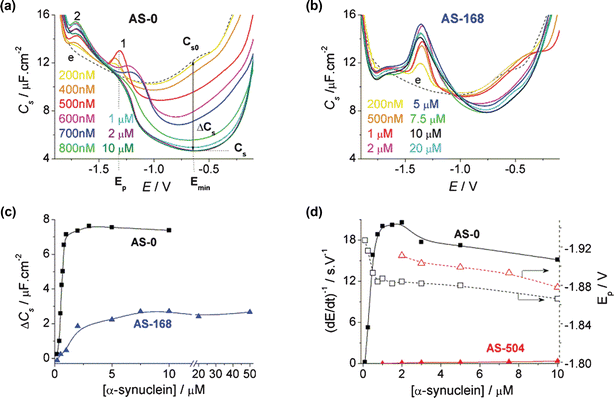 | ||
Fig. 1 Dependence of interfacial properties on the concentration of native and aggregated α-synuclein (AS). (a,b) Adsorptive transfer stripping (AdTS, ex situ) C–E curves of AS. Dependence on concentration of (a) native AS (untreated AS-0) and of (b) aggregated (168 h treated, AS-168). In (a) the schematic representations of C–E curves of AS (—), and e, the background electrolyte (![[dash dash, graph caption]](https://www.rsc.org/images/entities/b_char_e091.gif) ), are also shown. Emin is the potential at which the differential capacity Cs reaches its minimum value; Cs is the differential capacity of the AS-modified hanging mercury drop electrode (HMDE) at the given potential; Cs0 is the differential capacity of the bare HMDE in the background electrolyte; ΔCs = Cs – Cs0; Ep is the peak potential. (c) Dependence of ΔCs at –0.65 V on the concentration of AS-0 (–■–) and AS-168 (–▲–). (d) Dependence of height (—) and peak potential, Ep () of the chronopotentiometric peak H on the concentration of untreated AS-0 (black) and aggregated fibrils AS-504 (red). AS was adsorbed at the HMDE from a 5 µl drop for the accumulation time (tA) 60 s at open current circuit. The AS-modified HMDE was washed and transferred into an electrolytic cell containing 10 ml of a blank background electrolyte (50 mM Na phosphate, pH 7.0) and the C–E curve was measured. For details see the Experimental section. Conditions of aggregation: 100 µM AS in 0.1 M NaCl, 25 mM Tris-HCl, pH 7.3, was stirred at 300 rpm at 37 °C; samples were withdrawn at the indicated time intervals. ), are also shown. Emin is the potential at which the differential capacity Cs reaches its minimum value; Cs is the differential capacity of the AS-modified hanging mercury drop electrode (HMDE) at the given potential; Cs0 is the differential capacity of the bare HMDE in the background electrolyte; ΔCs = Cs – Cs0; Ep is the peak potential. (c) Dependence of ΔCs at –0.65 V on the concentration of AS-0 (–■–) and AS-168 (–▲–). (d) Dependence of height (—) and peak potential, Ep () of the chronopotentiometric peak H on the concentration of untreated AS-0 (black) and aggregated fibrils AS-504 (red). AS was adsorbed at the HMDE from a 5 µl drop for the accumulation time (tA) 60 s at open current circuit. The AS-modified HMDE was washed and transferred into an electrolytic cell containing 10 ml of a blank background electrolyte (50 mM Na phosphate, pH 7.0) and the C–E curve was measured. For details see the Experimental section. Conditions of aggregation: 100 µM AS in 0.1 M NaCl, 25 mM Tris-HCl, pH 7.3, was stirred at 300 rpm at 37 °C; samples were withdrawn at the indicated time intervals. | ||
Already with 200 nM AS, the C–E curve decreased below the curve of the background electrolyte between –0.1 and –0.85 V and the first signs of two peaks appeared: peak 1 at –1.35 V and a less well developed peak 2 at –1.72 V [Fig. 1(a)]. These peaks did not increase with AS concentration in the same manner. Peak 1 increased up to 500 nM AS, but greater concentrations led to a decrease of this peak and its shift to less negative potentials. Complete disappearance was observed at 800 nM AS. In contrast, peak 2 and ΔCs increased with AS concentration up to 1 µM and leveled off between 1 and 2 µM. On the in-phase a.c. voltammetric curve (more sensitive to reduction and oxidation processes and less to adsorption/capacitive phenomena)42 of 500 nM AS only a single peak was observed at –1.74 V (not shown).
Our results suggest that the observed peaks were of a capacitive nature, although participation of some reduction process in peak 2 could not be excluded. If we assume the presence of a single protein species in solution, peak 1 could reflect segmental desorption/reorientation of the protein at the surface, whereas peak 2 could indicate either segmental desorption/reorientation without complete detachment of the AS molecules or complete desorption of the protein from the surface. The presence of the peak H of native AS at more negative potentials (see below) would be consistent with the former possibility. On the other hand the relatively slow voltage scanning in C–E measurement could allow a slower desorption process to be completed, which may not occur in the much faster CPSA.
Electrocatalytic properties of AS
We also measured the peak H of AS [Fig. 1(d), 2(c), 3(b)]. This peak reflects the ability of proteins to catalyze hydrogen evolution at mercury electrodes.37,38,43 Briefly, the catalyst RH+ takes up an electron at the electrode, followed by evolution of hydrogen, and regeneration and protonation of the catalyst through the interaction with protons in the solvent (usually the buffer acid component). In peptides and proteins , aa side-chains bearing protonated amino groups such as arginine and lysine are among the most catalytically active residues. Cysteine residues may also play an important role in catalysis .34 There are no Arg and Cys residues in AS. Thus 15 Lys residues per monomer may constitute the main source of AS electrocatalytic activity. | ||
| Fig. 2
(a) Dependence height of peak H of AS-0 on the stripping current, Istr. (b) Dependence height (–●–) and area (○) of peak H of AS-0 on the accumulation potential, EA. Relative peak height and area are shown; their values at EA –0.1 V were taken as 1. (c) Reproducibility of 2 µM AS-0 peak H measurements (seven curves); Istr = –9 µA, tA = 60 s. Other details as in Fig. 1. | ||
 | ||
| Fig. 3 Time course of aggregation of AS. (a) Dependence of peak H height of AS and β-synuclein on incubation time. 2 µM AS (–●–) or 2 µM β-synuclein (Δ) were adsorbed on the HMDE for tA = 60 s. (b) Comparison of peaks H of 2 µM AS after different incubation times: AS-0 (black), AS-2 (green), AS-2.5 (orange), AS-4 (light magenta) and AS-50 (blue); Istr –9 µA was used for AS. For β-synuclein, the Istr of –7 µA was used to obtain peak heights comparable to those of AS. (c) Dependence of ΔCs of 2 µM AS on incubation time. (d) Comparison of C–E curves of 2 µM AS at different incubation times. C–E curves of mature fibrils (AS-168 and AS-504) differ strikingly from AS-50 and pre-aggregated AS. Other details as in Fig. 1. | ||
Dependence on stripping current, Istr
In CPSA, Istr should be carefully chosen to obtain well-developed peaks and optimum sensitivity for the given analysis. We followed the dependence of peak H of 2 µM native AS on Istr in the range from –5 to –15 µA. We observed a usual decrease of the peak with the increasing Istr [Fig. 2(a)] accompanied by a shift of potentials (Ep) of peak H to more negative potentials (not shown). At Istr < –5 µA it was difficult under the given conditions to measure the whole peak H, whereas at Istr > –13 µA peak H was too small. Fig. 2(c) shows seven subsequently measured CPSA curves of native AS obtained under these conditions with a standard deviation of 2.2%.Dependence of peak H on accumulation potential, EA
Most of our experiments were done at open current circuit during the accumulation of AS at the surface. We were interested in the effect of EA on the resulting AS signal. We adsorbed 1 µM AS from the electrolytic cell at various EA values and transferred the AS-modified electrode to another cell containing a blank background electrolyte. Ep, the height and area of peak H were little influenced by EA from –0.1 V to about –1.5 V [Fig. 2(b)]. Between EA +0.1 and –0.1 V the height of peak H decreased with shifting of EA to positive values whereas changes in peak area were much smaller, suggesting changes in the electrode processes, perhaps affected by a different protein orientation at the positively charged electrode surface. Between EA –1.60 and –1.72 V the area of peak H slightly increased and Ep shifted to positive values by about 30 mV. At more negative EA the Ep shifted to negative values (down to –1.82 V at EA –1.70 V) and at EA –1.80 V the peak decreased almost to zero. Our results suggest that at an EA more negative than EA –1.82 V, AS almost did not adsorb at the electrode, but once it became adsorbed at more positive EA values (e.g. at –1.4 V) it remained at the surface even at potentials more negative than –1.85 V, as indicated by an appearance of peak H with an Ep more negative than –1.9 V (not shown).Dependence on AS concentration
Potentials (Ep) of peak H shifted from –1.93 V at 100 nM AS to less negative values and the height of this peak increased linearly with the AS concentration (at tA 60 s) up to about 1 µM AS [Fig. 1(d)]. Between 1 and 2 µM AS, the peak height and area leveled off, in agreement with the a.c. impedance measurements [Fig. 1(a,c)] and suggesting that the electrode was fully covered. Up to 10 µM AS, the peak area and Ep of peak H, –1.88 V, did not change appreciably.Fully aggregated AS
We measured C–E curves of AS after 7 days (AS-168) of stirring at 37 °C, producing fibrils which yielded a typical circular dichroism spectrum and ThioTfluorescence (not shown). The C–E curve of 2 µM AS-168 differed greatly from that of native untreated AS (AS-0) [Fig. 1(a–c)]; AS-168 showed a much smaller decrease in the capacity of the electrode double-layer (ΔCs 1.85 µC cm–2). Already with 200 nM AS-168, a distinctive peak appeared: at –1.35 V (peak 1f), corresponding to the Ep of peak 1 of AS-0 [Fig. 1(a)]. A much smaller peak was observed at –1.65 V (peak 2f), corresponding approximately to the Ep of peak 2 of AS-0. Both peaks grew with increasing AS-168 concentration up to about 7.5 µM. ΔCs increased almost linearly with the concentration of AS-168 up to 2 µM, less steeply up to ca. 7.5 µM, and leveled off. The ΔCs value for 7.5 µM AS-168 was 2.71 µC cm–2, i.e. substantially lower than for 2 µM AS-0 [Fig. 1(c)].Under the conditions used to measure peak H of AS-0 (Istr –9 µA, 2 µM AS-168) no peak H was produced by 2 µM AS-168. Prolonged incubation of AS for 21 days (AS-504) resulted in a C–E curve very similar to that of AS-168 [Fig. 3(d)]. As in the case of AS-168, no peak was observed with AS-504 at 2 µM concentration and 10 µM AS-504 yielded a very small peak H (data not shown). We also measured a fibril sample obtained after 50 h aggregation (AS-50), which showed appreciable ThioTfluorescence . In contrast to AS-168, sample AS-50 displayed a peak H [Fig. 3(a)] albeit much smaller than that of AS-0. Thus, the interfacial behavior of mature fibrils differed not only from that of AS-0 but also from AS-50.
These results suggest that mature AS fibrils (AS-168 and AS-504) adsorb at the mercury electrode much more weakly than AS-0 and AS-50. Peak 1 of AS-168 and AS-504 probably represents a reorientation of the fibrils at the electrode because at potentials more negative than peak 1 the C–E curve dipped below the line of the background electrolyte, suggesting that the protein was still adsorbed at the surface. However, a partial desorption of the fibrils at the potentials of peak 1 cannot be excluded. Compared to the dramatic changes of AS-168 with mercury electrodes [Fig. 3(d)], a smaller decrease of the tyrosine oxidation peak Y was observed at carbon electrodes (not shown). The smaller decrease of peak Y can be related to the finding44 that out of four tyrosines in AS three of them, located in the AS C-terminal region, remain solvent-accessible even in mature fibrils.
We may conclude that in mature fibrils both the adsorbability at the HMDE and the ability to catalyze the hydrogen evolution (manifested by peak H) are strongly decreased. This is not surprising because in such fibrils a significant portion of both the hydrophobic groups (very important participants in protein adsorption at the hydrophobic mercury surfaces) and proton donor-containing groups should be hidden in the interior of the fibrils. Current models of AS fibrils suggest that half of Lys residues become buried after fibrilization. Moreover, in adsorbed fibrils the distance of some electroactive groups from the electrode surface may be too large to accommodate effective electron transfer. In addition, the rate of diffusion of large fibrils to the electrode should be greatly reduced (as compared to the AS-0 monomers), thus decreasing the measured signal under partial electrode coverage. Considering the known45 polymorphy of aggregated AS, it is incumbent to study their interfacial properties in greater detail.
Pre-aggregated AS
Recent data obtained by NMR and other techniques suggested that native AS adopts an ensemble of conformations stabilized by long-range interactions forming an intricate network.17,26,28,30,46 This network involves the N-terminus (aa 1–60) as well as the C-terminus (aa 109–140), which shields the highly amyloidogenic, hydrophobic NAC region (aa 61–95) from interaction with the solvent. In PD-linked mutations, such as A53T and A30P, the intricate network of the long-range interactions is perturbed. Thus, both A53T and A30P mutants are able to overcome the energetic barrier for self-association, resulting in a greater tendency to oligomerization . But how can the oligomerization be induced in a native wild-type (wt) AS? Differential scanning calorimetry did not reveal a thermal transition in wt AS over the temperature range of 15–110 °C.47 On the other hand, using NMR it was shown that residual dipolar couplings in the C-terminus of wt AS were greatly reduced upon heating to 37 °C.26 Numerous publications have dealt with the time course of AS fibrilization in vitro.16,17,20,21 Changes in AS properties preceding its fibrilization were detected in wt and pathologic AS mutants by sedimentation followed by gel filtration, indicating the disappearance of monomeric AS.27 Visualization by electron microscopy or by AFM showed oligomers and protofibrils of different shapes and sizes;48,49 the smallest detectable oligomeric species had a mass of 140 kDa, corresponding to a decamer.49 The presence of transient soluble oligomers during the lag period has been recently detected by various methods, including fluorescence resonance energy transfer (FRET) as well as acrylamide quenching, fluorescence anisotropy, dye binding, FTIR and dynamic light scattering.44,49–51 Most of these measurements were performed on AS mutants, such as Y125W/Y133F/Y136F, in which one of the tyrosine residues was replaced by tryptophan and two others by phenylalanine. The procedure led to a mutant AS containing a single tyrosine donor and a single tryptophan acceptor. However, such mutants aggregated significantly faster than the wild-type AS,44 making an exact comparison with our data difficult.We studied the ability of AS to catalyze hydrogen evolution at the mercury electrode and the dependence of the adsorption/desorption behavior of AS (Fig. 3) after incubation times much shorter than those leading to aggregation in vitro. A solution of 100 µM AS in 0.1 M NaCl, 25 mM Tris-HCl pH 7.3 was stirred at 37 °C (see experimental procedures for details) and samples were withdrawn at different time intervals up to 50 h of aggregation, that is in the time period encompassing the increase in the ThioTfluorescence indicative of AS aggregation (data not shown).
Early pre-aggregation changes in interfacial behavior of AS
Measurements were performed with 2 µM AS at tA 60 s, corresponding to full electrode coverage of AS-0 [Fig. 1(a,c,d)]. After 1 h of AS incubation (sample AS-1) we observed a shift in Ep of peak H to positive values (by almost 10 mV) and some increase of the peak height [Fig. 3(a,b)]. The latter continued during the next 90 min (samples AS-2 and AS-2.5) but the Ep of peak H gradually shifted to more negative potentials [region U, Fig. 3(a,c)] although it still remained more positive than the Ep of AS-0. The displacement of peak H to negative potentials continued between 2.5 and 4 h but the height of the peak decreased (region O1), reaching values and Ep measurements differing only slightly from those of AS-0. Between 6 and 25 h of incubation the height and Ep value of peak H did not change appreciably (region O2). β-Synuclein, which does not form fibrils under the given conditions,52 was treated in the same way as AS and peak H was measured with 2 µM β-synuclein at different time intervals up to 24 h. No significant changes in the height of peak H of β-synuclein were observed under these conditions [Fig. 3(a)].The increase in peak H of AS between 0 and 2.5 h (region U) was accompanied by an increase of ΔCs, which reached the value of ca. 9.1 µF cm–2. The subsequent decrease of ΔCs (down to 8.8 µF cm–2) during 2.5–6 h (region O1) resembled the change of peak H. Between 6 and 25 h (region O2) peak H was fairly constant but ΔCs continued to decrease up to 10 h [Fig. 3(c)]. After 25 h (region A) both ΔCs and peak H decreased, reflecting the protein aggregation. The DLS measurements showed a decrease in the monomer content and formation of some oligomers after 6 h of incubation (Fig. 4), i.e. at the end of region O1 [Fig. 3(a,c)]. A prevalence of oligomers over monomers was observed after 24 h, i.e. at the beginning of region A, derived from the electrochemical data (Fig. 3).
 | ||
| Fig. 4 Dynamic light scattering of AS. (a) The results suggest a population of oligomers preceding fibril formation. Histograms for the size-dependent scattering observed for AS during the early steps of aggregation. Size estimations (nm): monomer diameter, d ≈ 6, dimer d ≈ 12, low molecular weight oligomers 50 < d < 200, high molecular weight oligomers (protofibrils) 200 < d < 2000. (b) Structural diversity of intermediates in AS fibrilization probed by electrochemical methods. The intrinsically unstructured AS monomer presents inhibitory long-range interactions. A fully unfolded, aggregation-prone, conformation may be attained early in the aggregation pathway, exposing the hydrophobic NAC region and facilitating oligomerization . Consequently, high molecular oligomers are populated as a result of protein self-interaction before fibril formation. Later, β-sheet-rich oligomers nucleate fibrilization and AS monomers assemble into ordered amyloid-like fibrils. The cartoons were created using Pymol (DeLano Scientific LLC) according to published NMR structural data26,45,59 and may serve as illustration purposes only. | ||
Our results demonstrate that shortly after the beginning of the incubation, about 20 h before formation of the fibrils, the interfacial properties of AS start to change (Fig. 3). How can we interpret this phenomenon? We first focus on the increase of peak H and the shift of its Ep to more positive potentials (as compared to AS-0) accompanied by an increase of ΔCs, observed in the region U [Fig. 3(c)]. We believe that these changes may be related to the destabilization of the soluble monomer AS, resulting in a disturbance of long-range interactions and loss of the ordered structure. In the region O1 (in time intervals longer than 2.5 h) the decrease of peak H and ΔCs may be related to the initial stage of the oligomerization accompanied by a decrease of the monomer concentration. In this region, ΔCs and height of peak H are higher and the Ep of this peak is less negative than in AS-0, suggesting that the destabilization of the monomer still prevails over the oligomerization .
Recently it was shown that denaturation by urea or guanidium chloride of bovine serum albumin and other proteins possessing ordered structure resulted in a large increase of peak H.39 In contrast to the changes observed in region U [Fig. 3(a)] the increases in peak H height in denatured proteins were much larger, suggesting that the extent of the early changes in AS structure is much smaller than that accompanying protein denaturation. Particularly interesting is the increase of peak H in AS-2 and AS-2.5 [Fig. 3(a,b)] accompanied by a shift of Ep to negative values, not observed in other proteins . It may arise from two overlapping processes, such as the structure destabilization and changes in orientation of the protein molecule at the surface, making the electrode electrocatalysis process more difficult. At time intervals longer than about 6 h (region O2), the interfacial behavior of AS is determined by a fall in the monomer concentration and the formation of oligomers and protofibrils, detected also by dynamic light scattering [Fig. 4(a)]. Between 24 and 50 h (region A) fibril formation is manifested by a decrease of ΔCs, and of peak H height and a marked shift of its Ep to negative values. These changes in interfacial properties of AS are accompanied by an increase in ThioTfluorescence (data not shown).
Relation between intra- and inter-molecular interactions of pre-aggregated AS and its interfacial behavior
AS was described originally as an unfolded protein with no apparent ordered secondary structure detectable by various methods, such as far-UV circular dichroism , Fourier transform IR and NMR spectroscopy,4,53,54 although recent evidence indicates the existence of distinct, functionally relevant intramolecular interactions.25,26,30,46,55 In several amyloid-related neurodegenerative diseases, protein oligomerization and aggregation requires an initial destabilization of a monomeric protein .18 Disturbance of long-range interactions may destabilize the AS molecule, manifested by an increase of peak H [Fig. 3(a,b)]. Such a destabilization can be related to changes in the protein adsorption [Fig. 3(c,d)] and increase of peak H [Fig. 3(a,b)] to the release of more catalytically active groups, contributing to the electrocatalytic process. For example, it can be anticipated that abrogation of the interactions of the C-terminus with the hydrophobic NAC region will result in a strong adsorption of this region on the hydrophobic mercury surface, leading to an increased ΔCs [Fig. 3(c,d)]. Therefore, we tentatively infer that the increase in peak H in the region U [Fig. 3(a,b)] accompanied by the shift of Ep to positive potentials and increase in ΔCs [Fig. 3(c,d)], during the first hours of the AS incubation in vitro reflects destabilization of the AS molecule. At times longer than about 6 h (region O2) we observed opposite effects in both the electrocatalysis (peak H) and in the adsorbability of the protein (ΔCs) (Fig. 3). Both ΔCs and peak H decreased and the Ep of this peak shifted to negative values. These changes indicate decreased electrocatalytic activity and adsorbability of the protein . In this case the changes may reflect the beginning of the AS oligomerization , characterized by redistribution in the ensemble of AS conformations, involving decreased accessibility of electrocatalytic groups, and probably also of hydrophobic groups, for the electrode processes.Our results show that the mercury electrodes are best suited for studies of AS aggregation processes but that carbon electrodes may also find some use in such studies. Mercury and carbon electrodes can yield AS reduction or oxidation signals, respectively. Moreover, liquid mercury with its atomically flat surface is ideally suited for the a.c. impedance studies. It is possible that a.c. impedance spectroscopy instead of measurements at a single a.c. frequency used in this paper will provide additional data about the interfacial properties of monomeric and polymeric AS. Testing other electrodes and surfaces may extend the possibilities of AS electrochemical research. Quite recently, AS was covalently bound to a gold thin film and the effect of the concentration of urea on the denaturation /renaturation of the protein was studied by surface plasmon resonance.56Denaturation of the immobilized AS was observed at a lower urea concentration than with free AS in solution. However, it is unclear how the structure, and particularly the long-range interactions in native AS, were disturbed by the covalent binding of AS to the surface. We believe that non-covalent binding of AS to the surfaces is better suited for studies of AS properties in the course of its aggregation.
Experimental
Materials
Recombinant human wild-type AS and β-synuclein were expressed and purified by C. W. B. as described earlier.20 The concentration of the pure protein (judged by PAGE, ESI-MS, analytical gel filtration) was obtained from the absorbance at 280 nm using an extinction coefficient of 5960 M–1 cm–1. ThioT and the components of the background electrolytes (Na phosphate, Na acetate and Tris-HCl buffers) were from Sigma Chemical Co. (St. Louis, MO) and were of analytical grade. All solutions were prepared using deionized water (Millipore, Milli-Q).Aggregation protocol
Aggregation was performed by incubating 100 µM AS solutions in 0.1 M NaCl, 25 mM Tris-HCl, pH 7.3, at 37 °C (thermobox Heraeus, Kendro Laboratory Products, Germany) with constant stirring at 300 rpm with a magnetic bar Variomag Telesystem 15.40 (H + P Labortechnik AG, Germany) in 1.5 ml vials. Aliquots were removed at different time intervals and diluted by 50 mM Na phosphate, pH 7.0, for voltammetry, CPSA and a.c. impedance measurements.Thioflavin T binding
Aliquots (3.8 µl) were withdrawn from AS incubations and added to 1.5 ml of 5 µM ThioT in 50 mM Na phosphate, pH 7. The final protein concentration was 190 nM. Fluorescence measurements were carried out on and ISS PC1 photon counting spectrofluorometer using 3.5 ml quartz cuvettes (Hellma, Germany) with a 1 cm light-path. Fluorescence emission spectra were recorded from 465 to 600 nm, using excitation at 446 nm, an excitation bandwidth of 16 nm and an emission bandwidth of 8 nm.Dynamic light scattering
Aliquots of 50 µl were withdrawn from the aggregation assay at different time intervals and measurements were performed on a Malvern Zetasizer Nano instrument employing a 633 nm laser and the detector in the back-scattering configuration. Decays of photon correlation were fitted employing the built-in software with the CONTIN routine. The results were represented in the form of a histogram showing the contribution of each size interval to the overall scattering of the sample.Apparatus
Electrochemical measurements were performed with an AUTOLAB analyzer (EcoChemie, The Netherlands) connected to a VA-Stand 663 (Metrohm, Herisau, Switzerland) and using a standard three-electrode cell. The working electrode was a hanging mercury drop electrode (HMDE) or a carbon paste electrode. An Ag/AgCl/3 M KCl electrode was used as the reference electrode and a platinum wire electrode as the auxiliary electrode. All experiments were carried out at 25 ± 1 °C under air. The software GPES 4.9 supplied by EcoChemie was employed for smoothing and baseline correction in the case of measurements with carbon paste electrodes.Adsorptive transfer (ex situ) stripping
In this technique a protein -modified electrode is prepared, which is then washed and used for electrochemical measurement in an electrolytic cell containing a blank background electrolyte.33,57,58 The HMDE was immersed into a 5 µl droplet of the AS solution in 50 mM Na phosphate, pH 7, for an accumulation time (tA) of 60 s at open current circuit. The protein -modified electrode was then rinsed for 20 s with the background electrolyte (50 mM Na phosphate, pH 7), placed in the electrolytic cell containing the above blank background electrolyte, followed by application of the stripping current (Istr) to scan the E–t curve, which was automatically converted to (dE/dt)–1–E yielding peak H. Alternatively, C–E curves were recorded to obtain data on the protein adsorption/desorption behavior. Parameters: CPSA, initial potential, Ei –0.1 V, the potential limit, Ef –1.95 V, and Istr –9 µA (unless stated otherwise); C–E curves: Ei –0.1 V, Ef –1.9 V, step 5 mV, amplitude 50 mV, frequency 230 Hz. The specific capacity Cs was calculated for the HMDE area of 0.4 mm2.Adsorptive stripping on carbon paste electrodes
The carbon paste was made of 70% graphite powder (Aldrich) and 30% mineral oil (Sigma; free of DNase, RNase, and protease). This carbon paste was housed in a Teflon body with a 2.5 mm diameter disk surface. Prior to the measurements the electrode was polished on filter paper. A drop of 6 µl AS solution in 50 mM Na phosphate, pH 7.0 was pipetted onto the electrode surface. After tA 60 s, the latter was rinsed with the background electrolyte (50 mM Na phosphate, pH 7.0), placed in the electrolytic cell containing the above blank background electrolyte, and subjected to SWV. SWV: Ei +0.15 V, Ef +1.2 V, step 5 mV, amplitude 25 V, frequency 230 Hz.Conclusions
This paper demonstrates that interfacial properties of AS at mercury electrodes greatly change, not only during the AS aggregation and formation of mature fibrils but particularly during the early stages of AS incubation in vitro. The adsorbability and the ability of AS to catalyze hydrogen evolution increase during the first hours (region U, Fig. 3). Such behavior may correspond to a destabilization of the protein and disturbance of the AS long-range interactions. In the following hours (region O1) the changes in interfacial behavior of AS turn in the opposite direction, i.e. both the adsorbability and the electrocatalytic activity decrease with incubation time. Such behavior is attributable to the beginning of the AS oligomerization and disappearance of some electrocatalytically active and hydrophobic groups from the surface, which is related to the formation of oligomers . These results show a new way of studies of AS in the course of its transition from monomer to aggregated AS and open the door to better understanding of the mechanism of formation of oligomers and protofibrils suspected for their pathogenicity in PD.22,24 The proposed electrochemical methods are relatively simple and inexpensive.60 They offer a new path for testing the ability of various agents to stop or even reverse the pathologic process. Our preliminary results show large differences in the interfacial behavior between pre-aggregated wild-type and PD-linked mutant AS, indicating a faster course of pre-aggregation in mutant samples.60,61Acknowledgements
Technical assistance of Mrs Lída Římánková as well as the help of Dr Hana Černocká and Mr Marko Živanović are gratefully acknowledged. This work was supported by grants of the Academy of Sciences of the Czech Republic AVOZ50040507 and KAN400310651, of Grant Agency of the Czech Republic 301/07/0490 to E. P. and of Ministry of Education, Youth and Sports, CR, LC06035. C. W. B. was the recipient of a graduate study fellowship from an award of the Center for the Molecular Physiology of the Brain (Göttingen, Germany) to T. M. J.References
- C. Dobson, Trends Biochem. Sci., 1999, 24, 329 CrossRef CAS.
- R. Kruger, T. Muller and O. Riess, J. Neural Transm., 2000, 107, 31 CrossRef CAS.
- R. O. Weller, J. Pathol., 2001, 194, 1 CrossRef CAS.
- P. H. Weinreb, W. Zhen, A. W. Poon, K. A. Conway and P. T. Lansbury, Jr., Biochemistry, 1996, 35, 13709 CrossRef CAS.
- L. C. Serpell, J. Berriman, R. Jakes, M. Goedert and R. A. Crowther, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4897 CrossRef CAS.
- L. C. Serpell, Biochim. Biophys. Acta–Mol. Basis Dis., 2000, 1502, 16 CrossRef CAS.
- V. Uversky, J. Biomol. Struct. Dyn., 2003, 21, 211 CAS.
- L. Maroteaux, J. T. Campanelli and R. H. Scheller, J. Neurosci., 1988, 8, 2804 CAS.
- V. N. Uversky, J. Li, P. Souillac, I. S. Millett, S. Doniach, R. Jakes, M. Goedert and A. L. Fink, J. Biol. Chem., 2002, 277, 11970 CrossRef CAS.
- R. Kruger, W. Kuhn, T. Muller, D. Woitalla, M. Graeber, S. Kosel, H. Przuntek, J. T. Epplen, L. Schols and O. Riess, Nat. Genet., 1998, 18, 106 CrossRef CAS.
- M. H. Polymeropoulos, C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, E. S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T. Papapetropoulos, W. G. Johnson, A. M. Lazzarini, R. C. Duvoisin, G. DiIorio, L. I. Golbe and R. L. Nussbaum, Science, 1997, 276, 2045 CrossRef CAS.
- J. J. Zarranz, J. Alegre, J. C. Gomez-Esteban, E. Lezcano, R. Ros, I. Ampuero, L. Vidal, J. Hoenicka, O. Rodriguez, B. Atares, V. Llorens, E. G. Tortosa, T. del Ser, D. G. Munoz and J. G. de Yebenes, Ann. Neurol., 2004, 55, 164 CrossRef CAS.
- K. A. Conway, J. D. Harper and P. T. Lansbury, Biochemistry, 2000, 39, 2552 CrossRef CAS.
- T. Ding, S. Lee, J. Rochet and P. Lansbury, Biochemistry, 2002, 41, 10209 CrossRef CAS.
- H. A. Lashuel, D. Hartley, B. M. Petre, T. Walz and P. T. Lansbury, Nature, 2002, 418, 291 CrossRef CAS.
- W. Hoyer, T. Antony, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, J. Mol. Biol., 2002, 322, 383 CrossRef CAS.
- W. Hoyer, D. Cherny, V. Subramaniam and T. M. Jovin, Biochemistry, 2004, 43, 16233 CrossRef CAS.
- C. Dobson, Nature, 2003, 426, 884 CrossRef CAS.
- J. Harper and P. Lansbury, Annu. Rev. Biochem., 1997, 66, 385 CrossRef CAS.
- T. Antony, W. Hoyer, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, J. Biol. Chem., 2003, 278, 3235 CrossRef CAS.
- W. G. Hoyer, D. Cherny, V. Subramaniam and T. M. Jovin, J. Mol. Biol., 2004, 340, 127 CrossRef CAS.
- B. Caughey and P. T. Lansbury, Annu. Rev. Neurosci., 2003, 26, 267 CrossRef CAS.
- B. I. Giasson and V. M. Y. Lee, Cell, 2003, 114, 1 CrossRef CAS.
- K. K. Dev, K. Hofele, S. Barbieri, V. L. Buchman and H. van der Putten, Neuropharmacology, 2003, 45, 14 CrossRef CAS.
- C. W. Bertoncini, C. O. Fernandez, C. Griesinger, T. M. Jovin and M. Zweckstetter, J. Biol. Chem., 2005, 280, 30649 CrossRef CAS.
- C. W. Bertoncini, Y. S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430 CrossRef CAS.
- K. A. Conway, S. J. Lee, J. C. Rochet, T. T. Ding, R. E. Williamson and P. T. Lansbury, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 571 CrossRef CAS.
- M. M. Dedmon, K. Lindorff-Larsen, J. Christodoulou, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2005, 127, 476 CrossRef CAS.
- M. S. Goldberg and P. T. Lansbury, Nat. Cell Biol., 2000, 2, E115 CrossRef CAS.
- A. Binolfi, R. M. Rasia, C. W. Bertoncini, M. Ceolin, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernandez, J. Am. Chem. Soc., 2006, 128, 9893 CrossRef CAS.
- J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2005, 127, 16388 CrossRef CAS.
- G. R. Winkler, S. B. Harkins, J. C. Lee and H. B. Gray, J. Phys. Chem., 2006, 110, 7058 Search PubMed.
- M. Masařík, A. Stobiecka, R. Kizek, F. Jelen, Z. Pechan, W. Hoyer, T. M. Jovin, V. Subramaniam and E. Paleček, Electroanalysis, 2004, 16, 1172 CrossRef CAS.
- E. Paleček, in Electrochemistry of nucleic acids and proteins. Towards electrochemical sensors for genomics and proteomics., ed. E. Paleček, F. Scheller and J. Wang, Elsevier, Amsterdam, 1st edn, 2005, ch. 19, p. 690 Search PubMed.
- E. Paleček, F. Jelen, C. Teijeiro, V. Fucik and T. M. Jovin, Anal. Chim. Acta, 1993, 273, 175 CrossRef CAS.
- X. Cai, G. Rivas, M. A. P. Farias, H. Shiraishi, J. Wang and E. Paleček, Anal. Chim. Acta, 1996, 332, 49 CrossRef CAS.
- F. G. Banica and A. Ion, in Encyclopedia of analytical chemistry, ed. R. A. Meyers, John Wiley and Sons, Chichester, 2000, p. 11115 Search PubMed.
- M. Heyrovský, in Electrochemistry of nucleic acids and proteins. Towards electrochemical sensors for genomics and proteomics., ed. E. Paleček, F. Scheller and J. Wang, Elsevier, Amsterdam, 1st edn, 2005, ch. 18 p. 657 Search PubMed.
- V. Ostatná, B. Uslu, B. Dogan, S. Ozkan and E. Paleček, J. Electroanal. Chem., 2006, 593, 172 CrossRef CAS.
- M. Vestergaard, K. Kerman, M. Saito, N. Nagatani, Y. Takamura and E. Tamiya, J. Am. Chem. Soc., 2005, 127, 11892 CrossRef CAS.
- V. Vetterl and S. Hasoň, in Electrochemistry of nucleic acids and proteins. Towards electrochemical sensors for genomics and proteomics., ed. E. Paleček, F. Scheller and J. Wang, Elsevier, Amsterdam, 1st edn, 2005, ch. 2, p. 18 Search PubMed.
- A. M. Bond, Modern Polarographic Methods in Analytical Chemistry, Marcel Dekker, Inc., New York, 1980 Search PubMed.
- M. Tomschik, L. Havran, M. Fojta and E. Paleček, Electroanalysis, 1998, 10, 403 CrossRef CAS.
- J. Kaylor, N. Bodner, S. Edridge, G. Yamin, D. P. Hong and A. L. Fink, J. Mol. Biol., 2005, 353, 357 CrossRef CAS.
- H. Heise, W. Hoyer, S. Becker, O. C. Andronesi, D. Riedel and M. Baldus, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15871 CrossRef CAS.
- R. M. Rasia, C. W. Bertoncini, D. Marsh, W. Hoyer, D. Cherny, M. Zweckstetter, C. Griesinger, T. M. Jovin and C. O. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4294 CrossRef CAS.
- C. D. Syme, E. W. Blanch, C. Holt, R. Jakes, M. Goedert, L. Hecht and L. D. Barron, Eur. J. Biochem., 2002, 269, 148 CrossRef CAS.
- A. L. Fink, Acc. Chem. Res., 2006, 39, 628 CrossRef CAS.
- H. A. Lashuel, B. M. Petre, J. Wall, M. Simon, R. J. Nowak, T. Walz and P. T. Lansbury, J. Mol. Biol., 2002, 322, 1089 CrossRef CAS.
- H. A. Lashuel and P. T. Lansbury, Q. Rev. Biophys., 2006, 39, 167 CrossRef CAS.
- M. J. Volles, S. J. Lee, J. C. Rochet, M. D. Shtilerman, T. T. Ding, J. C. Kessler and P. T. Lansbury, Biochemistry, 2001, 40, 7812 CrossRef CAS.
- G. Yamin, L. A. Munishkina, M. A. Karymov, Y. L. Lyubchenko, V. N. Uversky and A. L. Fink, Biochemistry, 2005, 44, 9096 CrossRef CAS.
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 10737 CrossRef CAS.
- R. Bussell and D. Eliezer, J. Biol. Chem., 2001, 276, 45996 CrossRef CAS.
- C. O. Fernandez, W. Hoyer, M. Zweckstetter, E. A. Jares-Erijman, V. Subramaniam, C. Griesinger and T. M. Jovin, EMBO J., 2004, 23, 2039 CrossRef CAS.
- T. Kang, S. Hong, H. J. Kim, J. Moon, S. Oh, S. R. Paik and J. Yi, Langmuir, 2006, 22, 13 CrossRef CAS.
- E. Paleček and I. Postbieglová, J. Electroanal. Chem., 1986, 214, 359 CrossRef CAS.
- E. Paleček, Bioelectrochem. Bioenerg., 1992, 28, 71 CrossRef CAS.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, J. Biol. Chem., 2005, 280, 9595 CAS.
- E. Paleček and V. Ostatná, Electroanalysis, 2007 Search PubMed , in press.
- V. Ostatná, M. Živanovič, D. Krstič, C. Bertoncini, T. Jovin and E. Paleček, unpublished results.
Footnote |
| † Present address: Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. |
| This journal is © The Royal Society of Chemistry 2008 |