
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rectifying interphases for preventing Li dendrite propagation in solid-state electrolytes†
Xuhui
Yao‡
ab,
Xuekun
Lu‡
cd,
Yundong
Zhou
 b,
Tomáš
Šamořil
e,
Jinxin
Bi
a,
Mateus G.
Masteghin
a,
Huixing
Zhang
b,
Leslie
Askew
a,
JeongWon
Kim
f,
Fangyu
Xiong
g,
Jianan
Wang
h,
David C.
Cox
a,
Tan
Sui
bi,
Ian
Gilmore
b,
S. Ravi P.
Silva
a,
Liqiang
Mai
g,
Gareth
Hinds
b,
Paul R.
Shearing
*c,
Juyeon
Park
*b and
Yunlong
Zhao
*ab
b,
Tomáš
Šamořil
e,
Jinxin
Bi
a,
Mateus G.
Masteghin
a,
Huixing
Zhang
b,
Leslie
Askew
a,
JeongWon
Kim
f,
Fangyu
Xiong
g,
Jianan
Wang
h,
David C.
Cox
a,
Tan
Sui
bi,
Ian
Gilmore
b,
S. Ravi P.
Silva
a,
Liqiang
Mai
g,
Gareth
Hinds
b,
Paul R.
Shearing
*c,
Juyeon
Park
*b and
Yunlong
Zhao
*ab
aAdvanced Technology Institute, University of Surrey, Guildford, Surrey, GU2 7XH, UK. E-mail: yunlong.zhao@surrey.ac.uk
bNational Physical Laboratory, Teddington, Middlesex TW11 0LW, UK. E-mail: juyeon.park@npl.co.uk
cElectrochemical Innovation Lab, Department of Chemical Engineering, University College London, London, WC1E 7JE, UK. E-mail: p.shearing@ucl.ac.uk
dSchool of Engineering and Materials Science, Queen Mary University of London, London, E1 4NS, UK
eTESCAN ORSAY HOLDING, a.s., Libušina tř. 21, Brno, 623 00, Czech Republic
fKorea Research Institute of Standards and Science, 267 Gajeong-ro, Daejeon, 3411305-600, Republic of Korea
gState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, School of Materials Science and Engineering, Wuhan University of Technology, Wuhan, 430070, China
hDepartment of Environmental Science and Engineering, Xi’an Jiaotong University, Xi’an, 710049, China
iSchool of Mechanical Engineering Sciences, University of Surrey, Guildford, Surrey, GU2 7XH, UK
First published on 21st March 2023
Abstract
Solid-state electrolytes have emerged as the grail for safe and energy-dense Li metal batteries but still face significant challenges of Li dendrite propagation and interfacial incompatibility. In this work, an interface engineering approach is applied to introduce an electronic rectifying interphase between the solid-state electrolyte and Li metal anode. The rectifying behaviour restrains electron infiltration into the electrolyte, resulting in effective dendrite reduction. This interphase consists of a p-Si/n-TiO2 junction and an external Al layer, created using a multi-step sputter deposition technique on the surface of garnet pellets. The electronic rectifying behaviour is investigated via the asymmetric I–V responses of on-chip devices and further confirmed via the one-order of magnitude lower current response by electronic conductivity measurements on the pellets. The Al layer contributes to interface compatibility, which is verified from the lithiophilic surface and reduced interfacial impedance. Electrochemical measurements via Li symmetric cells show a significantly improved lifetime from dozens of hours to over two months. The reduction of the Li dendrite propagation behaviour is observed through 3D reconstructed morphologies of the solid-state electrolyte by X-ray computed tomography.
Broader contextRapid transportation electrification and growing market demand for portable consumer electronics call for revolutionary solid-state Li metal batteries that combine safety and performance to replace commercialised Li-ion batteries that are close to their theoretical limits. Solid-state Li metal batteries still face a challenging scenario in finding commercialisation solutions due to issues including Li dendrites and the associated short circuit hazards. The community has learned much about the dendrite mechanism in recent years, whilst dendrite growth challenges remain unsolved. Interface engineering methods create opportunities for physicochemical regulation on the basis of typical solid battery architectures. The fundamentals of dendrite generation involve the undesirable electrochemical reduction process of Li-ions by free electrons under thermodynamically favourable conditions. In this study, we engineered to break the basic requirements for dendrite generation, with the idea to spatially constrain the Li-ion deposition behaviour by controlling the electron flux in battery systems. The successful demonstration of this prototype involves asymmetric conductance realised by interface-engineered electronic rectifying interphases. Statistically, elimination of Li dendrites occurs faster than their generation, so dendrites do not propagate during the whole battery operation process. This idea offers a new route to overcoming the dendrite growth in solid-state electrolytes and realising viable solid-state Li metal batteries. |
Introduction
More than 190 nations have joined the Paris Agreement, committing to fight climate change by cutting carbon emissions.1 This global decarbonisation trend has led to research on shifting to renewable energy supplies instead of fossil fuels. Intermittently produced renewable energy technologies need a grid-scale energy storage system to ensure continuous supplies. The electrification of transport also calls for battery technologies to support electric vehicles to replace internal combustion engine cars.2 Therefore, electrochemical devices in the application scenario of rechargeable batteries have been in the research spotlight for decades. Some already mature technologies, such as Li-ion batteries, have become ubiquitous to power portable electronics and electric vehicles in daily life. However, the energy density, durability and safety of conventional Li-ion batteries cannot necessarily satisfy the strict net zero requirements in the future.3Solid-state batteries (SSBs) would be a promising candidate for surpassing the capabilities of conventional Li-ion batteries due to the utilisation of the more energy-dense Li-metal anodes and the safety characteristic of non-flammable inorganic solid-state electrolytes (SSEs).4 Research on SSBs began with the development of suitable SSEs that feature both high ionic conductivity (σLi > 0.1 mS cm−1) and dielectric properties (σe < 10−7 mS cm−1), and peaked with the revival of Li-metal anodes.3,5 Li metal is the ultimate anode with the highest gravimetric capacity and the lowest electrochemical potential surpassing all other anode candidates. Thus, successful implementation of Li-metal anodes could significantly increase the specific energy of today's Li-ion batteries and also pave the way for other promising battery systems (such as Li–S and Li–air) in the future.3 Li-metal anodes were first applied to batteries in the 1970s by one of the Li-ion battery pioneers, British chemist Stanley Whittingham, who was awarded the Nobel Prize in Chemistry in 2019 alongside Akira Yoshino and John B. Goodenough.6 Nevertheless, frequent unintended accidents of catching fire in that era brought safety concerns to public attention. Despite much effort from Moli Energy, NEC, Mitsui and so forth to address safety issues, Li-metal anodes were eventually replaced with more reliable carbonaceous anodes patented by Akira Yoshino in 1987.6–8 After years of development, today's cells with carbonaceous anodes approach the theoretical limit of their capabilities. The increasing demand for high-energy systems and concerns over safety call for renewed development of Li-metal anodes. Dendritic Li deposition (namely, Li dendrites or Li filaments) is now widely recognised as the primary cause of short-circuit failure and accidents involving conventional Li-ion batteries.9 When the Li dendrite grows and reaches the cathode, the internal short circuit triggers an uncontrollable self-discharge and a chain of exothermal reactions, resulting in large amounts of accumulated heat, possible ignition of flammable organic liquid electrolytes and eventually catastrophic thermal runaway. As a result, researchers have turned their attention to non-flammable inorganic SSEs, which can effectively ameliorate the ignition issue of electrolytes. Furthermore, it has been known for some time that high-density SSEs with a high elastic modulus should hopefully be able to resist dendrite penetration and prevent cell failures.10 However, research in recent decades has shown that Li dendrites can still penetrate SSEs despite attempts at modification by various means, such as doping, increasing density, or introducing higher ionic conductivity at grain boundaries.11–15 Despite the negligible ignition hazard, internal short-circuit due to dendrite growth can still cause the failure of SSBs. Dendrite penetration through SSEs could occur readily, because the measured critical current density (CCD, J*) of SSEs (<1 mA cm−2 for Li7La3Zr2O12 – LLZO) is normally lower than that of their liquid counterparts (>4 mA cm−2).11,16–19 Once the CCD is reached in a local area, the electrical neutrality of the plating progress is broken, leading to dendrite growth.3,20,21 Dendrite growth is also self-enhanced because the higher electric field at the hemispherical tip tends to attract more Li-ions.22–24 These mechanisms are theoretically valid for most cases, but the dendrite may also be detected in electrochemical cycling at low current densities.25 To address the dendrite issues, a significant number of investigations and modifications have been carried out in recent years. The inhomogeneous Li plating process has turned out to be related to dendrite formation because the high local current density can easily exceed the CCD. This is mainly attributed to insufficient interfacial contact and can be improved by achieving a conformal interface.11,15,26–28 Moreover, pre-existing defects, voids or cracks are also thought to promote dendrite formation as the dendrite tends to grow along the grain boundaries.13,29–31 Dendrite propagation along transgranular cracks and deposition-induced cracks has been observed in situ through a mesoscale SSB.32 Improving the mechanical properties of SSEs could effectively reduce the generation of cracks and resist dendrite penetration accordingly.33 Furthermore, the high electronic conductivity of SSEs has also been shown to favour dendrite growth. Visualisation of internal Li concentration by Wang et al. indicates direct deposition of Li inside the bulk SSEs, and the higher the electronic conductivity, the greater the extent of Li deposition.5 The electronic conductivity could arise from anion frameworks, grain boundaries, impurities and dopants in the SSEs.4 Decreasing electronic conductivity or, in other words increasing dielectric properties would also be one of the effective methodologies for dendrite suppression. Although significant advances have been achieved in developing SSBs, no particular theory or practice can reconcile every aspect to prevent dendrite formation. Further efforts are still required to prevent dendrite generation and successfully commercialise SSBs.
According to the fundamentals of electrochemistry, dendrites are typically generated when ion carriers are electrochemically reduced in the electrolyte under mass transport limitation.4 This occurs during the battery's charging process, where electrons flow from the external circuit into the anode, and the internal Li-ions migrate from the cathode towards the anode (Fig. 1a, left panel). When capturing free electrons becomes a thermodynamically favourable process, these Li-ions could be reduced easily and become metallic Li atoms in the allowable space. Since Li plating on the anode requires a negative potential to overcome surface overpotential, the thermodynamic condition is likely to be satisfied in the SSE near the anode side. In principle, therefore, the most straightforward and effective way to prevent dendrite propagation is to remove either Li-ions or free electrons from the SSE. Since free Li-ion carriers are fundamental to battery operation, the only viable option to prevent dendrite propagation is to eliminate free electrons from the SSE. Here, the electrons should be restrained from entering the SSE during the charging process, but during the discharging process, they should be allowed to flow out of the SSE to remove the possibility of dendrite growth caused by inevitable current leakage (Fig. 1a, right panel). This idea brings us to the rectifying behaviour (asymmetric conductance) of diodes in semiconductor electronic devices that works by eliminating one direction of current flow using a potential barrier. One of the typical rectifiers is the p–n junction diode, which is created between the positive (p-type) and the negative (n-type) semiconductors. The electrons can easily flow from the n-type semiconductor to the p-type semiconductor under forward bias, whilst reverse electron flow is limited by the increased resistance induced by reverse bias. In the case of the SSB charging process, the internal electrostatic field is from the cathode to the anode, with electrons tending to flow from the anode to the cathode via the electrolyte. In order to put the internal rectifying diode in the reverse-biased state, the n-type semiconductor terminal should be attached to the surface of the SSE, and the p-type terminal should face the Li-metal anode. Electron flow from the Li-metal anode to the SSE is thereby restrained by the reverse-biased junction.
 | ||
| Fig. 1 (a) Left panel: schematic diagram of the Li dendrite growth in SSBs. Right panel: schematic diagram of the rectifying interphase at the interface between the Li-metal anode and SSE, which prevents electron infiltration into the SSE and suppresses Li dendrite propagation. (b) Schematic diagram of the on-chip rectifying device. Inset: digital photograph of the fabricated on-chip devices array. (c) I–V curves of the on-chip rectifying device at different temperatures. | ||
In this work, we created a p-Si/n-TiO2 electronic rectifying interphase between the Li-metal anode and SSE, featuring electronic rectifying behaviour and improved interface compatibility. Tantalum (Ta) doped LLZO (Li6.4La3Zr1.4Ta0.6O12 – LLZTO) was used as an example of the SSE because of its chemical stability and high ionic conductivity. A sputter deposition technique was used to create this rectifying interphase on the surface of LLZTO pellets (namely, LLZTO-RI) prior to battery cell fabrication. An external Al layer was deposited to protect the junction from ambient air and also to provide better wettability against Li metal. As a result, the internal p–n junction prevents electrons from infiltrating the LLZTO, meaning that Li-ion carriers cannot be reduced to form Li dendrites inside the LLZTO. The improved interface compatibility also contributed to formation of a conformal interface with a lower interface impedance.
Results and discussion
In order to verify the rectifying behaviour of the p-Si/n-TiO2 junction, equivalent on-chip devices were designed and fabricated, as shown in Fig. 1b. Each wafer has six on-chip devices, as shown in the inset photograph. All thin-film components were prepared via a multi-step direct current sputter deposition approach, as shown in Fig. S1 (ESI†). In addition to a single-component layer, the compounds can be deposited using a compound target or via a reactive sputter process by pumping reactant gas into the chamber. The Ti thin film was deposited on a SiO2-coated wafer as the bottom electrode (BE) by sputtering with the Ti target. For the p-Si/n-TiO2 junction, the n-TiO2 was deposited first via sputtering of a Ti target in a mixed Ar and O2 atmosphere, whilst the p-Si layer was created via sputtering of a p-Si target in a pure Ar atmosphere. The Al layer was deposited on top of the Si as the top electrode (TE). The metal–semiconductor junction should be in ohmic contact, which was confirmed from the linear I–V characteristics of the single-layer-configuration devices (Fig. S2, ESI†). The I–V characteristics of this p-Si/n-TiO2 on-chip devices show an asymmetrical plot at different temperatures (Fig. 1c). The forward bias current starts and remains low up to a turn-on voltage, after which it increases exponentially as the voltage increases. In contrast, only a small leakage current can be observed under reverse bias, indicating that the p–n junction blocks most response currents in that direction. A knee voltage (built-in potential, Vbuilt-in) of 0.6 V is obtained from the inflexion point, and the breakdown reverse voltage is shown to be >2 V. As the potential at the anode–SSE interface is typically low, this voltage tolerance applies to the potential situation at the interface.This p-Si/n-TiO2 junction was then deposited on the surface of LLZTO pellets based on the same parameters as on-chip devices, followed by the deposition of an external Al layer (Fig. 2a). Chemical depth profiles of the as-deposited interphase were characterised by secondary ion mass spectrometry (SIMS) using an OrbiSIMS instrument34 operated in the time-of-flight (ToF) mass spectrometry mode, details in the ESI.† SIMS has a high sensitivity for Al, Si, Ti and La in the positive ion mode with oxygen sputtering.35 The surface topography of the LLZTO pellet (Fig. S3, ESI†) indicates a roughness in microns, which cannot support a precise depth measurement of the attached interphase due to the shadowing effect in SIMS analysis. Therefore, the as-deposited rectifying interphase on a Si wafer was characterised first since the wafer was a perfectly flat substrate (Fig. 2b). The secondary ion signal of 27Al+ appeared first, followed by the signal of 28Si+ and then 48Ti+ signals. This ToF-SIMS depth profile shows a defined three layers structure of the interphase, and the approximate depth scale agrees well with the values (Al: 25 nm, Si: 10 nm and TiO2: 10 nm) measured using a stylus profilometer (Fig. S4, ESI†). From the SIMS results for the rectifying interphase attached LLZTO pellet (Fig. 2c), strong representative secondary ion signals of 27Al+, 28Si+ and 48Ti+ from the interphase were obtained at the beginning of sputtering, followed by the emergence of the 139La+ signal from the LLZTO pellet after a long period of sputtering. A detailed depth profile shows the defined TiO2 layers, but the Al and Si cannot be clearly distinguished from the profile. X-Ray photoelectron spectroscopy (XPS) was used to investigate the composition and the chemical state of the as-deposited TiO2 thin film. Fig. 2d shows the high-resolution spectrum of the Ti 2p core level. The curve was fitted using symmetrical Gaussian and Lorentzian functions with a mixing ratio of M = 0.5 (where M = 1.0 represents 100% Gaussian). The separation between 3/2 and 1/2 components of the 2p core level was fixed to 5.70 eV, the corresponding area ratio was fixed to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, and the full width at half maximum (FWHM) was fixed for the same split orbital. The overall intensity was then deconvoluted into four peaks centred at around 458.06, 459.01, 463.76 and 464.71 eV, corresponding to Ti3+ and Ti4+ in the unit cell, respectively.36 The unsaturated coordination of Ti3+ manifests oxygen deficiencies (vacancies) or interstitial titanium atoms, and both of these defects are donor-type defects. Therefore, the as-deposited TiO2 thin film should display n-type semiconducting properties. The mechanical property changes of the pellets after deposition have been examined using the nanoindentation technique (Fig. S5, ESI†). There was a slight decrease in Young's modulus after the deposition, which could mainly be attributed to the relatively low modulus of Al.
:2, and the full width at half maximum (FWHM) was fixed for the same split orbital. The overall intensity was then deconvoluted into four peaks centred at around 458.06, 459.01, 463.76 and 464.71 eV, corresponding to Ti3+ and Ti4+ in the unit cell, respectively.36 The unsaturated coordination of Ti3+ manifests oxygen deficiencies (vacancies) or interstitial titanium atoms, and both of these defects are donor-type defects. Therefore, the as-deposited TiO2 thin film should display n-type semiconducting properties. The mechanical property changes of the pellets after deposition have been examined using the nanoindentation technique (Fig. S5, ESI†). There was a slight decrease in Young's modulus after the deposition, which could mainly be attributed to the relatively low modulus of Al.
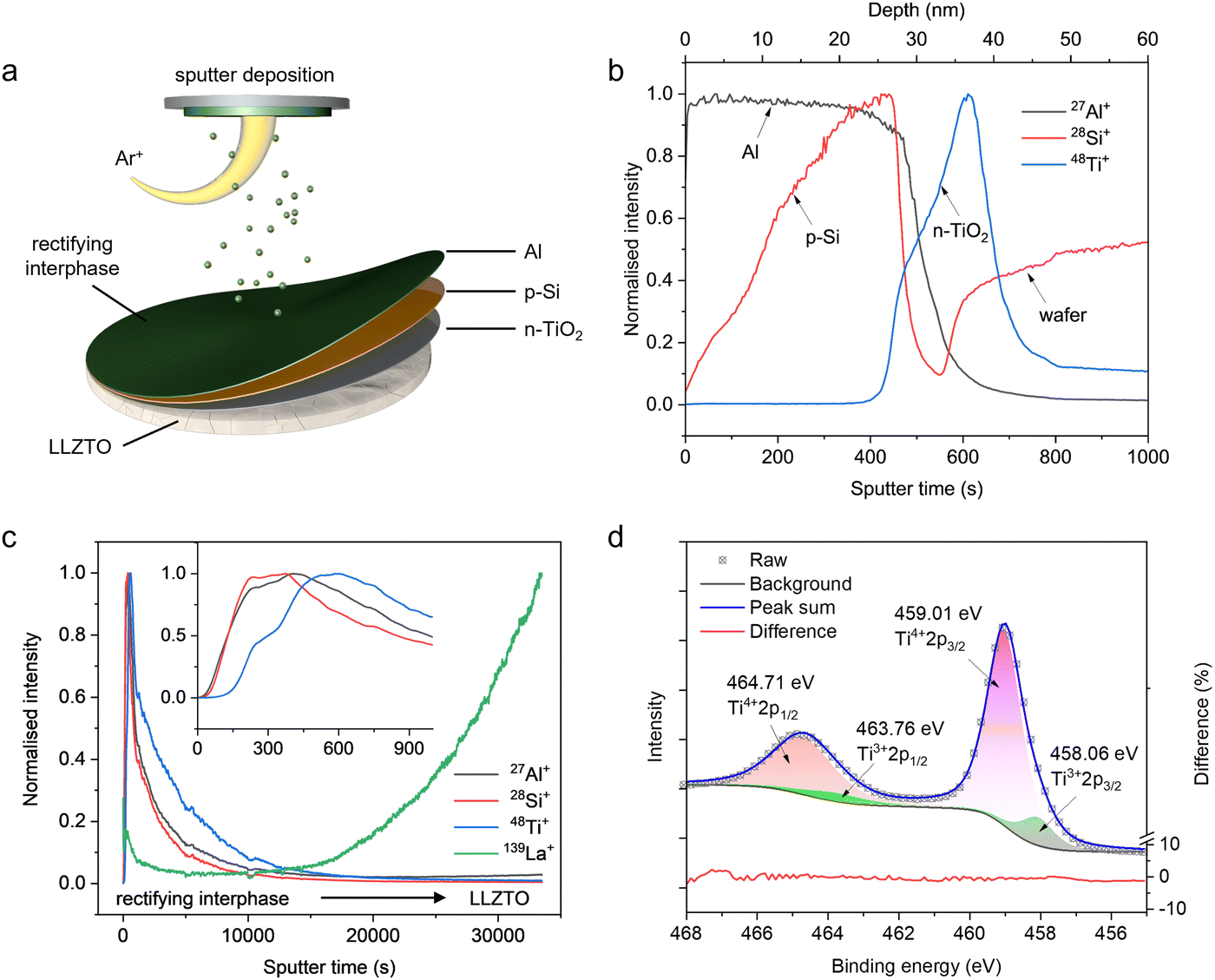 | ||
| Fig. 2 (a) Schematic diagram of the sputter deposition process for the rectifying interphase on the surface of LLZTO pellets. (b) ToF-SIMS depth profile of the rectifying interface on Si wafers, showing a defined three layers of the interphase. (c) ToF-SIMS depth profile of rectifying interface attached LLZTO pellet. The inset shows details of the rectifying interphase. (d) High-resolution Ti 2p core level spectrum of reaction sputtering deposited TiO2. | ||
The electronic conductivity of the LLZTO pellets was evaluated through a modified Wagner–Hebb method under a constant polarisation of 1 V at different temperatures. Chronoamperometry curves were obtained via Au/SSE/Au cells featuring two Au ionic blocking electrodes. The rectifying interphase was deposited on one side of the pellet, and 300 nm Au electrodes were deposited on both sides of the pellet using an E-beam evaporator. In theory, the chronoamperometry curves should show a large current response initially, followed by decay to a steady-state current response. The large initial current indicates the capacitive contribution of the ionic blocking electrodes, corresponding to the polarised Li-ion concentration (Fig. 3a). The steady-state current is attributed only to the intrinsic electronic conductance, and the corresponding electronic conductivity can be calculated according to Ohm's law. The corresponding equivalent circuit for the LLZTO-RI pellet is a combination of a resistor and a rectifying diode. The experimental chronoamperometry curves at different temperatures indicate relatively large equilibrium current responses from the LLZTO pellet (Fig. 3b–d and Fig. S6, ESI†). The LLZTO sample exhibits hysteretic current response, which is attributed to a small amount of Li metal plating in the near-surface region of the SSE. This plating process is kinetically limited by charge/mass transfer. So the corresponding current response is hysteretic compared to the capacitive contribution. For LLZTO-RI, the equilibrium current responses are of the same magnitude as those of the LLZTO pellet when the rectifying interface is forward-biased, whilst the responses remain fairly low when the interface is reverse-biased. The electronic conductivity of the LLZTO system is calculated to be 3.25 × 10−8 S cm−1 at room temperature, which agrees with previous reports of garnet-type SSEs.5,37 The electronic conductivity of the LLZTO-RI system is 2.57 × 10−9 S cm−1 in the reversed-biased state at room temperature. It is worth noting that the electronic conductivities of both samples increase with temperature, but the reverse leakage current increase in LLZTO-RI is significantly lower due to the thermal stability of the rectifying interphase confirmed using the on-chip devices (Fig. 3e). It should be mentioned here that the conventional Wagner–Hebb measurement requires an ionic blocking electrode and a Li-metal electrode.38 Our measurement via a dual ionic blocking electrode system can instead provide the upper limit of electronic conductivity, which is commonly used in SSE research as the SSE may not strictly be thermodynamically stable against Li metal.5,39,40 The Li-ion transportation across the rectifying interphase has been verified through an in-situ SEM characterisation (Fig. S7, ESI†). The results demonstrate that Li-ions can migrate from the bulk to the surface by crossing the interphase. The ionic conductivity of the pellets was measured via electrochemical impedance spectroscopy (EIS) analysis through the same ionic blocking electrode systems. Both Nyquist plots consist of a linear low-frequency tail and part of a high-frequency arc (Fig. S8, ESI†). The low-frequency tail corresponds to the capacitive behaviour of the ionic blocking electrodes. The real axis intercept in the high-frequency region is assigned to the bulk ionic resistance, whilst the depressed arc is associated with the grain boundary response. The total resistance contributions can be calculated via a low-frequency intercept of 268 Ω, and the corresponding ionic conductivity for both pellets is calculated to be 6 × 10−4 S cm−1.
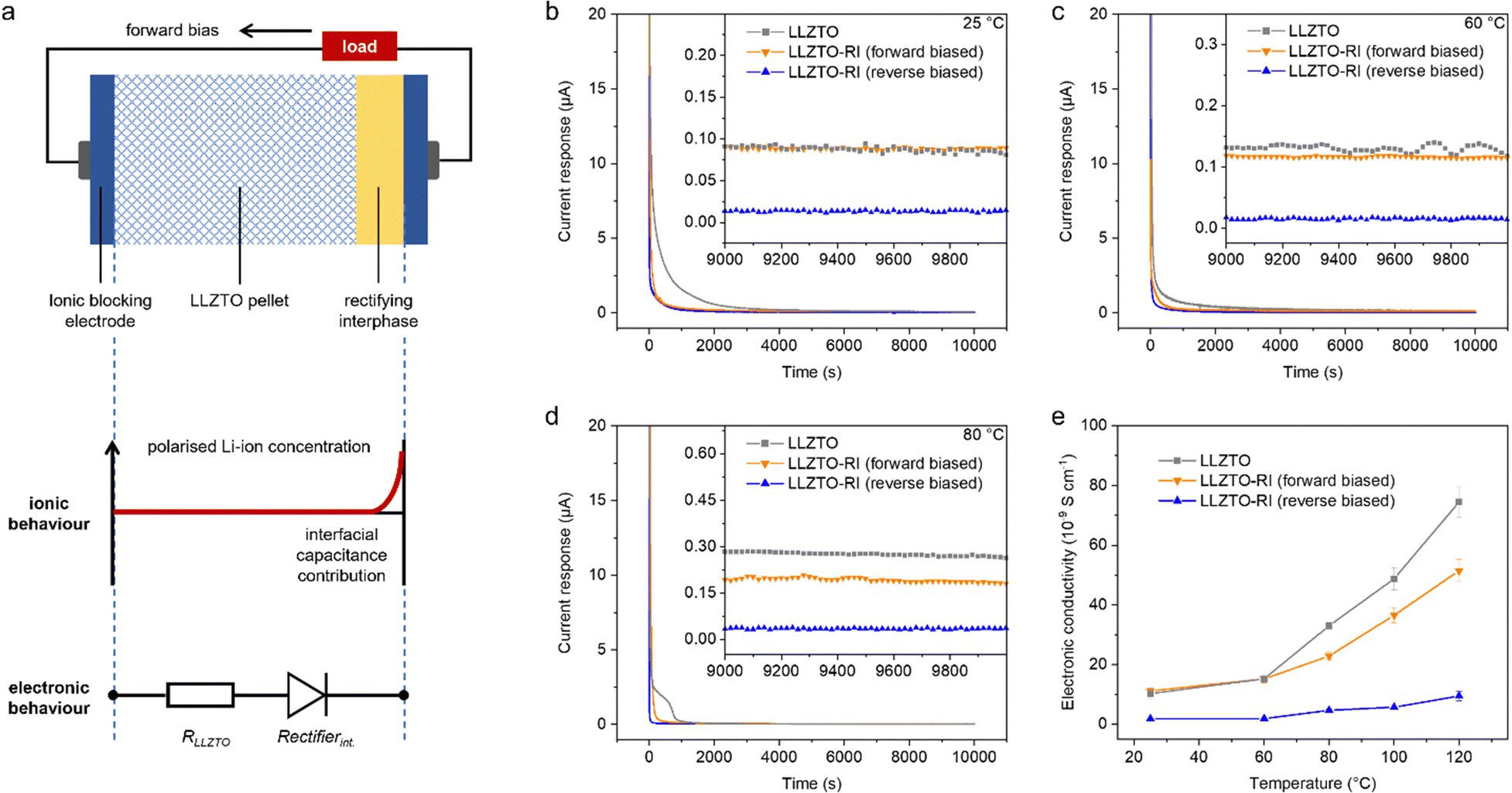 | ||
| Fig. 3 (a) Schematic diagram of the ionic behaviour and electronic behaviour for the LLZTO-RI pellet with two ionic blocking electrodes. (b–d) Chronoamperometry curves under a constant direct current polarisation of 1 V. Inset: current response at the equilibrium state. (e) Electronic conductivities at different temperatures. Error bars represent the variance of current fluctuations during the steady states. | ||
Previous research reveals that garnet-type SSEs with Al or Al2O3 coating have a tight adhesion property (wettability) against Li metal, contributing to forming a conformal contact.15,41,42 Considering that the as-deposited external Al layer might be oxidised during the transfer process, XPS depth profiling was used to characterise the degree of oxidation and the composition of the Al layer. The specific spectra at sputter times of 0 s, 1400 s and 6000 s show an evolution of components as a function of sputter time (Fig. 4a). At the beginning of the sputter, only Al2O3 can be found, corresponding to the surface oxidation layer. With increasing sputter time, mixed phases of Al2O3 and Al metal appear, followed by a pure Al metal phase in the bulk. These results indicate that the external Al layer is sufficient to protect the rectifying interphase from oxidation during the transfer process. According to equilibrium phases in the Li–Al–O ternary phase diagram (Fig. 4b),43,44 the spontaneous reaction with Li metal generates thermodynamically favourable Li9Al4 and LiO2 binaries. The existence of the metallic alloy phase would lead to a mixed ionic and electronic conducting interphase, which means that this deposited Al layer will merge into the Li-metal anode completely. To investigate the wettability of the mixed Al/Al2O3 layer against Li metal, droplets of molten Li were applied to the pellets. As shown in Fig. 4c, the drops instantly form a molten Li ball with an obtuse contact angle on the surface of the pure LLZTO pellet, whilst the molten Li spreads out and substantially covers the LLZTO-RI pellet surface. The interface impedance against Li was evaluated via EIS in Li symmetric cells (Li/SSE/Li). The cells were pre-heated to 200 °C on a hot plate in an Ar-filled glovebox to ensure good contact, and subsequent measurements were performed in an Ar-filled glovebox at room temperature. The area-specific resistance (ASR) value is calculated based on the pellet diameter. The resistance values from two sides of the LLZTO pellet do not overlap because of the contact differences in two non-wetting interfaces that cannot be eliminated by thermal treatment. The cell assembled with the LLZTO pellet shows a resistance of 1750 Ω cm2, which is much higher than the 500 Ω cm2 value of the LLZTO-RI cell (Fig. 4d). This significantly reduced resistance is attributed to the improved wettability against Li metal via the Al/Al2O3 surface layer. By subtracting the garnet resistance value (bulk and grain boundary), the interfacial charge transfer resistance is calculated to be 131 Ω cm2 for two symmetric interfaces.
 | ||
| Fig. 4 (a) XPS depth profiling analysis for Al 2p core level, which shows that the external surface is in the form of Al2O3. (b) Phase diagram for the Li–Al–O system. (c) Digital photograph of melting Li metal on the surface of LLZTO and LLZTO-RI pellets. (d) Interfacial impedance spectra measured from the Li symmetric cells. | ||
The Li plating/stripping stability of both pellets was evaluated by galvanostatic cycling of the Li symmetric cells. Fig. 5a shows the time-dependent voltage profile of the cells at a current density of 0.2 mA cm−2. Both negative and positive potentials denote Li plating/stripping processes on both sides of the pellets. The symmetric cells were periodically cycled for 5 min until an abrupt potential drop due to the short circuit corresponding to the dendrite penetration. The LLZTO-RI cells exhibited excellent cycling stability for over two months until they reached the upper cycling number limit of the instrument. A potential of ±0.1 V for each cycle corresponds to a resistance of 500 Ω cm2 according to Ohm's law. In contrast, an initially stable voltage profile of ±0.4 V followed by an abrupt potential drop after 80 h cycles was observed in the LLZTO cells (Fig. 5b). The abrupt potential drop corresponds to a short circuit caused by dendrite penetration. For reference, the performance of pellets with pure Al layer deposition (namely, LLZTO-Al) was also measured. It is worth noting that the initial potentials are relatively low, but a continuous overpotential increase occurs at 150 h, followed by a sharp potential drop after 230 h of cycling. This result reveals that even if conformal contact could be achieved at the beginning, inhomogeneous charge transfer still leads to progressive interface deterioration. The CCD values of the LLZTO-RI and LLZTO pellets were measured in Li symmetric cells under increasing current density from 0.02 mA cm−2 to 0.8 mA cm−2. The voltage profile shows stable cycling of the LLZTO-RI cells even at a current density of 0.8 mA cm−2 (Fig. 5c). The CCD value of the LLZTO cells was measured to be 0.48 mA cm−2, shortly after which the short circuit occurred (Fig. 5d). The voltage profile of the LLZTO-Al cell exhibits a similar behaviour to that of the LLZTO-RI cell. This result indicates that the increase in the CCD value is mostly attributed to the improved wettability by Al layer deposition. Energy dispersive X-ray spectroscopy (EDS) chemical mapping and XPS analysis (Fig. S9 and S10, ESI†) were performed to investigate the rectifying interphase after cycling. The cycled cells were disassembled in a glovebox, and the Li metal electrodes could only be removed by heating to 200 °C using a hot plate. Both EDS and XPS results reveal the existence of Ti and Si, whilst no Al signal could be detected after cycling due to the spontaneous reaction between Al and Li metal. The Si 2p core level spectrum indicates the existence of Si, and the SiO2 should result from oxidation during the transfer process as there was no external Al layer protection against ambient air. The Ti 2p core level spectrum reveals unsaturated coordination of Ti3+, suggesting that TiO2 retains n-type semiconducting characteristics after cycling. The performance of the LLZTO-RI pellet in the full cell was demonstrated with LiNi0.8Mn0.1Co0.1O2 (NMC811) as the cathode material in a hybrid solid-state system (liquid electrolyte was added on the cathode side for wetting the porous electrode). Only one side of the pellet attaching to the Li-metal anode was deposited with the rectifying interphase, and details of the full cell preparation are shown in the Experimental section. The full cell delivers an initial capacity of 160 mA h g−1 and very stable Coulombic efficiency (Fig. 5e). Over 2000 cycles with a capacity retention of 70% are achieved at the 2C rate. The voltage profiles at different cycles are shown in Fig. S11 (ESI†). The increase in polarisation could be mainly attributed to the degradation of cathode material.
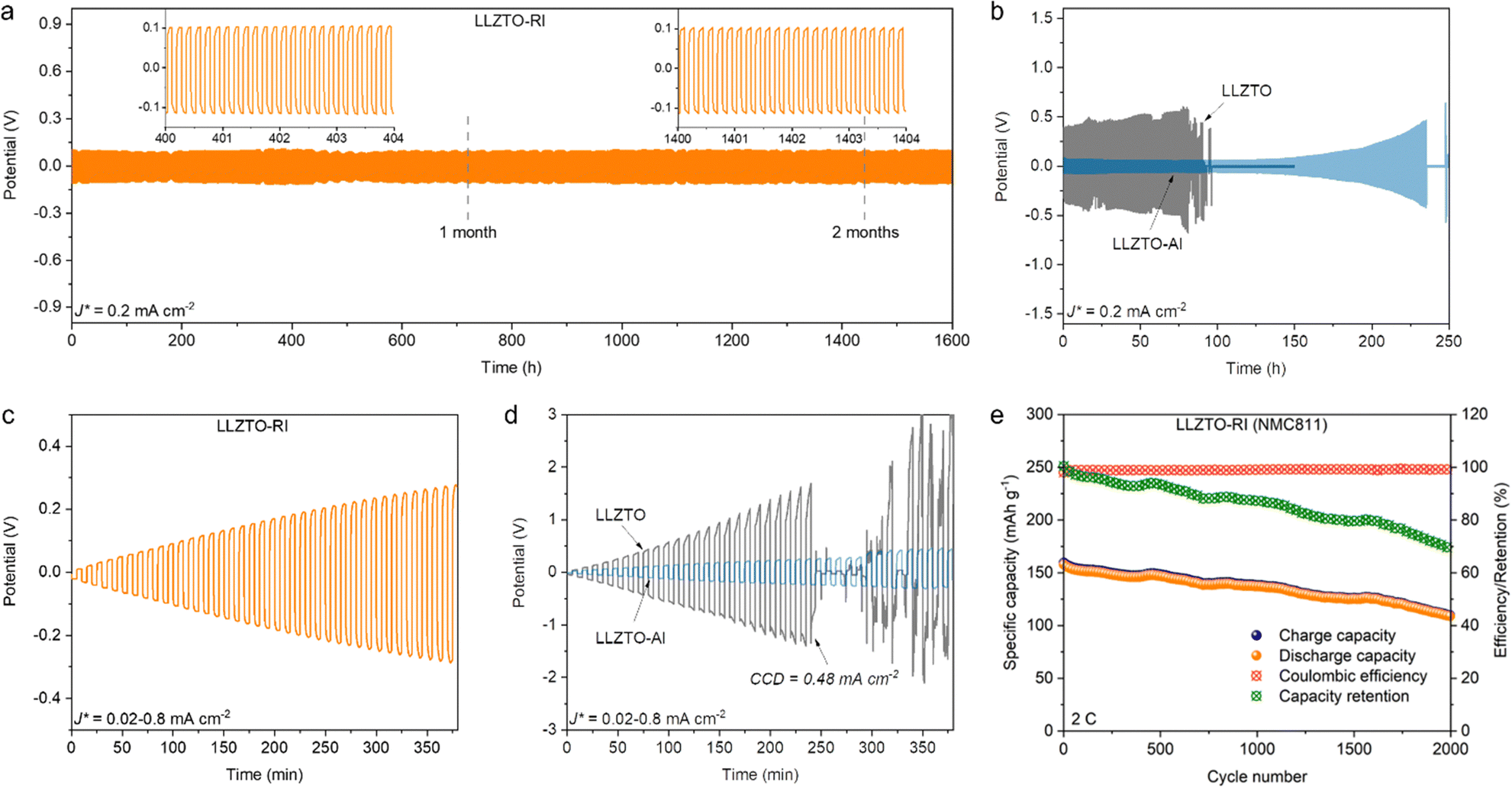 | ||
| Fig. 5 Galvanostatic cycling curves of the Li symmetric cells assembled with (a) LLZTO-RI, (b) LLZTO-Al and LLZTO pellets. (c and d) CCD tests for the Li symmetric cells with increasing current densities. (e) Cycling performance of the hybrid full cell with NMC811 cathode. | ||
Apart from the electrochemical methods, the distinct difference in dendrite growth resistance between the LLZTO and LLZTO-RI samples can be substantiated by the direct observation of porosity and crack formation after cycling. Many characterisation techniques, such as optical microscopy,13,45 scanning electron microscopy,46,47 neutron depth profiling5,48 and nuclear magnetic resonance,49 have been used to observe and investigate the mechanism of Li dendrites. However, most of these techniques normally reach the surface or subsurface of the sample only, providing 1D or 2D information. It is challenging to visualise the 3D morphology of internal cracks or Li dendrites because of the buried nature inside the SSEs. Here, we used laboratory-based X-ray computed tomography (CT) to reveal the 3D microstructural evolution of the SSEs before and after cycling at a spatial resolution of 0.95 μm. By comparing the pristine (Fig. 6a) and the cycled LLZTO (Fig. 6b), it was found that the porosity of the LLZTO increases significantly from 3% to 10.4% after cycling, mainly residing at the grain boundaries and forming a porous network that weakens the mechanical strength of the electrolyte. Besides, transgranular cracks are observed in the cycled LLZTO sample (red arrows in Fig. 6b and the zoom-in view in Fig. S12, ESI†). Both the intergranular porosity and transgranular cracks are attributed to the nucleation and growth of Li dendrites. The histogram of the pore size distribution displays that the formation of pores and cracks creates discrete spikes. Meanwhile, the neighbouring columns decrease in height, indicating that the coalescence of the newly formed pores and cracks with the pre-existing ones is the major mechanical failure mechanism of the electrolyte. It is shown that the dominant pore size of the cycled LLTZO electrolyte is approx. 8 μm (Fig. 6e). In contrast, almost no crack can be seen in the cycled LLZTO-RI sample (Fig. 6c). As a consequence, the histogram of the pore size distribution (Fig. 6f) resembles the pristine sample without fluctuations observed in Fig. 6e, except for a mild rise of the overall column heights due to the increase of porosity. The characteristic size of the newly formed pores is a merely approx. 2 μm plus no cracks are visible after cycling, implying a much higher resistance to the nucleation and growth of Li dendrites.
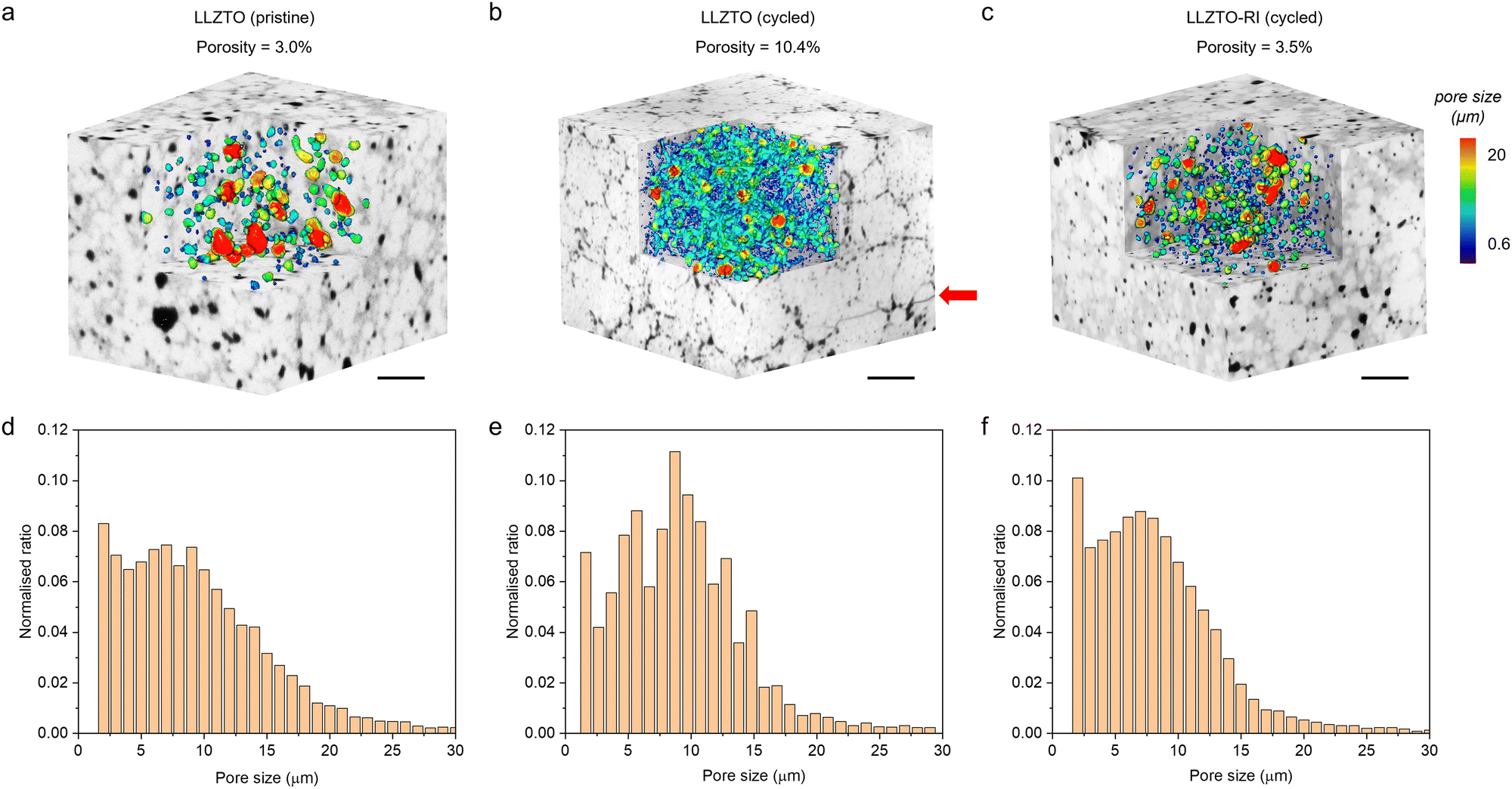 | ||
| Fig. 6 The reconstructed 3D volumes of the LLZTO and LLZTO-RI samples by X-ray CT before and after cycling are shown in the top row (a–c). The black texture represents the pore and crack phases due to their low X-ray attenuation coefficient. The pore morphology is visualised by the corner cut, with the colour coding indicating (d–f) the pore size distribution, which is quantified in the bottom row. Scale bar: 200 μm. | ||
Conclusion
In summary, we introduced a rectifying interphase between the Li metal anode and the SSE, featuring an electronic rectifying behaviour and improved interface compatibility. The rectifying behaviour was investigated via the asymmetric I–V responses of on-chip devices and further confirmed by electronic conductivity measurements on SSE pellets. The SSE pellets with rectifying interphases in the reversed-biased state exhibited one order of magnitude lower current response than those in the forward-biased state and intrinsic responses. This asymmetric conductance induces different rates of dendrite formation and elimination, that is, slow reductive generation but rapid oxidative elimination. The improved interface compatibility was verified by contact angle measurements and reduced interfacial impedance. The electrochemical measurements showed a significantly improved lifetime of over two months for LLZTO-RI compared with only 80 h for bare LLZTO pellets in Li symmetric cells. The CCD value also increased from 0.48 mA cm−2 to >0.8 mA cm−2. The X-ray CT indicates virtually no crack propagation or porosity increase in the LLZTO-RI sample after electrochemical cycling, whilst the LLZTO sample shows a significant increase in porosity and cracks.This work provides an effective approach to address the challenge of Li dendrite propagation in SSEs. We believe more functional buffer layers will be developed in the same spirit to stabilise the SSE and pave the way for viable solid-state Li metal batteries. Furthermore, the concept of electronic rectifying interphases may be extended to commercialised liquid cells by integration with conventional separators. Future research efforts may consider concentrating on creating a free-standing rectifying interphase that can be readily attached to the substrate, enabling Li metal anodes in various cell structures. It is suggested to consider the behaviour of electrons and ions and their coupling simultaneously during the development of batteries.
Author contributions
Y. Zhao, J. Park and P. Shearing conceived the presented idea. X. Yao performed sample preparation, device fabrication, cell assembly and electrochemical characterisation studies. X. Lu performed X-ray CT and XPS experiments, and J. Kim contributed to XPS analysis. Y. Zhou and T. Šamořil performed the ToF-SIMS experiment. J. Bi performed the X-ray diffraction experiment. M. Masteghin, X. Yao and D. Cox contributed to in-situ SEM analysis. H. Zhang performed the nanoindentation test. X. Yao wrote the manuscript in consultation with Y. Zhao, J. Park and F. Xiong. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks to Dr Steven Hinder at the School of Mechanical Engineering Sciences, University of Surrey, for help with the XPS analysis. Thanks to Dr Pierre Kubiak and Dr James Blakesley at the National Physical Laboratory (UK) for giving advice and suggestions. Thanks to Dr Jose Anguita at the Advanced Technology Institute, University of Surrey, for technical support in the cleanroom. Y. Zhao acknowledges support from the Royal Society project (IEC\NSFC\211074) and EPSRC project (EP/V002260/1). Y. Zhao, J. Park, G. Hinds and Y. Zhou acknowledge support from the National Measurement System of the UK Department of Business, Energy & Industrial Strategy.References
- C.-F. Schleussner, J. Rogelj, M. Schaeffer, T. Lissner, R. Licker, E. M. Fischer, R. Knutti, A. Levermann, K. Frieler and W. Hare, Nat. Clim. Change, 2016, 6, 827–835 CrossRef.
- A. Olabi and M. A. Abdelkareem, Renewable Sustainable Energy Rev., 2022, 158, 112111 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Materials for sustainable energy: a collection of peer-reviewed research and review articles from Nature Publishing Group, World Scientific, 2011, pp. 171–179 Search PubMed.
- K. Brandt, Solid State Ionics, 1994, 69, 173–183 CrossRef CAS.
- A. Jana, S. I. Woo, K. Vikrant and R. E. García, Energy Environ. Sci., 2019, 12, 3595–3607 RSC.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396 CrossRef CAS.
- A. Sharafi, H. M. Meyer, J. Nanda, J. Wolfenstine and J. Sakamoto, J. Power Sources, 2016, 302, 135–139 CrossRef CAS.
- R. Sudo, Y. Nakata, K. Ishiguro, M. Matsui, A. Hirano, Y. Takeda, O. Yamamoto and N. Imanishi, Solid State Ionics, 2014, 262, 151–154 CrossRef CAS.
- L. Porz, T. Swamy, B. W. Sheldon, D. Rettenwander, T. Frömling, H. L. Thaman, S. Berendts, R. Uecker, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2017, 7, 1701003 CrossRef.
- F. Yonemoto, A. Nishimura, M. Motoyama, N. Tsuchimine, S. Kobayashi and Y. Iriyama, J. Power Sources, 2017, 343, 207–215 CrossRef CAS.
- C.-L. Tsai, V. Roddatis, C. V. Chandran, Q. Ma, S. Uhlenbruck, M. Bram, P. Heitjans and O. Guillon, ACS Appl. Mater. Interfaces, 2016, 8, 10617–10626 CrossRef CAS PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- R. Raj and J. Wolfenstine, J. Power Sources, 2017, 343, 119–126 CrossRef CAS.
- A. Sharafi, C. G. Haslam, R. D. Kerns, J. Wolfenstine and J. Sakamoto, J. Mater. Chem. A, 2017, 5, 21491–21504 RSC.
- N. J. Taylor, S. Stangeland-Molo, C. G. Haslam, A. Sharafi, T. Thompson, M. Wang, R. Garcia-Mendez and J. Sakamoto, J. Power Sources, 2018, 396, 314–318 CrossRef CAS.
- J. Chazalviel, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 7355–7367 CrossRef CAS PubMed.
- C. Brissot, M. Rosso, J.-N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81, 925–929 CrossRef.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2003, 150, A1377 CrossRef CAS.
- M. Rosso, T. Gobron, C. Brissot, J.-N. Chazalviel and S. Lascaud, J. Power Sources, 2001, 97, 804–806 CrossRef.
- L. Cheng, W. Chen, M. Kunz, K. Persson, N. Tamura, G. Chen and M. Doeff, ACS Appl. Mater. Interfaces, 2015, 7, 2073–2081 CrossRef CAS PubMed.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS.
- F. Han, J. Yue, X. Zhu and C. Wang, Adv. Energy Mater., 2018, 8, 1703644 CrossRef.
- K. Kerman, A. Luntz, V. Viswanathan, Y.-M. Chiang and Z. Chen, J. Electrochem. Soc., 2017, 164, A1731 CrossRef CAS.
- B. Wu, S. Wang, J. Lochala, D. Desrochers, B. Liu, W. Zhang, J. Yang and J. Xiao, Energy Environ. Sci., 2018, 11, 1803–1810 RSC.
- T. Swamy, R. Park, B. W. Sheldon, D. Rettenwander, L. Porz, S. Berendts, R. Uecker, W. C. Carter and Y.-M. Chiang, J. Electrochem. Soc., 2018, 165, A3648 CrossRef CAS.
- J. Zhao, Y. F. Tang, Q. S. Dai, C. C. Du, Y. Zhang, D. C. Xue, T. W. Chen, J. Z. Chen, B. Wang, J. M. Yao, N. Zhao, Y. S. Li, S. M. Xia, X. X. Guo, S. J. Harris, L. Q. Zhang, S. L. Zhang, T. Zhu and J. Y. Huang, Energy Environ. Mater., 2021, 5, 524–532 CrossRef.
- Y. Qi, C. M. Ban and S. J. Harris, Joule, 2020, 4, 2599–2608 CrossRef CAS.
- M. K. Passarelli, A. Pirkl, R. Moellers, D. Grinfeld, F. Kollmer, R. Havelund, C. F. Newman, P. S. Marshall, H. Arlinghaus and M. R. Alexander, Nat. Methods, 2017, 14, 1175–1183 CrossRef CAS PubMed.
- R. Wilson, Int. J. Mass Spectrom. Ion Processes, 1995, 143, 43–49 CrossRef CAS.
- S. Gong, Z. Jiang, S. Zhu, J. Fan, Q. Xu and Y. Min, J. Nanopart. Res., 2018, 20, 1–13 CrossRef CAS.
- Y. Song, L. Yang, W. Zhao, Z. Wang, Y. Zhao, Z. Wang, Q. Zhao, H. Liu and F. Pan, Adv. Energy Mater., 2019, 9, 1900671 CrossRef.
- Y. Su, J. Falgenhauer, A. Polity, T. Leichtweiß, A. Kronenberger, J. Obel, S. Zhou, D. Schlettwein, J. Janek and B. K. Meyer, Solid State Ionics, 2015, 282, 63–69 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- K. K. Fu, Y. Gong, B. Liu, Y. Zhu, S. Xu, Y. Yao, W. Luo, C. Wang, S. D. Lacey and J. Dai, Sci. Adv., 2017, 3, e1601659 CrossRef PubMed.
- X. Han, Y. Gong, K. K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang and G. Rubloff, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, Adv. Sci., 2017, 4, 1600517 CrossRef PubMed.
- D. H. Snydacker, V. I. Hegde and C. Wolverton, J. Electrochem. Soc., 2017, 164, A3582 CrossRef CAS.
- E. Kazyak, R. Garcia-Mendez, W. S. LePage, A. Sharafi, A. L. Davis, A. J. Sanchez, K.-H. Chen, C. Haslam, J. Sakamoto and N. P. Dasgupta, Matter, 2020, 2, 1025–1048 CrossRef.
- M. Golozar, P. Hovington, A. Paolella, S. Bessette, M. Lagacé, P. Bouchard, H. Demers, R. Gauvin and K. Zaghib, Nano Lett., 2018, 18, 7583–7589 CrossRef CAS PubMed.
- F. M. Pesci, R. H. Brugge, A. O. Hekselman, A. Cavallaro, R. J. Chater and A. Aguadero, J. Mater. Chem. A, 2018, 6, 19817–19827 RSC.
- Q. Li, T. Yi, X. Wang, H. Pan, B. Quan, T. Liang, X. Guo, X. Yu, H. Wang and X. Huang, Nano Energy, 2019, 63, 103895 CrossRef CAS.
- L. E. Marbella, S. Zekoll, J. Kasemchainan, S. P. Emge, P. G. Bruce and C. P. Grey, Chem. Mater., 2019, 31, 2762–2769 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee04006a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |