Radical reactions with 3H-quinazolin-4-ones: synthesis of deoxyvasicinone, mackinazolinone, luotonin A, rutaecarpine and tryptanthrin†
W. Russell Bowman*, Mark R. J. Elsegood, Tobias Stein and George W. Weaver*
Department of Chemistry, Loughborough University, Loughborough, Leics. UK LE11 3TU. E-mail: g.w.weaver@lboro.ac.uk; Fax: 44(0) 1509 223925; Tel: 44(0) 1509 222584
First published on 3rd November 2006
Abstract
Alkyl, aryl, heteroaryl and acyl radicals have been cyclised onto the 2-position of 3H-quinazolin-4-one. The side chains containing the radical precursors were attached to the nitrogen atom in the 3-position. The cyclisations take place by aromatic homolytic substitution hence retain the aromaticity of the 3H-quinazolin-4-one ring. The highest yields were obtained using hexamethylditin to facilitate cyclisation rather than reduction without cyclisation. The alkaloids deoxyvasicinone 2, mackinazolinone 3, tryptanthrin 4, luotonin A 5 and rutaecarpine 8 were synthesised by radical cyclisation onto 3H-quinazolin-4-one.
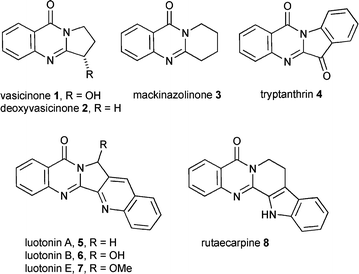
The 3H-quinazolin-4-one ring system is important to the biological activity of both naturally occurring alkaloids, biosynthesised from anthranilic acid, and pharmaceuticals. The alkaloids include vasicinone 1 and deoxyvasicinone 2,1 mackinazolinone 3,2 tryptanthrin 4,3 luotonins A 5, B 6 and E 74 and rutaecarpine 8.5 3H-Quinazolin-4-one alkaloids have been recently reviewed.6 All the 3H-quinazolin-4-one natural products have interesting biological activity and have therefore been extensively investigated for useful pharmaceutical activity. The 3H-quinazolin-4-one ring is regarded as a ‘privileged structure’ in combinatorial synthesis.7 These are structures which represent molecules that are capable of binding at multiple sites with high affinity and facilitate more rapid discovery of useful medicinally active compounds.7
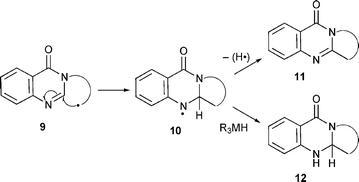
Our study involved the development of protocols involving radical cyclisation for the synthesis of polycyclic 3H-quinazolin-3-ones (Scheme 1). The protocols have also been used for the synthesis of novel polycyclic quinazolinones including the natural products deoxyvasicinone 2, mackinazolinone 3, tryptanthrin 4, luotonin A 5 and rutaecarpine 8.
 | ||
| Scheme 1 General synthetic protocol. | ||
Radical cyclisation onto heteroarenes has been developed in recent years to considerable advantage for the synthesis of novel polycyclic heteroarenes. Examples of these cyclisations include: a. alkyl radicals onto pyrroles,8,9 imidazoles,8 pyrazoles,10 indoles,9,11,12 1,2,3-triazoles,13 pyridinium salts,14 and quinolones;15 b. acyl radicals onto pyrroles,16 quinolines,17 pyridines18 and arenes;19 c. aryl radicals onto indoles,20,21 pyrroles,9 pyridones,22 and 5-amino- and 5-hydroxyuracils,23 2-quinolones,24 quinolines25 and pyridines.26
All of the above cyclisations are ‘oxidative’ i.e. the intermediate π-radicals are not reduced by triorganometal hydrides [e.g. tributyltin hydyride (Bu3SnH)] as normally observed for these reagents. The cyclisations proceed by aromatic homolytic substitution with abstraction of hydrogen in a rearomatisation process. Aromatic homolytic substitution has been recently reviewed27 and the mechanism of Bu3SnH mediated ‘oxidative’ cyclisation elaborated.28 The pyrimidin-4-one ring of the quinazolin-4-ones has some aromaticity and therefore aromatic homolytic substitution could be predicted, and was observed in our studies, as shown in Scheme 1 (9 to 11via the π-radical 10). However, the lower aromaticity could favour reductive cyclisation in which the intermediate π-radical 10 is intercepted by reagents such as Bu3SnH. Our prediction that radical cyclisation onto the quinazolin-4-one ring would be ‘oxidative’ was supported by the ‘oxidative’ radical cyclisation onto related ring systems, e.g. pyrimidine-2,4-diones,23,24 quinolinones15 and pyridinones.22,29,30 However, the lower aromaticity in the pyrrole ring of indole, facilitates both reductive and oxidative cyclisation depending on the conditions.11,12,31 The radical intermediate 10, whether a π-radical or not, is still a strongly stabilised anilyl radical and therefore the rate of reduction by Bu3SnH to yield 12 is probably too low to be competitive with loss of hydrogen to yield the ‘oxidised’ product. The reactions could also be regarded as exo-cyclisations onto imines which are well known.
We used two general methodologies to synthesise the radical precursors. Firstly, heteroarenes containing an NH group facilitate N-alkylation and provide a suitable synthetic route to radical precursors for cyclisation. The radical leaving group is introduced as part of the N-alkyl substituent. Secondly, there is a wide variety of protocols for the synthesis of 3-substituted quinazolinones using ring synthesis from anthranilic acid derivatives, which were employed as required.
Aryl radical cyclisation
Aryl radicals have been successfully cyclised onto pyrroles, indoles and pyrazoles.32 Aryl radicals are very reactive and the most likely to cyclise onto quinazolinones. Therefore, suitable radical precursors for 5- and 6-exo cyclisation were prepared (Scheme 2). The 5-membered ring cyclisation with the precursor 16 using Bu3SnH gave only the reduced uncyclised product 19 (Scheme 3). Even syringe pump addition of Bu3SnH, with either Et3B (r.t.) or AIBN (110 °C) as initiators, gave only 19. We considered that the lack of cyclisation could be due to 1,5-hydrogen abstraction from the 2-position of the quinazolinone as shown in Scheme 3 (route b: 18 to 21). This possibility was eliminated by repeating the reaction with Bu3SnD which gave only deuteriation on the aryl radical position, and none on the 2-position of the quinazolinone, and these results indicated that the rate of cyclisation was not favourable compared to reduction with Bu3SnH. Therefore, hexamethylditin [(Me3Sn)2] was used so that the intermediate radical (cf.9 in Scheme 1) was not reduced. The reaction was repeated with (Me3Sn)2 which resulted in a small amount of the cyclised product 20 (18%) as well as uncyclised 19 (65%) as the major compound.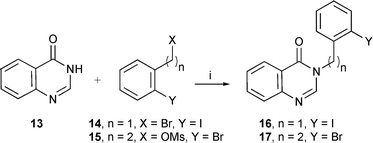 | ||
| Scheme 2 Reagents and conditions: i, tert-BuOK, DMF, 63% (16 from 14), 38% (17 from 15). | ||
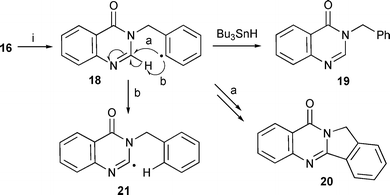 | ||
| Scheme 3 Reagents and conditions: i, Et3B, PhMe, r.t., Bu3SnH (fast addition) 96% (19), slow addition, 30% (19); AIBN, Bu3SnH (slow addition), PhMe, reflux, 45% (19); Et3B, TTMSS, 35% (19); (Me3Sn)2, tert-BuPh, reflux, 18% (20), 65% (19). | ||
The analogous six-membered ring aryl radical cyclisation using precursor 17 gave an excellent yield of the cyclised product 22 (92%) with no reduction using (Me3Sn)2 (Scheme 4). The reaction with Bu3SnH again gave largely the uncyclised reduced product 23 (55%) but did also yield a small amount of the cyclised product 22 (8%). We have observed before that 5-ring cyclisation of radicals onto heteroarenes is difficult due to strain whereas 6-ring cyclisation is more favourable.8,10,16,32 Therefore, the higher yields of cyclisation from 17 relative to 16 are expected. The results indicate that the oxidative route is dominant (i.e. loss of hydrogen from the π-radical intermediate 10) for cyclisation onto quinazolinones as observed for other heteroarenes.
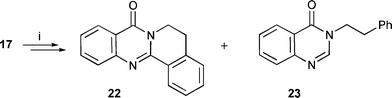 | ||
| Scheme 4 Reagents and conditions: i, (Me3Sn)2, tert-BuPh, reflux, 92% (22), 0% (23); Et3B, PhMe, r.t., Bu3SnH (fast addition), 8% (22), 55% (23). | ||
3H-4-Oxoquinazolin-2-yl radical cyclisation
As shown in Scheme 5 we envisaged an alternative route to polycyclic quinazolinones by cyclisation of 3H-4-oxoquinazolin-2-yl radicals onto pendant side chains attached to the 3-position instead of cyclisation of side chain radicals onto the quinazolinone moiety. 3H-4-Oxoquinazolin-2-yl radicals,33 generated photolytically from 3-(but-1-en-4-yl)-3H-quinazolin-4-one, have been reported to undergo 5-exo cyclisation onto the pendant alkene.33a The procedure however was not suitable for synthetic application. During our studies, a synthesis of luotonin A analogues using 3H-4-oxoquinazolin-2-yl radicals was reported.33b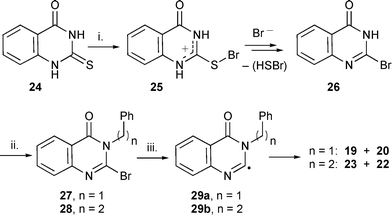 | ||
| Scheme 5 Reagents and conditions: i, Br2, EtOH, 50% (26); ii, NaH, DMF, BnBr, 46% (27); NaI, PhCH2CH2Br, 6% (28); iii, 27: Bu3SnH, Et3B, slow addition, 62% (19); Bu3GeH, Et3B, slow addition, 41% (19); (Me3Sn)2, tert-BuPh, reflux, 41% (19), 27% (20); 28: (Me3Sn)2, tert-BuPh, reflux, 0% (23), 97% (22). | ||
The plan of our protocol was to alkylate 2-bromo-3H-quinazolin-4-one to provide suitable radical precursors (Scheme 5). 2-Bromo-3H-quinazolin-4-one 26 was prepared by an adapted literature procedure from 2-mercapto-3H-quinazolin-4-one 24.34 Alkylations with activated halides, e.g. benzyl bromide, allyl bromide and methyl iodide were successful but alkylations with unactivated halides were very poor or failed. Alkylation with activated propargyl halides has also recently been reported.33b A maximum yield (6%) of alkylation was obtained with 2-phenylethyl bromide and alkylation with 3-phenylpropyl bromide failed. Alkylation with the corresponding triflates failed to give improved results. We suggest that the anion of 2-bromo-3H-4-oxoquinazoline is too stabilised and hence not nucleophilic enough to react with unactivated halides.
Various protocols were attempted to circumvent the alkylation problem. For instance, the successful bromination suggested that other 2-thioxo-2,3-dihydro-1H-quinazolin-4-ones with the 3-alkyl side chain in place could also be converted to the corresponding 2-bromo compounds. In order to test this hypothesis, 2-(phenylethyl)-2-thioxo-2,3-dihydro-1H-quinazolin-4-one 30 was prepared by ring synthesis using the reaction between 2-(methoxycarbonyl)phenyl isothiocyanate and amines.35 The structure of 30 was confirmed by X-ray crystallography (Fig. 1). However, the bromination procedure yielded only the corresponding disulfide instead of the 2-bromoquinazolinone 28.
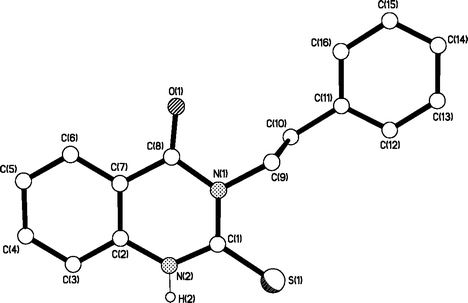 | ||
| Fig. 1 X-Ray structure of 3-(2-phenylethyl)-2-thioxo-2,3-dihydro-1H-quinazolin-4-one 30 with atom labelling. | ||
The mechanism of these brominations is unknown. The most likely intermediate is the sulfenyl bromide 25 which can either lose a proton and undergo substitution with bromide, or undergo direct substitution with bromide. The intermediate sulfenyl bromide from 2-(phenylethyl)-2-thioxo-2,3-dihydro-1H-quinazolin-4-one cannot lose a proton which is possibly significant in directing the reaction towards disulfide formation. Examples from the literature show bromoimine formation for thioureas with N,N′-substitution (i.e. can lose a proton)36 and disulfides and sulfides from thioamides.37
We carried out radical cyclisations with the precursors 27 and 28 (Scheme 5). As observed for the cyclisation of precursor 16, the 5-membered ring cyclisation from 27 was unfavourable with both Bu3SnH and Bu3GeH and yielded only the reduced uncyclised product 19. Radical abstraction of hydrogen from Bu3GeH is 20 times slower than from Bu3SnH which would favour cyclisation over reduction,38 but still only reduction was observed from the intermediate 29a. When (Me3Sn)2 was used, a moderate yield of cyclisation product (27%) was obtained but reduction was still the major route (41%). The 6-membered ring cyclisation from precursor 28via29b gave a quantitative yield of cyclisation to 22 with none of the reduced compound 23 formed. This cyclisation could either proceed by 5-exo cyclisation followed by a neophyl rearrangement or directly by 6-endo cyclisation. The results show that radical cyclisation can be used onto, or from, the quinazolinone moiety, but that a useful method of synthesis of the 2-bromoquinazolinones is still required. Addition of 3H-4-oxoquinazolin-2-yl radicals onto isonitriles has also recently been reported.33b
Alkyl radical cyclisation
Several methods have been used for the synthesis of both deoxyvasicinone 2 and mackinazolinone 3 but none involving radicals.39 Our protocol using cyclisation of an alkyl radical onto 3H-quinazolin-4-one rings was aimed at the synthesis of both natural products (Scheme 6). Routine alkylation yielded the required radical precursors 31a,b. An initial study with Bu3SnH using precursor 31a yielded only reduced uncyclised product 33a (78%). The rate of cyclisation of the alkyl radical is obviously slow, and it is intercepted by Bu3SnH to yield the respective reduced uncyclised product 33a. Therefore, (Me3Sn)2 was used to facilitate cyclisation which gave moderate yields of deoxyvasicinone 2 and mackinazolinone 3.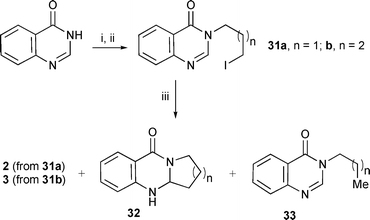 | ||
| Scheme 6 Reagents and conditions: i, NaH, DMF, a. 1-chloro-3-iodopropane, 51%; b. 1-chloro-4-iodobutane, 65%; ii, NaI, acetone, reflux, 53% (31a), 67% (31b); iii, (Me3Sn)2, tert-BuPh, reflux, hν: a. 31a gave 20% (2), 13% (32a) and 6% (33a); b. 31b gave 30% (3), 23% (32b) and 0% (33b); Et3B (20 equiv.), tert-BuPh, air (yields by 1H NMR analysis): a. 31a gave 40% (2), 10% (32a) and 30% (33a); b. 32b gave 61% (3), 23% (32b) and 0% (33b). | ||
The use of Et3B mediated reactions gave better yields of both 2 and 3 (Scheme 6). We suggest that the ethyl radical generated from the reaction between Et3B and oxygen is able to abstract iodine from the radical precursors 31a,b to yield alkyl radicals and ethyl iodide, which is lost from the reaction. The hydrogen in the π-radical intermediates (cf.10) is also most likely abstracted by ethyl radicals to facilitate rearomatisation.40 Unusually, some cyclised reduced material 32a,b was obtained suggesting disproportionation was also taking place. GCMS analysis of the crude product from the Et3B facilitated reaction with 31a indicated traces (ca. 5%) of 2-ethyl-3-propyl-3H-quinazolin-4-one and 3-pentyl-3H-quinazolin-4-one indicating addition of ethyl radicals to intermediates. No uncyclised reduced material 33b was observed for the 6-ring cyclisation of 31b. Again, this provides evidence that 5-ring cyclisation onto heteroarenes is strained and that 6-ring cyclisation (84% of cyclised products) is more favourable.
Heteroaryl radical cyclisation
During our studies, Pd(0) catalysed cyclisations were reported for 2-bromoindole41 and 2-bromoquinoline41,42 moieties onto the 2-position of quinazolin-4-one to yield rutaecarpine in poor yield (24% maximum) and luotonin A in good yield (86%) respectively.The synthesis of luotonin A 5 using 2-quinolinyl radicals was carried out as a further example of cyclisation onto the quinazolinone moiety (Scheme 7). The use of 2-quinolinyl radicals in cyclisation has two literature precedents; synthesis of camptothecin using cyclisation of 2-quinolinyl radicals onto a pyridone moiety29 and synthesis of 10,11-methylene-14-azaxamptothecin and 14-azacamptothecin using cyclisation of 2-quinolinyl radicals onto 3H-pyrimidin-4-one moieties.30 These cyclisations also proceeded by ‘oxidative cyclisation’ as observed for the 3H-quinazolin-4-ones.
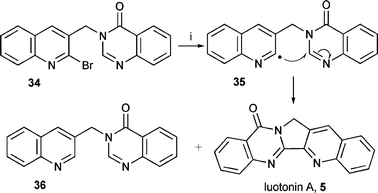 | ||
| Scheme 7 Reagents and conditions: i, (Me3Sn)2, tert-BuPh, reflux, hν: 51% (5), 15% (36); Et3B (20 equiv.), PhMe, r.t., air: Bu3SnH (fast addition), 0% (5), 53% (36) Bu3SnH (slow addition), 14% (5), 32% (36); Bu3GeH (fast addition), 18% (5), 11% (36). | ||
The required starting material 34 was prepared by a literature procedure from 2-chloroquinoline-2-carbaldehyde46 and reacted under several radical conditions. Unusually, the yield from 5-exo cyclisation was significant. The use of (Me3Sn)2 gave a reasonable yield (53%) of luotonin A 5 but also some of the reduced product 36. The yield was high in comparison to the 5-exo cyclisation of the phenyl analogue 16. Luotonin A was even obtained with Bu3SnH and Bu3GeH, clearly indicating that the cyclisation is more favourable for 2-quinolinyl radicals as compared to phenyl radicals. The yield was not as high as the Pd(0)-catalysed cyclisation42 but further optimisation could improve the yield.
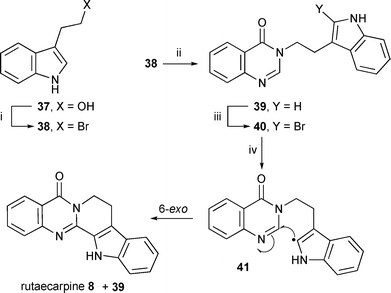 | ||
| Scheme 8 Reagents and conditions: i, PBr3, DCM, 56% (38); ii, tert-BuOK, DMF, 3H-quinazolin-4-one, 39% (39); iii, NBS, DCM, 0 °C, 30 min, 38% (40); iv, (Me3Sn)2, tert-BuPh, reflux, hν: 55% (8), 0% (39); Et3B (20 equiv.), PhMe. r.t., air: Bu3SnH (slow addition), 15% (8), 57% (39). | ||
The preparation of the 2-bromoindole part of the precursor proved troublesome even with protection of the side chain and the NH. The use of NBS has been reported in the literature but the reaction is very sensitive, and can be adversely affected by the NH and side chain groups.48,49,51 We finally used an adapted literature procedure41 whereby the 3H-quinazolin-4-one moiety was used as the ‘protective group’ for the indole side chain (i.e.39). The bromination was rapid and short reaction times and low temperature gave the best yields of the 2-bromoindole precursor 40. As reported in the literature for 2-bromoindoles,48,49,51,52 decomposition of the product was a problem until it was purified. The free indole-NH did not interfere and was therefore not protected. A blank reaction between NBS and 3-methylquinazolin-4-one gave no reaction after two days indicating that in the NBS reaction with 39, bromination of the quinazolinone ring is unlikely to be the cause of the decomposition.
3-[2-(1H-Indol-3-yl)ethyl]-4(3H)-quinazolinone 39 was prepared by alkylation of 4(3H)-quinazolinone with 3-(2-bromoethyl)-1H-indole 38 which was prepared by bromination of tryptophol 37.
Radical cyclisation of the precursor 40 gave the predicted 6-exo cyclisation of the intermediate radical 41 to yield rutaecarpine 8. Only cyclisation was obtained when (Me3Sn)2 was used and even reductive conditions with Bu3SnH yielded a small amount of cyclisation product. The 6-exo cyclisation yields were similar to the equivalent cyclisation with aryl radicals (see Scheme 4). Longer reactions times led to decomposition.
Acyl radicals—synthesis of tryptanthrin
Tryptanthrin has been synthesised by a range of protocols.3,6,53We prepared an authentic sample of tryptanthrin 4 by a literature procedure in order to obtain full spectroscopic data for comparison.53
Aromatic acyl radical cyclisation has recently been shown to be a useful synthetic technique.17–19,54 Although 5-membered ring cyclisation was reported to be unsuccessful,19 we have shown that 5-ring cyclisation onto the 3H-quinazolin-4-one moeity was possible (e.g. the luotonin synthesis). Therefore, we carried out the syntheses as shown in Scheme 9. The aryl-CO bond in the intermediate acyl radical 44 is strong enough to avoid decarbonylation which is a rapid reaction for alkyl-CO radicals.55 However, it is possible that we failed to isolate products resulting from CO loss.
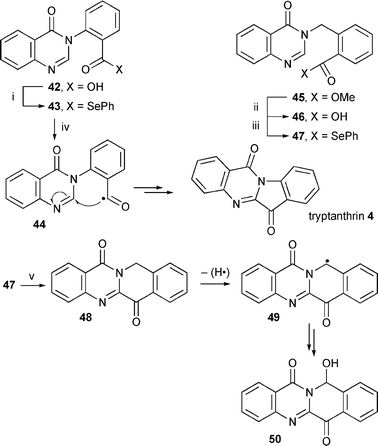 | ||
| Scheme 9 Reagents and conditions: i, Bu3P, PhSeSePh, DCM, 73% (42); ii, LiOH, EtOH, H2O; iii, Bu3P, PhSeSePh, DCM, 68% (47); iv, (Me3Sn)2, tert-BuPh, reflux, hν, 3% (4); PhH, r.t., hν, 12 h, 15% (4); PhH, reflux, hν, 10 h, 11% (4); AIBN, PhH, reflux, hν, 0% (4); v, (Me3Sn)2, PhH, reflux, hν, 39% (50). | ||
The starting material 42 was prepared in one step by a literature procedure56 and converted to the acyl selenide 43 by standard procedures.16 Several conditions were used based on literature reports.17–19,54 The highest yield (15%) was obtained by photolysis at r.t. Although the yield is poor, we believe this is the first example of a 5-exo acyl radical cyclisation onto a heterocycle. When AIBN was added to the reaction, an intractable mixture was obtained. Heating under reflux was not required and UV photolysis alone was enough to facilitate the reaction, presumably by homolysis of the carbonyl-SePh bond. In a blank reaction, heating under reflux in benzene yielded only unaltered starting material after 24 h. The mechanism is unclear, other than 5-exo acyl radical cyclisation followed by hydrogen abstraction from the resulting π-radical intermediate. Large amounts of diphenyl diselenide were isolated indicating CO–Se bond homolysis.
We also investigated the 6-membered ring cyclisation because these had proved to be more successful than the 5-ring cyclisations (Scheme 9). The acyl radical precursor 47 was prepared by standard procedures. The ester 45 was synthesised by alkylation of 3H-quinazolin-4-one with methyl 2-(bromomethyl)-benzoate. Cyclisation using Et3B only, AIBN only, UV photolysis only and slow addition of TTMSS with Et3B as initiator all gave intractable mixtures. However, reaction with (Me3Sn)2 gave a moderate yield of the unusual product 50 along with an intractable mixture of other products. The structure of the hydroxylated product 50 was confirmed by X-ray crystallography (Fig. 2). The product indicated 6-ring cyclisation as expected but that the product 48 was unstable to the reaction conditions and was readily oxidised introducing an OH group onto the newly formed ring.
![X-Ray structure of 11-hydroxy-11H-isoquinolino[3,2-b]quinazolin-6,13-dione 50 with atom labelling.](/image/article/2007/OB/b614075k/b614075k-f2.gif) | ||
| Fig. 2 X-Ray structure of 11-hydroxy-11H-isoquinolino[3,2-b]quinazolin-6,13-dione 50 with atom labelling. | ||
Analogous reactions with 2-indolyl acyl radicals have yielded a range of unexpected products which included a ‘quinone’ product caused by oxidation of the benzylic methylene during the reaction.17,18 2-Cyanoprop-2-yl radicals from the breakdown of AIBN were proposed as the abstracting radicals. In our reaction, no AIBN is present so we suggest that any of the intermediate radicals (e.g. intermediate acyl or π-radical, PhSe˙) can abstract the benzylic hydrogen, which is a favourable process, to form a stable radical intermediate 49. Although air was excluded, traces of oxygen are likely to be present and to react rapidly with trimethylstannyl radicals to form a peroxyl radical (Me3SnOO˙).45,57 Combination of the peroxyl radical with the benzylic radical 49 would yield a peroxide which could easily break down to yield 50. Alternatively, a standard auto-oxidation mechanism with the traces of oxygen present could explain this unusual product.
Conclusions
Our results show that radical cyclisation on the quinazolinone moiety can be used for synthesis. The results show that radical cyclisation is also favourable for radicals centred on the 2-position of the quinazolinone moiety but that a better method for the synthesis of 2-bromoquinazolinones is still required.Experimental
General
Commercial dry solvents were used in all reactions except for light petroleum and ethyl acetate which were distilled from CaCl2 and dichloromethane which was distilled from CaH2. Light petroleum refers to the bp 40–60 °C fraction. Sodium hydride was obtained as 60% dispersion in oil. A 2.5 M solution of n-butyl lithium in hexane was used. A solution of Et3B in hexane (1.0 M) was used in all cases. Melting points were determined on a Stuart Scientific SMP3 melting point apparatus and are uncorrected. Elemental analyses were determined on a Perkin Elmer 2400 CHN Elemental Analyser. Infrared spectra were recorded on a Perkin-Elmer Paragon 1000 FT-IR spectrophotometer on NaCl plates. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a Bruker DPX-400 instrument, or 1H (250 MHz) spectra were obtained using a Bruker AC-250 spectrometer. Spectra were obtained from solutions in CDCl3 with TMS as the internal standard for 1H NMR spectra and deuteriochloroform as the standard for 13C NMR spectra unless otherwise specified. Chemical shifts are given in parts per million (ppm) and J values in hertz (Hz). Assignments were made using a combination of COSY and HMQC analysis. Mass spectra were recorded on a Jeol SX102 high resolution mass spectrometer or were run by the EPSRC MS Service at the University of Wales, Swansea. GCMS was carried out on a Fisons 8000 series mass spectrometer using a 15 m × 0.25 mm DB-5 GC column. TLC using silica as absorbent was carried out with aluminium backed plates coated with silica gel (Merck Kieselgel 60 F254), and TLC using alumina as absorbent was carried out with aluminium backed plates coated with neutral aluminium oxide (Merck 150 F254, TypeT). Silica gel (Merck Kieselgel 60 H silica) was used for column chromatography unless otherwise specified. Column chromatography using alumina was carried out with Aldrich aluminium oxide, activated neutral, Brockmann 1, STD Grade, 150 mesh size. Prep-TLC was carried out using aluminium oxide (Merck 60 PF254, Type E). Bu3GeH was prepared from GeCl4 by a known procedure.38General procedure for Bu3SnH reactions using Et3B as initiator. A solution of Bu3SnH (0.76 g, 2.6 mmol) and 3-(2-iodobenzyl)-3H-quinazolin-4-one (0.44 g, 1.2 mmol) in dry toluene (40 cm3) was deoxygenated under an atmosphere of nitrogen and stirred for 1 h. Triethylborane (3.6 cm3, 3.6 mmol) was added via a needle through a septum; the needle was then left open to allow air (oxygen) to enter the reaction and the mixture stirred for 1 h. More triethylborane (3.6 cm3, 3.6 mmol) was added and the reaction mixture stirred for a further 10 h. Dilute hydrochloric acid was added to extract the protonated quinazolone products into the aqueous layer. The aqueous layer was washed with light petroleum to remove tributyltin residues. The aqueous layer was basified with sodium hydroxide to pH 14 and extracted with DCM. The combined organic layers were dried and evaporated under reduced pressure yielding the reduced product 3-benzyl-3H-quinazolin-4-one 19 as colourless crystals (0.27 g, 1.2 mmol, 96%), mp 116–117 °C (lit.,58 117–118 °C); νmax(thin film)/cm−1 3419, 1675, 1609, 1559, 1473, 1369, 1332, 1161, 1026, 773, 709, 694 and 645; δH 5.08 (2 H, s, CH2), 7.24–7.23 (5 H, m), 7.48 (1 H, ddd, J 8.2 6.3 1.8, 6-H), 7.74–7.68 (2 H, m, 7,8-H), 8.11 (1 H, s, 2-H) and 8.32 (1 H, dd, J 8.2 2.0, 5-H); δC 49.6 (CH2), 122.2 (4a-C), 126.9 (5-H), 127.4 (6-H), 127.5 (8-H), 128.0 (CH), 128.3 (CH), 129.0 (CH), 134.3 (7-H), 135.8 (C), 146.4 (2-H), 148.0 (8a-C) and 161.1 (4-C). The data were identical to that in the literature.58 The procedure was repeated using TTMSS in place of Bu3SnH to yield 3-benzyl-3H-quinazolin-4-one 19 (35%).
Hexamethylditin. The general procedure for reactions using photolysis and hexamethylditin yielded 5,6-dihydroisoquino[1,2-b]quinazolin-8-one 22 as colourless crystals (92%), mp 195–197 °C (lit.,60 196 °C); Found: M+, 249.1025. C16H12N2O requires 249.1022; νmax(thin film)/cm−1 3421, 1682, 1590, 1556, 1471, 1150, 762 and 692; δH 3.06 (2 H, t, J 6.4, CH2), 4.35 (2 H, t, J 6.4, NCH2), 7.22–7.13 (1 H, m, ArH), 7.43–7.37 (3 H, m, ArH), 7.71–7.69 (2 H, m, ArH), 8.25 (1 H, d, J 7.6, ArH) and 8.43–8.40 (1 H, m, ArH); δC 27.4 (CH2), 39.5 (NCH2), 120.7 (C), 126.5 (CH), 126.8 (CH), 127.5 (CH), 127.6 (CH), 127.9 (CH), 129.5 (C), 131.7 (CH), 134.2 (CH), 137.0 (C), 147.7 (C), 149.3 (C) and 161.6 (CO); m/z (EI) 249 (M+, 17%), 247 (100), 128 (20), 116 (46), 104 (30), 90 (34), 89 (62), 77 (85), 76 (97), 63 (53) and 39 (54). One CH signal was coincident or could not be detected.
Bu3SnH and Et3B. The general procedure for Bu3SnH reactions using Et3B as initiator yielded an inseparable mixture of 3-[2-(phenylethyl)]-3H-quinazolin-4-one 23 (55%) and 5,6-dihydroisoquino[1,2-b]quinazolin-8-one 22 (8%). The yields were estimated using 1H NMR spectroscopic analysis.
Bu3SnH and Et3B. The general procedure for Bu3SnH reactions using Et3B as initiator were used with Bu3SnH added by syringe pump over 6 h and the reaction stirred for a further 5 h. Work-up yielded 3-benzyl-3H-quinazolin-4-one 19 as colourless crystals (0.15 g, 0.62 mmol, 62%). The data were identical to authentic material. A repeat experiment using Bu3GeH in place of Bu3SnH gave 3-benzyl-3H-quinazolin-4-one 19 in reduced yield (47%).
Hexamethylditin. The general procedure for reactions using photolysis and hexamethylditin yielded an inseparable mixture of 12H-isoindolo-[1,2-b]quinazolin-10-one 20 (27%) and 3-benzyl-3H-quinazolin-4-one 19 (41%). The yields were estimated using 1H NMR spectroscopic analysis.
Hexamethylditin. The general procedure for reactions using photolysis and hexamethylditin (24 h) yielded 5,6-dihydroisoquino[1,2-b]quinazolin-8-one 22 as colourless crystals (97%). The data were identical to authentic material.
Hexamethylditin. The general procedure for reactions using photolysis and hexamethylditin with 3-(3-iodopropyl)-3H-quinazolin-4-one 31a (24 h) yielded: 2,3-dihydro-1H-pyrrolo[2,1-b]quinazolin-9-one 2 as colourless crystals (20%), mp 190–192 °C (lit.,61 196–198 °C); νmax(thin film)/cm−1 3418, 2355, 1651, 1621, 1470, 1385, 1285, 775, 695 and 667; δH 2.35–2.23 (2 H, m, CH2), 3.18 (2 H, t, J 12.6, CH2), 4.21 (2 H, t, J 11.5, NCH2), 7.47–7.62 (1 H, m, ArH), 7.76–7.62 (2 H, m, ArH) and 8.28 (1 H, dd, J 12.6, 2.4, ArH); δC 19.5 (CH2), 32.5 (CH2), 46.5 (NCH2), 120.4 (C), 126.2 (CH), 126.4 (CH), 126.8 (CH), 134.2 (CH), 149.1 (C), 159.5 (C) and 161.0 (C). 2,3,3a,4-Tetrahydro-1H-pyrrolo[2,1-b]quinazolin-9-one 32a as colourless crystals (12%), mp 187–188 °C (lit.,62 184–185 °C); νmax(thin film)/cm−1 3421, 1633, 1503, 1470, 1434, 1337, 1151, 752 and 695; δH 2.32–2.29 (2 H, m, 2-CH2), 3.19 (1 H, t, J 8.0, 1-H), 3.75–3.67 (2 H, m, CHCH2), 4.21 (1 H, dd, J 8.0, 7.2, 1-H), 4.5 (1 H, bs, NH), 5.03 (1 H, dd, J 7.2, 5.6, 3a-H), 6.69 (1 H, d, J 12.9, ArH), 6.92–6.88 (1 H, m, ArH), 7.27–7.24 (1 H, m, ArH) and 7.91–7.87 (1 H, dd, J 12.3, 9.9, ArH); δC 21.7 (CH2), 33.2 (CHCH2), 44.3 (NCH2), 69.9 (CH), 114.9 (CH), 118.4 (CH), 119.9 (C), 126.8 (CH), 132.9 (CH), 149.1 (C) and 162.2 (C). 3-Propyl-3H-quinazolin-4-one 33a as colourless crystals (6%). The data were identical with those from an independently prepared authentic sample.
Bu3SnH and Et3B. 3-Propyl-3H-quinazolin-4-one 33a (78%) was the only product.
Et3B only. The general procedure for Bu3SnH and Et3B reactions were used except that the Bu3SnH was not added. 1H NMR spectral analysis showed 2,3-dihydro-1H-pyrrolo[2,1-b]quinazolin-9-one 2 (40%), 2,3,3a,4-tetrahydro-1H-pyrrolo[2,1-b]quinazolin-9-one 32a (10%) and 3-propyl-3H-quinazolin-4-one 33a (30%).
(Me3Sn)2 and photolysis. The general procedure for reactions using photolysis and hexamethylditin (24 h) yielded: 6,7,8,9-tetrahydropyrido[2,1-b]quinzolin-11-one 3 as colourless crystals (30%), mp 94–95 °C (lit.,63 96–97 °C); Found: MH+, 201.1020. C12H13N2O requires 201.1022; νmax(thin film)/cm−1 3423, 2948, 2110, 1655, 1614, 1565, 1477, 1173, 1102, 990, 870, 771 and 583; δH 2.18–1.91 (4 H, m, CH2CH2), 3.00 (2 H, t, J 6.6, NCCH2), 4.09 (2 H, t, J 6.3, NCH2), 7.44–7.40 (1 H, m, ArH), 7.59 (1 H, dd, J 8.3, 0.5, ArH), 7.73–7.69 (1 H, m, ArH) and 8.26 (1 H, dd, J 8.0 1.5, ArH); δC 19.3 (CH2), 22.1 (CH2), 31.9 (CH2), 42.3 (CH2), 120.4 (C), 126.1 (CH), 126.4 (CH), 126.6 (CH), 134.1 (CH), 147.4 (C), 154.9 (C) and 162.2 (C); m/z (EI) 200 (M+, 50%), 199 (40), 90 (33), 76 (34), 63 (33), 50 (34) and 41 (90). 5,5a,6,7,8,9-Hexahydropyrido[2,1-b]quinazolin-11-one 32b as colourless crystals (23%), mp 128–130 °C (lit.,2 132–133 °C); Found: MH+, 203.1180. C12H15N2O requires 203.1179; νmax(thin film)/cm−1 3398, 3058, 2938, 2224, 1632, 1518, 1416, 1218, 1153, 1026, 754 and 692; δH 1.52–1.49 (2 H, m), 1.91–1.75 (4 H, m), 2.66–2.59 (1 H, m, 9-H), 4.25 (1 H, m, NH), 4.64–4.59 (1 H, m, 9-H), 4.78 (1 H, dd, J 13.5, 3.1, 5a-H), 6.57 (1 H, d, J 7.1, ArH), 6.79 (1 H, ddd, J 7.8, 2.9, 1.0, ArH), 7.27–7.22 (1 H, m, ArH) and 7.90 (1 H, dd, J 7.8 1.6, ArH); δC 22.3 (CH2), 24.2 (CH2), 33.4 (CH2), 42.0 (CH2), 68.5 (CH), 113.7 (CH), 115.2 (C), 118.8 (CH), 128.7 (CH), 133.4 (CH), 145.9 (C) and 164.3 (C); m/z (EI) 202 (M+, 30%), 146 (42), 119 (48), 92 (42), 55 (79) and 41 (100).
Et3B only. The general procedure for Bu3SnH and Et3B reactions were used except that the Bu3SnH was not added. 1H NMR spectral analysis showed 6,7,8,9-tetrahydro-pyrido[2,1-b]quinzolin-11-one 3 (61%) and 5,5a,6,7,8,9-hexahydropyrido[2,1-b]quinazolin-11-one 32b (23%).
(Me3Sn)2 and photolysis. The general procedure for reactions using photolysis and hexamethylditin (24 h) yielded: luotonin A 5 (51%). All data were the same as reported.45 3-[(Quinolin-3-yl)methyl]-3H-quinazolin-4-one 36, colourless crystals, mp 252–253 °C; Found: MH+, 288.1130. C18H13N3O requires 288.1131; νmax(thin film)/cm−1 3420, 2357, 2107, 1646, 1558, 1450, 1361, 1332, 510 and 454; δH 5.39 (2 H, s, CH2), 7.58–7.53 (2 H, m, ArH), 7.81–7.70 (4 H, m, ArH), 8.10 (1 H, dd, J 8.9, 0.4, ArH), 8.17 (1 H, d, J 1.8, ArH), 8.35–8.32 (1 H, m, ArH) and 8.99 (1 H, d, J 1.8, ArH); δC 47.7 (CH2), 122.1 (CH), 126.9 (CH), 127.3 (CH), 127.7 (2 × CH), 127.9 (CH), 128.6 (2 × C), 129.3 (CH), 130.0 (CH), 134.6 (CH), 135.4 (CH), 145.9 (CH), 147.9 (C), 148.0 (C), 150.1 (CH) and 161.1 (C); m/z (EI) 287 (M+, 45%), 270 (42), 132 (100), 115 (70), 89 (20), and 63 (21).
Bu3SnH and Et3B. The general procedure for Bu3SnH reactions using Et3B as initiator were used with Bu3SnH added by syringe pump over 5 h and the reaction stirred for a further 5 h to yield luotonin A 5 (14%) and 3-[(quinolin-3-yl)methyl]-3H-quinazolin-4-one 36 (32%). When the Bu3SnH was added at the beginning of the reaction only 3-[(quinolin-3-yl)methyl]-3H-quinazolin-4-one 36 was obtained (53%).
Bu3GeH and Et3B. Luotonin A (18%) and [(3-quinolin-3-yl)methyl]-3H-quinazolin-4-one 36 (11%) were obtained.
Bu3SnH addition. The general procedure for Bu3SnH reactions using Et3B as initiator were used with Bu3SnH added by syringe pump over 12 h to yield rutaecarpine 8 (15%), mp 254–255 °C (lit.,41 256–257 °C) and 3-[2-(1H-indol-3-yl)ethyl]-4(3H)-quinazolinone 39 (57%), both as colourless crystals. The data for rutaecarpine were the same as those in the literature.41 The data for 39 were the same as obtained in the previous synthesis.
(Me3Sn)2 and photolysis. The general procedure for reactions using photolysis and hexamethylditin (6 h) yielded rutaecarpine (55%) as the only product.
UV and reflux. 2-(4-Oxo-4H-quinazolin-3-yl)selenobenzoic acid Se-phenyl ester 43 (0.24 g, 0.59 mmol) was heated under reflux in benzene (20 cm3) for 12 h under UV irradiation. The solvent was removed under vacuum and the crude product was purified by column chromatography using silica gel as absorbent and light petroleum–EtOAc (2 : 1) yielding tryptanthrin 4 as yellow crystals (0.02 g, 0.07 mmol, 13%), mp 213–214 °C (lit.53 215–217 °C); Found: MH+, 249.0659. C15H9N2O2 requires 249.0660; δH 7.43 (1 H, t, J 8.0, ArH), 7.68 (1 H, t, J 8.0, ArH), 7.79 (1 H, t, J 8.0, ArH), 7.86 (1 H, t, J 8.0, ArH), 7.92 (1 H, d, J 8.0, ArH), 8.04 (1 H, d, J 8.0, ArH), 8.44 (1 H, d, J 8.0, ArH) and 8.64 (1 H, d, J 8.0, ArH); δC 118.0 (CH), 122.0 (C), 123.8 (C), 125.4 (CH), 127.2 (CH), 127.6 (CH), 130.2 (CH), 130.7 (CH), 135.1 (CH), 138.3 (CH), 146.4 (C), 146.7 (C), 158.1 (C) and 171.1 (2 × CH); m/z (EI) 248 (M+, 51%), 220 (22), 130 (28), 102 (54), 90 (100), 76 (81), 63 (55) and 50 (74). The data were identical to the literature data53 and those of authentic material. Repeating the reaction for 10 h gave a yield of 11% of 4. The procedure was repeated using the same conditions but with the addition of AIBN (2.0 equiv.) for 8 h. An intractable mixture was obtained. A blank reaction with no sunlamp photolysis or added AIBN, in benzene heated under reflux for 24 h gave a quantitative recovery of unaltered starting material 43.
(Me3Sn)2, sunlamp. The reaction between 2-(4-oxo-4H-quinazolin-3-yl)selenobenzoic acid Se-phenyl ester 47 (0.29 g, 0.72 mmol) and hexamethylditin (0.70 g, 2.15 mmol) was carried out in tert-butylbenzene (20 cm3). The mixture was heated under reflux and irradiated with a sunlamp for 4 h. Dilute hydrochloric acid was added to the cooled reaction mixture to extract the protonated quinazolone products into the aqueous layer. The aqueous layer was washed with light petroleum to remove tin residues. The aqueous layer was basified with sodium hydroxide to pH 14 and extracted with DCM. The combined organic layers were dried and evaporated under reduced pressure. The residue was purified by column chromatography using silica gel as absorbent and light petroleum–EtOAc (2 : 1) yielding tryptanthrin 4 (5 mg, 0.02 mmol, 3%).
Sunlamp. When the sunlamp irradiation alone was used for 30 min, the highest yield of tryptanthrin (15%) was obtained.
X-Ray crystallography
Data were collected at 150(2) K on a Bruker SMART 1000 diffractometer with sealed tube source64 for 30 and Bruker-Nonius CCD diffractometer with rotating anode source65 for 50. The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the SHELXTL suite of programs.66 All the non-hydrogen atoms were refined with anisotropic atomic displacement parameters and hydrogen atoms were inserted at calculated positions using a riding model except for H(2) for which coordinates were freely refined in both structures. In 30, molecules form into chains via intermolecular H-bonds N(2)–H(2)⋯O(1′). The structure of 50 additionally contains one molecule of CHCl3 in the asymmetric unit and molecules are linked into chains via strong intermolecular H-bonds O(2)–H(2)⋯N(2′). Off-set π–π stacking is also dominant with separations in the range 3.33–3.5 Å.Crystal data for 30: C16H14N2OS, M = 282.35, monoclinic, Cc, a = 4.6405(7), b = 25.329(4), c = 12.1910(17) Å, β = 97.047(2)°, U = 1422.1(4) Å3, T = 150(2) K, Z = 4, µ(Mo–Kα) = 0.224 mm−1, 5350 reflections measured, 3005 unique (Rint = 0.0368) which were used in all calculations, wR = 0.1590 for all data, R1 = 0.0602 for 2351 data with F2 ≥ 2σ(F2). Absolute structure parameter = 0.13(13)—thus reliably determined.
Crystal data for 50: C17H11Cl3N2O3, M = 397.63, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 6.6511(3), b = 8.6274(3), c = 14.5395(7) Å, α = 90.123, β = 94.525(3), γ = 99.594(3)°, U = 819.97(6) Å3, T = 120(2) K, Z = 2, µ(Mo–Kα) = 0.579 mm−1, 11250 reflections measured, 3715 unique (Rint = 0.0370) which were used in all calculations, wR = 0.0939 for all data, R1 = 0.0353 for 2898 data with F2
≥ 2σ(F2).‡
, a = 6.6511(3), b = 8.6274(3), c = 14.5395(7) Å, α = 90.123, β = 94.525(3), γ = 99.594(3)°, U = 819.97(6) Å3, T = 120(2) K, Z = 2, µ(Mo–Kα) = 0.579 mm−1, 11250 reflections measured, 3715 unique (Rint = 0.0370) which were used in all calculations, wR = 0.0939 for all data, R1 = 0.0353 for 2898 data with F2
≥ 2σ(F2).‡
Acknowledgements
We thank the EPSRC (DTA award) and Loughborough University for a Postgraduate Studentship (T. S.), and the EPSRC Mass Spectrometry Unit, Swansea University, Wales for mass spectra. We also thank the EPSRC National Crystallography Service at Southampton University, UK for collecting X-ray diffraction data for compound 50.References
- A. Al-Shamma, S. Drake, D. L. Flynn, L. A. Mitscher, Y. H. Park, G. S. R. Rao, A. Simpson, J. K. Swayze, T. Veysogly and S. T.-S. Wu, J. Nat. Prod., 1981, 745–747 CrossRef CAS.
- S. R. Johns and J. A. Lamberton, J. Chem. Soc., Chem. Commun., 1965, 267 Search PubMed; J. S. Fitzgerald, S. R. Johns, J. A. Lamberton and A. H. Redcliffe, Aust. J. Chem., 1966, 19, 151–158 CAS.
- M. Hamburger, Phytochem. Rev., 2002, 1, 333 Search PubMed.
- Z.-Z. Ma, Y. Hano, T. Nomura and Y.-J. Chen, Heterocycles, 1997, 46, 541–546 CrossRef CAS.
- Y. Asahina, Acta Phytochim., 1922, 1, 67 Search PubMed; Y. Asahina and K. Kashiwaki, J. Pharm. Soc. Jpn, 1915, 35, 1293.
- J. P. Michael, Nat. Prod. Rev., 2004, 21, 650–668 Search PubMed.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS.
- F. Aldabbagh, W. R. Bowman and E. Mann, Tetrahedron Lett., 1997, 38, 7937–7940 CrossRef CAS; F. Aldabbagh, W. R. Bowman, E. Mann and A. M. Z. Slawin, Tetrahedron, 1999, 55, 8111–8128 CrossRef CAS.
- Y. Antonio, E. De La Cruz, E. Galeazzi, A. Guzman, B. L. Bray, R. Greenhouse, L. J. Kurz, D. A. Lustig, M. L. Maddox and J. M. Muchowski, Can. J. Chem., 1994, 72, 15–22 CAS.
- S. M. Allin, W. R. S. Barton, W. R. Bowman and T. McInally, Tetrahedron Lett., 2002, 43, 4191–4193 CrossRef CAS.
- F. E. Ziegler and M. Belema, J. Org. Chem., 1997, 62, 1083–1094 CrossRef CAS and references therein.
- C. J. Moody and C. L. Norton, J. Chem. Soc., Perkin Trans. 1, 1997, 2639–2643 RSC.
- J. Marco-Contelles and M. Rodríquez-Fernández, Tetrahedron Lett., 2000, 41, 381–384 CrossRef CAS; J. Marco-Contelles and M. Rodríquez-Fernández, J. Org. Chem., 2001, 66, 3717–3725 CrossRef CAS.
- J. A. Murphy and M. S. Sherburn, Tetrahedron, 1991, 47, 4077–4088 CrossRef CAS.
- Y. Z. Osornio, L. D. Miranda, R. Cruz-Almaza and J. D. Muchowski, Tetrahedron Lett., 2004, 45, 2855–2858 CrossRef CAS.
- S. M. Allin, W. R. S. Barton, W. R. Bowman and T. McInally, Tetrahedron Lett., 2001, 42, 7887–7890 CrossRef CAS.
- M.-L. Bennasar, T. Roca and F. Ferrando, Org. Lett., 2006, 8, 561–564 CrossRef CAS.
- M.-L. Bennasar, T. Roca and F. Ferrando, J. Org. Chem., 2005, 70, 9077–9080 CrossRef CAS.
- M.-L. Bennasar, T. Roca and F. Ferrando, Tetrahedron Lett., 2004, 45, 5605–5609 CrossRef CAS.
- S. R. Flannagan, D. C. Harrowven and M. Bradley, Tetrahedron Lett., 2003, 44, 1795–1798 CrossRef CAS.
- T. C. T. Ho and K. Jones, Tetrahedron, 1997, 53, 8287–8294 CrossRef CAS.
- A. Nadin and T. Harrison, Tetrahedron Lett., 1999, 40, 4073–4076 CrossRef.
- K. C. Mujumbar and P. P. Mukhopadhyay, Synthesis, 2003, 920–924; K. C. Mujumbar, P. K. Basu, P. P. Mukhopadhyay, S. Sarkar, S. K. Ghosh and P. Biswas, Tetrahedron, 2003, 59, 2151–2157 CrossRef.
- K. Orito, Y. Satoh, H. Nishizawa, R. Harada and M. Tokuda, Org. Lett., 2000, 2, 2535–2537 CrossRef CAS.
- D. C. Harrowven, B. J. Sutton and S. Coulton, Tetrahedron, 2002, 58, 3387–3400 CrossRef CAS; D. C. Harrowven, B. J. Sutton and S. Coulton, Tetrahedron Lett., 2001, 42, 2907–2910 CrossRef CAS.
- D. C. Harrowven, B. J. Sutton and S. Coulton, Tetrahedron Lett., 2001, 42, 9061–9064 CrossRef CAS; D. C. Harrowven, B. J. Sutton and S. Coulton, Org. Biomol. Chem., 2003, 4047–4057 RSC.
- Review: A. Studer and M. Bossart, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 2001, vol. 2, pp. 62–80 Search PubMed.
- A. L. J. Beckwith, W. R. Bowman, V. W. Bowry, E. Mann, J. Parr and J. M. D. Storey, Angew. Chem., Int. Ed., 2004, 43, 95–98 CrossRef.
- D. L. Comins, H. Hong and G. Jianhua, Tetrahedron Lett., 1994, 35, 5331–5334 CrossRef CAS.
- M. A. Elban, W. Sun, B. M. Eisenhauer, R. Gao and S. M. Hecht, Org. Lett., 2006, 8, 3513–3516 CrossRef CAS; N. J. Rahier, K. Chen, R. Gao, B. M. Eisenhauer and S. M. Hecht, Org. Lett., 2003, 5, 835–837 CrossRef CAS.
- J. B. Bremner and W. Sengpracha, Tetrahedron, 2005, 61, 941–953 CrossRef CAS.
- S. M. Allin, W. R. Bowman, M. R. J. Elsegood, V. McKee, R. Karim and S. S. Rahman, Tetrahedron, 2005, 61, 2689–2696 CrossRef CAS.
- (a) C. Kaneko, K. Kasai, N. Katagiri and T. Chiba, Chem. Pharm. Bull., 1986, 34, 3672–3681 CAS; (b) R. Tangirala, S. Anthony, K. Agami, Y. Pommier and D. P. Curran, Synlett, 2005, 2843–2846 CAS.
- R. Hull and M. L. Swain, J. Chem. Soc., Perkin Trans. 1, 1976, 653–659 RSC.
- J. Azizian, A. A. Mohammadi and A. R. Karimi, Synth. Commun., 2003, 33, 415–420 CrossRef CAS.
- A. Andrzejewska, M. A. Pagano, F. Meggio, A. M. Brunati and Z. Kazimierczuk, Bioorg. Med. Chem., 2003, 11, 3997–4002 CrossRef; W. M. David, D. Kumar and S. M. Kerwin, Bioorg. Med. Chem. Lett., 2000, 10, 2509–2512 CrossRef CAS; J. W. Ellingboe, W. Spinelli, M. W. Winkley, T. T. Nguyen, R. A. Parsons, I. F. Moubarak, J. M. Kitzen, D. Von Engen and J. F. Bagli, J. Med. Chem., 1992, 35, 705–716 CrossRef CAS; C. R. Petrie, H. B. Cottam, P. A. McKernan, R. K. Robins and G. R. Revanker, J. Med. Chem., 1985, 28, 1010–1016 CrossRef; M. C. Huang, T. L. Avery, R. L. Blakley, J. A. Secrist and J. A. Montgomery, J. Med. Chem., 1984, 27, 800–802 CrossRef CAS; R. E. Holmes and R. K. Robins, J. Am. Chem. Soc., 1964, 86, 1242–1245 CrossRef CAS; A. G. Beaman, J. F. Gerster and R. K. Robins, J. Org. Chem., 1962, 27, 986–990 CrossRef CAS.
- M. K. Ali and M. McDermott, Tetrahedron Lett., 2002, 43, 6271–6273 CrossRef CAS; M. Joshaghani, A. R. Khosropour, H. Jafary and I. Mohammadpoor-Baltork, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 117–123 CrossRef CAS.
- W. R. Bowman, S. L. Krintel and M. B. Schilling, Org. Biomol. Chem., 2004, 585–592 RSC; W. R. Bowman, S. L. Krintel and M. B. Schilling, Synlett, 2004, 1215–1218 CrossRef CAS.
- J.-F. Liu, P. Ye, K. Sprague, K. Sargent, D. Yohannes, C. M. Baldino, C. J. Wilson and S.-C. Ng, Org. Lett., 2005, 7, 3363–3365 CrossRef CAS; E. S. Lee, J.-G. Park and Y. Jahng, Tetrahedron Lett., 2003, 44, 1883–1886 CrossRef CAS; A. Kamal, K. V. Ramana, H. B. Antaki and A. V. Ramana, Tetrahedron Lett., 2002, 43, 6861–6863 CrossRef CAS; K. G. Nazerenko, T. I. Shyrokaya and A. A. Tolemkev, Synth. Commun., 2003, 33, 303 CrossRef CAS.
- W. R. Bowman, M. R. J. Elsegood, A. J. Fletcher, P. J. Lovell and J. M. Pedersen, J. Org. Chem., 2005, 70, 10615–10618 CrossRef; W. R. Bowman, A. J. Fletcher, P. J. Lovell and J. M. Pedersen, Synlett, 2004, 1905–1908 CrossRef CAS.
- T. Harayama, A. Hori, G. Serban, Y. Morikami, T. Matsumoto, H. Abe and Y. Takeuchi, Tetrahedron, 2004, 60, 10645–10649 CrossRef CAS.
- T. Harayama, Y. Morikami, Y. Shigeta, H. Abe and Y. Takeuchi, Synlett, 2003, 847–848 CrossRef CAS.
- A. Cagir, S. H. Jones, R. Gao, B. M. Eisenhauer and S. M. Hecht, J. Am. Chem. Soc., 2003, 125, 13628–13629 CrossRef CAS.
- H. Twin and R. A. Batey, Org. Lett., 2004, 6, 4913–4916 CrossRef CAS; S. B. Mhaske and N. P. Argade, J. Org. Chem., 2004, 69, 4563–4566 CrossRef CAS; S. P. Chavan and R. Sivappa, Tetrahedron, 2004, 60, 9931–9935 CrossRef CAS; D. Osborne and P. Stevenson, Tetrahedron Lett., 2002, 43, 5469–5470 CrossRef CAS; M. Toyota, C. Komori and M. Ihara, Heterocycles, 2002, 56, 101–103 CrossRef CAS; S. Dallavalle and L. Merlini, Tetrahedron Lett., 2002, 43, 1835–1837 CrossRef CAS; P. Molina, A. Tárraga and A. González-Tejero, Synthesis, 2000, 1523–1525 CrossRef CAS; Z.-Z. Ma, Y. Hano, T. Nomura and Y.-J. Chen, Heterocycles, 1999, 51, 1593–1596 CrossRef CAS; H. Wang and A. Ganesan, Tetrahedron Lett., 1998, 39, 9097–9098 CrossRef CAS.
- W. R. Bowman, M. O. Cloonan, A. J. Fletcher and T. Stein, Org. Biomol. Chem., 2005, 3, 1460–1467 RSC.
- (a) M. A. Ciufolini and F. Roschangar, Tetrahedron, 1997, 53, 11049–11060 CrossRef CAS; (b) H. Abe, T. Harayama, A. Hori, T. Matsumoto, Y. Morikami, G. Serban and Y. Takeuchi, Tetrahedron, 2004, 60, 10645–10649 CrossRef CAS.
- Selected syntheses: (a) T. Harayama, A. Hori, G. Serban, Y. Morikami, T. Matsumoto, H. Abe and Y. Takeuchi, Tetrahedron, 2004, 60, 10645–10649 CrossRef CAS; (b) S. P. Chavan and R. Sivappa, Tetrahedron Lett., 2004, 45, 997–999 CrossRef CAS; P. K. Mohanta and K. Kim, Tetrahedron Lett., 2002, 43, 3993–3996 CrossRef CAS; S. H. Lee, S. I. Kim, J. G. Park, E. S. Lee and Y. Jahng, Heterocycles, 2001, 55, 1555–1559 Search PubMed; A. Ikuta, H. Urabe and H. Nashimura, J. Nat. Prod., 1998, 61, 1012–1014 CrossRef CAS; J. Bergman and S. Bergman, J. Org. Chem., 1985, 50, 1246–1255 CrossRef CAS; J. Kokosi, I. Hermecz, G. Szaz and Z. Meszaros, Tetrahedron Lett., 1981, 22, 4861–4862 CrossRef CAS; Y. Asahina, R. H. F. Manske and R. Robinson, J. Chem. Soc., 1927, 1708–1710 RSC.
- A. P. Dobbs, K. Jones and K. T. Veal, Tetrahedron Lett., 1995, 36, 4857–4860 CrossRef CAS.
- A. Fiumana and K. Jones, Tetrahedron Lett., 2000, 41, 4209–4211 CrossRef CAS.
- G. W. Gribble, H. L. Fraser and J. C. Badenock, Chem. Commun., 2001, 805–806 RSC.
- R. Liu, P. Zhang, T. Gan and J. M. Cook, J. Org. Chem., 1997, 62, 7447–7456 CrossRef CAS; U. Schmidt and S. Weinberger, Synthesis, 1996, 28–30 CrossRef CAS.
- A. R. Katritzky and K. Akutagava, Tetrahedron Lett., 1985, 5935–5938 CrossRef CAS.
- R. Ajaykumar, P. A. Bapu, D. S. Deevi, G. S. Kumar, K. B. S. Kumar, N. V. S. R. Mamidi, C. P. Narasimhulu, P. Prasanna, R. C. Puranik, C. S. Rao, C. V. L. Rao, S. Rajaopal, R. Rajagopalan, B. Renuka, K. V. A. Seshu, V. M. Sharma, D. Subramanyam and A. Venkateswarlu, Bioorg. Med. Chem. Lett., 2002, 12, 2303–2307 CrossRef CAS.
- M.-L. Bennasar, T. Roca and F. Ferrando, J. Org. Chem., 2006, 71, 1746–1749 CrossRef CAS.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2069 CrossRef CAS.
- S. Johne and M. Süsse, J. Prakt. Chem., 1984, 326, 1027–1033 CrossRef.
- J. A. Howard and J. C. Tait, Can. J. Chem., 1979, 57, 2761 CAS; S. B. Tong, I. P. Beletskaya, A. S. Sigeev, V. A. Kuzmin and A. S. Tatikolov, J. Chem. Soc., Perkin Trans. 2, 2000, 107 RSC.
- M. Baghbanzadeh, M. Dabiri, A. Mohammadi and P. Salehi, Synth. Commun., 2005, 279–287.
- S. Petersen and E. Tietze, Justus Liebigs Ann. Chem., 1959, 623, 166–176 CrossRef CAS; K. Nesmerak, H. Pelouchova, V. Vsetecka, I. Nemec and J. Gabriel, Folia Microbiol. (Prague), 1998, 43, 39–41 Search PubMed.
- L. Jung, I. Kranmacher and J.-F. Stambach, Heterocycles, 1990, 31, 2131–2138 Search PubMed.
- T. Kametani, C. Van Loc, T. Higa, M. Koizumi, M. Ihara and K. Fukumoto, J. Am. Chem. Soc., 1977, 2306–2308 CrossRef CAS.
- V. Gatta and R. L. Vittory, Gazz. Chim. Ital., 1969, 59.
- J. S. Yadav and B. V. Reddy, Tetrahedron Lett., 2002, 43, 1905–1908 CrossRef CAS.
- SMART and SAINT software for CCD diffractometers, Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
- R. W. W. Hooft, COLLECT data collection software, Nonius B.V., 1998 Search PubMed.
- G. M. Sheldrick, SHELXTL user manual, version 6.10, Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation and analytical details of 3H-quinazolin-4-one radical precursors and products. See DOI: 10.1039/b614075k |
| ‡ CCDC reference numbers 622451 and 622452. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b614075k |
| This journal is © The Royal Society of Chemistry 2007 |