The Heck–Mizoroki cross-coupling reaction: a mechanistic perspective
Jonathan P.
Knowles
and
Andrew
Whiting
*
Department of Chemistry, Durham University, Science Laboratories, South Road, Durham, UK DH1 3LE. E-mail: andy.whiting@durham.ac.uk
First published on 9th November 2006
Abstract
The Heck–Mizoroki cross-coupling reaction is an important part of the synthetic chemist's toolbox, and it has been applied to a huge variety of different substrates. In contrast, the mechanism of the process is much less studied, and consequently less understood. There have been numerous studies reported over recent years, both experimental and theoretical, aimed at uncovering the inner working of this palladium-mediated coupling process. This perspective aims to review and compare these works and to provide an up-to-date view of this reaction.
 Jonathan P. Knowles Jonathan P. Knowles | Jon carried out his undergraduate Master's degree studies at Durham University, graduating in 2005. He is currently studying for his PhD at Durham University in the Whiting group in the area of natural product synthesis. His main interests are in the field of highly stereoselective polyene synthesis using palladium cross-coupling reactions involving vinylboronate esters, and iodo-deboronation methodology. |
 Andrew Whiting Andrew Whiting | Andy carried out PhD studies with Professor R. J. Stoodley at Newcastle University, working on β-lactam chemistry, before moving on to postdoctoral research at Boston College, with Professor T. Ross Kelly working on natural product synthesis and the development of chiral Diels–Alder Lewis-acid catalysts. After a short period in industry with Ciba-Geigy Central Research, he moved to his first academic position as Lecturer in Chemistry at UMIST, and in 2001, moved to a Readership at Durham University. |
1 Introduction
Palladium catalysed cross-coupling reactions have proved extremely powerful synthetic tools and their scope continues to increase year on year. Alongside the well-established Heck–Mizoroki,1,2 Kumada,3,4 Negishi,5,6 Sonogashira–Hagihara,7,8 Stille9,10 and Suzuki–Miyaura11,12 reactions, palladium catalysis of aryl and vinyl hydroamination,13 sulfination,14 dechlorination,15 dialkoxylation,16 intramolecular arylation17 and C–H bond alkenylation18 have more recently been discovered.Although the mechanisms of many of these reactions are thought to be relatively well understood, their nature often prevents easy observation of the highly reactive intermediates often associated with homogeneous catalysis . Whilst accurate knowledge of the mechanism in operation is not necessarily a prerequisite for the application of this reaction in organic synthesis, knowledge of the mechanism may assist the optimisation of reaction conditions, improve regio-, stereo- and chemo-selectivity, and produce a concomitant reduction of side-reactions.
The majority of the palladium catalysed cross-couplings are thought to share a similar mechanism.19 For the Suzuki, Sonogashira, Stille, Negishi, Hayama20 and Kumada reactions, the proposed mechanism involves initial oxidative addition of the halide to a palladium(0) catalytic species to form a palladium(II) species, transmetallation of the organometallic reagent by palladium and reductive elimination of the product from this species to regenerate the active palladium(0) catalyst.
The Heck–Mizoroki (HM) reaction has evolved significantly from its original guise as the arylation of olefins with aryl mercury compounds.21–23 The independent discovery by both Mizoroki1 and Heck2 that aryl iodides could be used as a substitute for aryl mercury compounds, and that this modification maintained the oxidation state of palladium, allowing for the use of catalytic palladium in the absence of reoxidants was particularly significant. The reaction has been further developed over the years to allow the coupling of less reactive halides, such as bromides,24 chlorides,25 pseudo-halides such as triflates,26 tosylates,27 mesylates,28 and aryl diazonium salts .29 In addition, the reaction is not limited to arylation; it can be used to add vinyl halides to olefins30 and can involve the use of chiral ligands on palladium for the generation of chiral centres with a high degree of enantiocontrol for certain substrates.31–33 Recent developments involve the replacement of the halide with an organoboron reagent in the presence of stoichiometric reoxidant,34 although this is closely related to the original use of aryl mercury compounds.
Consequently, in contrast to the previously mentioned cross-couplings, the HM reaction, having no organometallic reagent, does not include a transmetallation step in its mechanism. This review will examine the current mechanistic view of the HM reaction and factors which impact upon it.
2 Basic mechanism
There has been a broadly accepted understanding of the mechanism operating in the HM reaction for many years,35 generally thought to involve an initial oxidative addition of the halide to a palladium(0) catalyst. Despite various claims for a possible palladium(II/IV) cycle in the mechanism,36,37 the evidence for this is poor, since it has been shown that in the majority of cases, the palladacycles involved act as reservoirs of palladium,38–44 some of which is reduced to palladium(0). Further evidence against this mechanism comes from gas phase computational studies which indicate that the rate determining step in a palladium(II/IV) cycle involving iodobenzene would be the oxidative addition of iodobenzene to palladium.45 Since the actual rate determining step in the HM reaction of aryl iodides is not oxidative addition 46 (vide infra) this indicates that a palladium(II/IV) cycle is not in operation. Hence, the mechanism of the HM process can be represented by Scheme 1, involving a palladium(0) species 1 undergoing oxidative addition to generate a palladium(II) species 2, which reacts with the olefin component 3, possibly following initial η2-coordination to the palladium atom. This results in a carbometallation reaction to generate palladium(II) alkyl complex 4. Elimination of palladium hydride from complex 4 furnishes the product 5 and base assisted elimination of HX from palladium(II) complex 6 regenerates the active palladium(0) catalyst 1.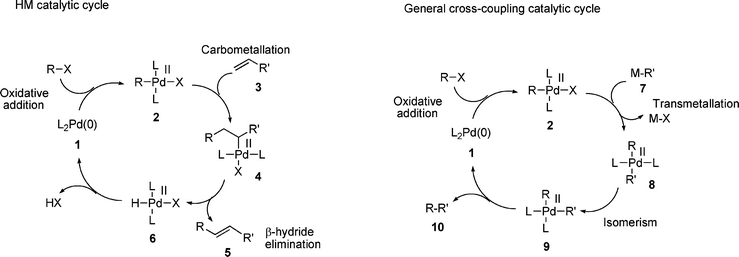 | ||
| Scheme 1 Comparison of HM mechanism with that of a generalised palladium catalysed cross-coupling reaction. | ||
3 Reaction conditions
A number of palladium sources are used in the HM reaction, which are either sources of palladium(0) such as Pd(PPh3)4, Pd(dba)2 and Pd2(dba)3, or sources of palladium(II) such as Pd(OAc)2 and PdCl2(MeCN)2. A wide range of solvents can be also used in the HM reaction and elevated temperatures compared with other cross-coupling reactions are frequently required (compared with Sonogashira reactions, for example, which often proceed rapidly at room temperature8). High boiling point solvents such as DMF, DMA and toluene are often used. Additionally, inorganic bases such as NaOAc are often used and polar solvents can be preferred to achieve homogeneity.47,48 The use of non-polar solvents also becomes convenient when organic bases, such as trialkylamines, are used. Recently, ionic liquids have also have been shown to be useful solvents in HM reactions.49The use of ligand free HM reactions is possible, and indeed, the early examples of HM reactions were performed under these conditions.2 However, it is more generally found that the addition of palladium-stabilising ligands is highly beneficial in terms of providing increased reactivity, stability and selectivity of the catalyst.24 The most widely used ligands are phosphines, such as triphenylphosphine and tri(o-tolyl)phosphine,50 however, nitrogen,51 arsine,52 sulfur53 and carbene54 derived ligands can be used. A wide variety of bidentate ligands have also been developed for palladium, these including P,P, P,N,55–59 N,N,60–68 N,S69 and P,S70,71 ligands as well as tridentate ligands which can switch to bidentate coordination to allow a substrate to bind.72 Bidentate diphosphine ligands that can switch between phosphorus and nitrogen coordination have also been developed.73,74 The use of chiral, chelating ligands can give rise to high levels of asymmetric induction for certain substrates (vide infra).
In addition to the use of palladium-stabilising ligands, HM reactions have often been subjected to empirical treatment with various additives, generally claimed to promote selectivity or reactivity. These have included saturation of the reaction with chloride ion30,75 (Jeffery protocol), addition of phase transfer catalysts,76 and the addition of either silver77 and thallium78 salts. Whilst this approach is often successful in terms of achieving the desired product, the exact mode of action of these additives is not always well understood.
4 Catalyst generation
As in most cases, the catalyst used in the HM reaction is generated in situ, which effectively means that the first step of the reaction has to be the reduction of the palladium(II) precursor to provide the active palladium(0) catalytic species. Although there have been various claims made for palladium(II/IV) catalytic cycles,37,38 and indeed, some palladium(IV) species have been isolated,79 all current evidence seems to point to a palladium(0/II) cycle, and this necessarily requires the reduction to the active palladium(0) catalyst.There is a range of methods for the in situ reduction of palladium(II) salts to palladium(0) species, including treatment with sodium borohydride,80 hydrazine,81 phenyl or methyl lithium (to give biphenyl or ethane and lithium chloride respectively),82n-butyllithium (to give butane, butene, octane and lithium chloride)82 and electrochemical methods.83 However, the most common procedure is the use of triphenylphosphine as the reducing agent and the mechanism of this reduction has been investigated independently by the groups of Jutand84 and Hayashi,85 both of which propose the mechanism shown in Scheme 2. This involves initial ligation of the palladium(II) complex, for example, palladium(II) acetate in this case, to give complex 7, which can eliminate acetoxytriphenylphosphonium acetate to generate a monotriphenylphosphinylpalladium(0) complex, which can coordinate further phosphines.
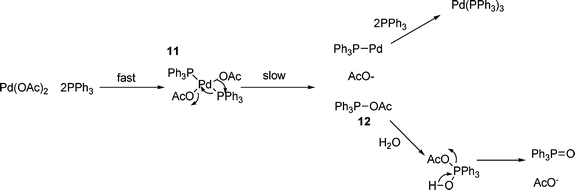 | ||
| Scheme 2 | ||
This mechanism explains the isotopic labelling observed by Hayashi85 and the independence of the rate of reaction on phosphine concentration observed by Jutand.84 It has been demonstrated that the presence of one equivalent of water is necessary for the reduction step to proceed (causing hydrolysis of acetoxytriphenylphosphonium acetate12, see Scheme 2),85 however, given the low catalyst loadings used, this is rarely a problem even under rigorously anhydrous conditions! Further evidence for this mechanism comes from the observation of similar behaviour of analogues of Pd(OAc)2, such as Pd(TFA)286 and related, sulfur bridged species.87
Given that this method of reduction only works for Pd(OAc)2 and related species, and not for palladium(II) halide salts,84 it appears that the thermodynamic driving force for this reaction is the formation of the strong phosphorus–oxygen bond in triphenylphosphine oxide. This reduction is faster for electron poor phosphines, as demonstrated by a positive Hammett parameter for para-substituted tri-aryl phosphines.88 This is also in agreement with the proposed mechanism of reduction. In certain cases, such as when using tri(o-tolyl)phosphine, reduction of palladium by this mechanism (Scheme 2) does not operate. Indeed, this led to the initial proposal of palladium(II/IV) cycles36,37 (vide supra) for the resulting palladacycle catalysts. However, other methods for the reduction of such species, including reaction with olefins and via a palladium amide species can account for a reductive catalyst generation process.89 Indeed, for the majority of palladacycles, and other supported palladium species, it is usually found that the active species involved in catalysis are palladium(0) species,38,39 often resulting from degradation of the ligand.40–44 The formation of active catalytic species from palladacycle ‘reservoirs’ can give rise to complicated kinetics.90
In cases where none of these potential reducing agents are present, it is possible that organic amine bases are also able to perform the reduction of palladium(II) to palladium(0),91 although this has been found to be slow compared to reduction by triphenylphosphine.88 In addition, the olefin may reduce the palladium(II) species, either by a Heck-type reaction for palladacycles,89 or by a Wacker-type process when the olefin is an allylic alcohol.92 In addition, another process that may be involved in catalyst generation is the dissociation of dimeric catalysts. This is sometimes responsible for the observed induction period in certain cases.93,94 EXAFS studies have shown that the active catalytic species in these systems are monomeric and that the equilibrium favours there species at high dilution.95 It could be argued that this is not a true catalyst generation step, but part of an equilibrium governing the oxidative addition step.
5 Oxidative addition
Despite being perhaps the easiest step to investigate due to it being the first step in the catalytic cycle (and having received the most attention), the mechanism of the oxidative addition step of aryl and vinyl halides and tosylates to palladium(0) species is still unclear. Early investigations of the mechanism of oxidative addition involved the reaction of alkyl and aryl iodides reacting with iridium complexes,96–98 aryl iodides, bromides and chlorides with Ni(PEt3)4,99 and the oxidative addition of benzyl chlorides to Pd(PPh3)4.100 Although these studies, particularly those involving nickel, shed some light on the oxidative addition process involved, it became clear that studies on palladium systems were required.The first such study101 was the oxidative addition of aryl iodides to Pd(PPh3)4, which proved that electron withdrawing groups on the aryl iodide accelerated the reaction with a clear positive Hammett correlation (ρ = +2.0). The reaction was found to be first order in both palladium and aryl iodide, but negative first order in added triphenylphosphine. Since at this time it was known that in solution, Pd(PPh3)4 dissociates to PPh3 and Pd(PPh3)3102,103 it was proposed that the active species in the oxidative addition was probably Pd(PPh3)2, formed from an unfavourable equilibrium dissociation of a further PPh3 from Pd(PPh3)3, which has actually been isolated as a solid. The build-up of negative charge on the aryl ring in the transition state of the oxidative addition process (demonstrated by the positive Hammett parameter) has been explained by a three centre transition state 13 (Scheme 3), which collapses to the oxidative addition product 14.101
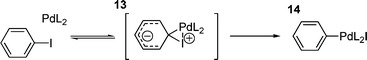 | ||
| Scheme 3 | ||
This study also explains the relative lack of reactivity of aryl bromides and chlorides, suggesting that the electropositive iodine is a better ligand for palladium than either bromine or chlorine.101 It has also been suggested that a transition state such as 13 occurs following initial η2-coordination of the aryl ring to palladium.104 This study also observed the disappearance of the palladium(0) starting material, but did not isolate the resulting product or provide information on the structure of oxidative addition species. However, based on the proposed transition state 13, the cis-isomer would be expected for product 14.
It has been found that co-ordinately unsaturated palladium (i.e. one triphenylphosphine per palladium) is unstable in solution and reacts readily with aryl iodides.84 This provides further support for the suggestion that the active species is co-ordinately unsaturated and that it is in equilibrium with the inactive, saturated species. A subsequent study of oxidative addition of aryl iodides to Pd(PPh3)4 in less polar solvents105 (toluene) gave a similar Hammett parameter (ρ = +2.3) and kinetics, the lack of effect of the change in solvent polarity indicating little charge development in the transition state . Because of this it was suggested that transition state 13 was unlikely due to the development of charge105 but a similar three centre transition state must occur as the alternative SNAr clearly requires a greater development of charge.
In contrast, the oxidative addition of aryl chlorides to palladium(0) species has been found to proceed through a highly charged transition state ;106 a Hammett parameter of +5.2 being found for the addition of aryl chlorides to Pd(dippp)2. Again, oxidative addition is predicted to proceed through an unsaturated palladium(0) species [i.e. Pd(dippp)] and involves a three-centre late-transition state charged species, which is essentially analogous to structure 13 (Scheme 3), but involving chloride ion.
Although oxidative addition is generally accepted to be an irreversible process, it has been shown that for sterically crowded systems, the process is reversible, and reductive elimination can be induced by the addition of bulky, electron-rich phosphines.107 Thus, reaction of tri-tert-butylphosphine with a palladium(II) oxidative addition product of mono-ligated tri-ortho-tolylphosphinyl complex produces reversal of the oxidative addition , regenerating the aryl halide and deriving a bis(tri-tert-butylphosphinyl)palladium(0) complex.107
As mentioned previously, the catalysts employed in the HM reaction are typically generated in situ from palladium(II) precursors. The investigation of the oxidative addition process involving Pd(PPh3)4 has tended to ignore the possible influences of any associated anions. When using Pd(OAc)2 as the catalyst precursor, two equivalents of acetic acid are generated during the palladium reduction process, however, since HM reactions are performed in the presence of stoichiometric bases, this results in the formation of two equivalents of acetate anion, and, in addition, two equivalents of the tributylammonium cation are also generated (for example). It had been found using 31P NMR that there are signals for palladium–phosphine complexes in solution, that their chemical shift depends on the anions present,82 and that their reactivity is dependent upon the palladium(II) precursor used. Additional investigations have shown that only one equivalent of acetic acid per palladium is actually generated,88 and investigation into the effect of added chloride ion on the rate and mechanism of oxidative addition has shown that the reaction is far more complex than previously thought.108 It appears that in the presence of chloride ions, a number of palladium(0) species are present in solution, all of which disappear to give the oxidative addition product upon addition of iodobenzene.108 This shows that either all species are active in the oxidative addition , or they are in rapid equilibrium with each other (or both). Although various anionic palladium species with chloride ligands have been proposed, there is little chemical evidence for the existence of any of them, and indeed, they have been proposed on the basis of ‘general chemical expectations’ rather than hard evidence.108 That said, it has been demonstrated that the transition state that occurs in the oxidative addition step involving iodobenzene and palladium(0) in the presence of chloride ions is different to the one that occurs in their absence. This has been clearly demonstrated by comparison of the Hammett parameters for two relevant processes, which produced Hammett parameters of ρ = +2.7 and ρ = 2.0 respectively. This indicates that there is a greater degree of negative charge in the transition state when chloride is added compared to chloride free, and is consistent with a chloride ligated anionic palladium species being involved. The presence of chloride was also found to accelerate the rate of oxidative addition ; two equivalents per palladium giving the greatest level of acceleration compared to the absence of chloride.83 A further addition of chloride ion was also shown to retard the reaction.108 The acceleration of the oxidative addition step by increasing the negative charge on palladium (to a point) is in some ways unsurprising, especially in the light of the fact that more electron donating phosphines also increase the rate of oxidative addition 88 or the overall reaction73 in HM reactions. It has also been found that the nature of the palladium(0) species changes upon the addition of acetate anions to the solution.109 This was again rationalised by the suggestion of anionic, acetate-ligated palladium species, however, in this case, a substantial increase in rate of oxidative addition was not observed.109 It was also found that the final product of the oxidative addition reaction, in the presence of acetate, was trans-ArPdL2OAc and not the expected trans-ArPdL2I, and that the acetate was reactive towards olefins whilst the iodide was not.109 These results led to the proposal of five-coordinate, anionic palladium intermediates in the oxidative addition .109,110 Hence, the HM process may be formulated as proceeding through the mechanism outlined in Scheme 4. An anionic complex of type 15 can react by oxidative addition in the presence of, for example, chloride, resulting in the formation of the proposed trans-ArPdL2I five-coordinate species 16. This structure is consistent with the observation of two equivalent phosphines by 31P NMR, and also accounts for the reported release of a second equivalent of chloride upon formation of the product.110 A similar five-coordinate intermediate has been proposed involving acetate,88,109 and therefore, in both chloride and acetate cases, a revised mechanism of oxidative addition can be proposed, in which a three-coordinate palladium anion adds to the aryl iodide to give a five-coordinate palladium anion (as outlined in Scheme 4).
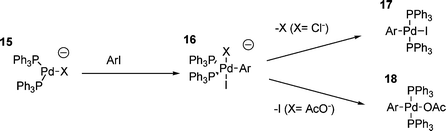 | ||
| Scheme 4 | ||
In contrast to the preceding discussion, the proposed five-coordinate intermediates such as 16 might appear to be unlikely, since the reluctance of palladium to form five-coordinate complexes is documented,111,112 five-coordinate species only being isolated when polydentate ligands are used. This reluctance of palladium to be five-coordinate can also be inferred from the lack of reactivity of chelated palladium(II) aryl halide complexes in the carbometallation step of the HM reaction (vide infra), and dissociation is necessary for the olefin to bind. Additionally, for such observed intermediates, such as 16 where X is chloride, this addition compound appears to be relatively stable, having a lifetime of over one hour (observed using electrochemical methods). This is clearly not consistent with a five coordinate palladium anion, because if such a species were to form, it would be expected to rapidly dissociate to restore the preferred four-coordinate geometry.
Curiously, it has also been claimed that an acetate ligated three-coordinate anion is less reactive than Pd(PPh3)2 and that this reduction in the rate of the oxidative addition step is responsible for the overall acceleration of the reaction by bringing the rates of the fastest and slowest steps closer together.113 However, the addition of acetate has been shown to have little or no effect on the rate of oxidative addition ,109 therefore, if its presence does accelerate the reaction, this is clearly not the reason.
It is interesting that recent DFT studies114–116 including solvation examining the anionic HM reaction mechanism have reached similar conclusions on the overall mechanism. A previous DFT study including solvation had suggested that three-coordinate palladium anions were stable species in solution.117 These calculations suggested that the addition of acetate to palladium to form a three-coordinate anion should increase the rate of oxidative addition .114 However, it is known that oxidative addition of aryl iodides is not rate limiting,46 indeed, such reactions occur rapidly at room temperature101 and it has been shown that it is not rate limiting even for some bromides.118 More importantly, both studies showed that oxidative addition of the three-coordinate anion to an aryl halide did not give the expected five-coordinate anionic palladium species, rather a four-coordinate anionic species was produced involving the halide of the aryl halide.114,115 This species undergoes an oxidative addition reaction with concurrent loss of halide to form a neutral four-coordinate product,114,115 with the dissociated halide electrostatically bound to a phosphine ligand.116 This mechanism rationalises some of the experimental observations such as an intermediate that undergoes slow release of chloride without resorting to less plausible five-coordinate palladium species.
Another point of interest is the geometry of the product obtained in the oxidative addition process. From the proposed three centre transition state (i.e.13, Scheme 3), the expected geometry of the resulting palladium(II) complex should be cis with respect to the Ar and X groups on palladium. However, the product that is invariably isolated when using monodentate phosphines is the trans-isomer; the cis product only having been found once.119 A clue as to the reason behind this comes from the observation that the isolated products of oxidative addition from stoichiometric reactions often react more slowly in subsequent steps than the apparently identical species under catalytic conditions.120 It was noted that if the isomerisation of a cis-oxidative addition product, i.e.20 (Scheme 5), was slower than transmetallation (traditional cross-coupling reactions such as Suzuki and Negishi, for example), then the reaction could proceed directly via a reductive elimination from 21.38 However, if isomerism is fast, transmetallation yields the trans-palladium species 23 and a second isomerism is required before reductive elimination can occur (Scheme 5). For stoichiometric reactions in which the product of oxidative addition is isolated, isomerism to the trans-product 22 is ensured, as this is the thermodynamically favoured product.38
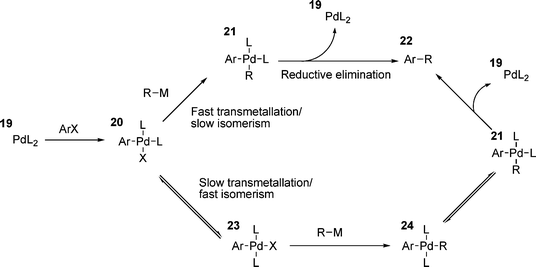 | ||
| Scheme 5 | ||
The mechanism of the isomerism of the cis-products of oxidative addition has been investigated.121 The mechanistic details are complex, however, after isolating an oxidative addition product, it was possible to study the rearrangement process. Four separate pathways were proposed to be in operation; two dependant on triphenylphosphine concentration and involving associative replacement of phosphines with iodide, either THF mediated, or iodide bridged species; and two independent, with both dissociative and associative pathways proposed, depending on the solvent and involving Berry pseudorotation processes.121
The oxidative addition of aryl triflates has also been investigated and appears to occur by a similar mechanism to that of the halides. Again, the oxidative addition is accelerated by the presence of electron withdrawing groups on the aryl group ; a Hammett parameter of ρ = +2.55 being found in the reaction of aryl triflates with Pd(PPh3)4.26 As had been previously established,70,122–130 the product of oxidative addition is ionic and the palladium triflate bond is fully dissociated in moderately polar solvents.26 In non-polar solvents, however, although no covalent bonding is present (IR spectroscopy), the ions exist in the form of tight ion pairs.26 Again, addition of chloride ion was found to accelerate the reaction but in this particular case, only when large excesses (150 fold) were added,26 while other systems show severely inhibited activity.131 It was also observed that the addition of chloride could enable regeneration of a neutral palladium halide species.82 This has subsequently been verified and applied synthetically.132
The magnitude of the Hammett parameter found for the oxidative addition of aryl electrophiles to palladium(0) species can be related to the reactivity of the aryl species towards oxidative addition . The experimentally observed order of reactivity is: I > OTf > Br > Cl, and the Hammett parameters found for iodides, triflates and chlorides are 2.0,101 2.5526 and 5.6106 respectively. Although the Hammett parameter for the oxidative addition of aryl bromides to palladium(0) has not been determined, the Hammett parameters for the oxidative additions of aryl iodides, bromides and chlorides to Ni(PEt3)4 are 2.0, 4.4 and 5.4 respectively.99 It therefore appears that the smaller the Hammett parameter found for the oxidative addition of a group of aryl electrophiles, the more facile the process is.
So far, only monodentate phosphines have been discussed. Whilst the majority of phosphines used in HM reactions are monodentate, bidentate (or chelating) phosphines are important because of their ability to activate unreactive halides, particularly chlorides,25 and for their ability, when chiral, to impart enantioselectivity in certain HM reactions.133 The use of bidentate phosphines has several implications for the oxidative addition : i.e. that the product of oxidative addition is necessarily cis; the chelating ligand not permitting the formation of trans-complexes;38 and the bite angle of the phosphine having a significant impact on the reactivity of the palladium(0) species.134,135 The first issue surrounding the use of bidentate phosphines arises when the catalyst is being formed in situ by reduction of a palladium(II) precursor. Since the oxidation of the bidentate phosphine effectively yields a monodentate phosphine, this gives rise to a scenario that is somewhat more complex than that found for the monodentate systems. Since it is clearly necessary to use at least two equivalents of the bidentate phosphine, species such as 25 tend to result136 with predictable mechanistic complications. This situation can be avoided in several ways: firstly the use of three equivalents of the bidentate ligand forces the formation of 2685,136 by means of the chelate effect; secondly, palladium(II) precursors can be avoided by the use of palladium(0) sources such as Pd2(dba)3, however, dba can coordinate palladium and impede oxidative addition ;137,138 and thirdly, addition of acetate promotes the formation of three coordinate species such as 27.136
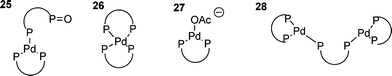
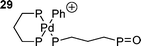
When three equivalents of bidentate phosphine are used the rate determining step is dissociative, involving a dimeric palladium species such as 28 to give a reactive di-coordinate palladium species.139 Oxidative addition of the halide to this gives the expected cis-product; one solvation included theoretical study finding that this occurred by initial η2 complexation of the iodo-arene to palladium.139 When the catalyst is generated from a palladium(0) dba species, dba dissociation is generally required prior to oxidative addition although the dba coordinated species show some activity in oxidative addtion.137 Three coordinate anionic palladium species with bidentate phosphines have been ‘characterised’ in solution by DFT calculations117 and have been shown to be active catalysts, giving either the cis-aryl acetate product or cationic species such as 29.
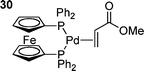
Another interesting feature of chelated palladium catalysts is that in some cases they allow the isolation of palladium-olefin complexes.140 The complex (dppf)Pd(methylacrylate) 30 has been isolated and characterised, and the complex was stable to dissociation of methylacrylate although this process could be promoted by the addition of Lewis acids.140 Addition of PhI or PhOTf led to oxidative addition with displacement of the alkene.140
As alluded to in the last section, the generation of palladium catalysts from palladium(0) precursors can have effects on the oxidative addition step. Such palladium(0) species often incorporate dba and this has been found to impact the reactivity of the active catalysts. Despite the common assumption that dba is a weak ligand for palladium, the presence of dba has been found to inhibit the oxidative addition of PhI to Pd(PPh3)4.141 Comparison of the rate constants for oxidative addition of PhI to preformed Pd(PPh3)4 and Pd(PPh3)n generated from Pd(dba)2 showed that the presence of dba decreases the rate of reaction by a factor of ten,141 and the oxidative addition to chelated palladium catalysts is similarly inhibited.137 Studies involving substituted dba analogues have shown that the dissociation of dba can govern the rate of oxidative addition to aryl iodides142 and in some cases the presence of dba can completely inhibit reaction by preventing oxidative addition taking place.138
Oxidative addition to amine ligated palladium–phosphine complexes has also been proposed,143 however, subsequent studies have shown that amine decomplexation is required before oxidative addition can take place.144,145
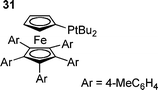
The coordination number of the active palladium species has also received recent attention, with two theoretical studies including solvation suggesting palladium–monophosphine species are involved.104,146 It was suggested that whilst oxidative addition of PhI to Pd(PPh3)2 occurs with an energy barrier, oxidative addition to PdPPh3 required no activation. Although the dissociation of PPh3 from Pd(PPh3)2 is endothermic, it was suggested that in solution, a favourable entropic contribution gives a sufficient concentration of the active catalytic species.83 Further evidence for this mechanism comes from a recent investigation of the oxidative addition of aryl iodides, bromides and chlorides to a palladium diphosphine complex with a bulky monodentate phosphine (Q-phos derivitive 31).147 The investigation revealed three distinct mechanisms in operation depending on the identity of the halide.147 Aryl iodides reacted by oxidative addition with concurrent dissociation of a ligand, whereas aryl bromides were found to follow a mechanism involving rate determining ligand dissociation followed by rapid oxidative addition , and aryl chlorides were found to react by reversible dissociation of a phosphine followed by rate limiting oxidative addition .147 Although these results are in agreement with those of DFT studies, it should be noted that the great steric bulk of the ligands involved is likely to promote reaction by ligand dissociation, although another experimental study has implied the participation of monoligated palladium triphenylphosphine species.148
Oxidative addition of alkyne ligated palladium complexes in solution has also been investigated theoretically.149 It has been suggested that acetylene is an excellent ligand for palladium and that oxidative addition of aryl iodides to such species is a favourable process. The addition occurs with initial iodide coordination, followed by concerted iodide dissociation and metal–carbon bond formation.149
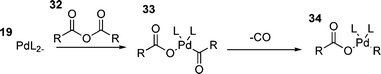
Finally, the oxidative additions of a number of other species have been investigated, including benzoic anhydride150 and acetic anhydride.151 Although rare, these species can be used as electrophiles in the HM reaction if a decarbonylation step is added between the oxidative addition and carbometallation steps.150 The mechanism of oxidative addition of these species involves insertion of palladium into one of the carbon–oxygen single bonds of 32 to generate species 33 from which loss of carbon monoxide generates a palladium alkyl or aryl acetate or benzoate 34 (Scheme 6).146,150 It has been suggested that the presence of chloride is necessary for this reaction as palladium benzoates are unreactive.91
 | ||
| Scheme 6 | ||
6 Carbometallation, β-hydride elimination and HX elimination
Since these steps follow oxidative addition they are significantly more difficult to investigate and are best discussed together rather than as separate steps. Although the products of oxidative addition are sometimes isolable, it has often been found that they show different reactivity to those generated under catalytic conditions.120 This has been ascribed to cis–trans-isomerism and makes experimental studies of carbometallation both difficult and potentially meaningless. Additionally, palladium alkyl compounds with β-hydrogens are generally sufficiently unstable to prevent their isolation,48,151 which has precluded the study of stoichiometric β-hydride eliminations.Before investigations of carbometallation, studies on the mechanism of insertion of various species into a transition metal carbon σ-bond served as a model for the carbometallation step. Investigation of the insertion of para-substituted styrenes into a rhodium hydride bond152 showed that the process produced a negative Hammett parameter, ρ = –0.9. This shows that electron donating substituents on the olefin accelerate the migratory insertion . The same study also demonstrated that the migration is accelerated by electron donating phosphine ligands.152 Interestingly, a second study on the kinetics and mechanism of the insertion of a range of olefins into a niobium hydride bond showed an identical Hammett parameter (ρ = –0.90) suggesting that this effect may be more general as the two hydride bonds involved are very different electronically.153 The postulated mechanism to explain these results involves cyclic transition state 35 in which there is build up of positive charge on the olefin being stabilised by electron donating groups.153
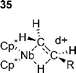
The bond breaking–forming process was suggested to be concerted,153 however, the electronic effects in these reactions appear to be complex. In another system it was found that electron withdrawing groups accelerated the migration;154 this being ascribed to stabilisation of the ground state with the effect of increasing the olefin binding constant. The migratory insertion of olefins into palladium alkyl bonds has also been investigated and it was found that electron poor olefins underwent a faster insertion process although electron rich olefins bound to the metal more strongly.155 It was found that syn insertion occurred for the insertion of olefins into palladium(II) acyl bonds; these reactions being faster for cationic palladium species than for neutral. The mechanism of insertion was suggested to involve the dissociation of either solvent (cationic pathway) or phosphine (neutral pathway), to allow the coordination of the olefin.156 Also, the investigation of the intramolecular insertion of alkynes and olefins into palladium acyl bonds showed a dissociative equilibrium existed between a phosphine ligated species and the less coordinately saturated active species. The existence of five-coordinate intermediates in the reaction was disproved.157
The nature of the product obtained from the oxidative addition step has a great influence on the rest of the catalytic cycle (see Scheme 7). Cationic palladium species formed from the oxidative addition of triflates130 and diazonium salts 158 behave differently to the neutral species generated from halides.129 Additionally, the nature of the phosphine (monodentate or bidentate) has a marked effect on subsequent steps, some bidentate phosphines chelating so strongly that they render the oxidative addition product unreactive.25
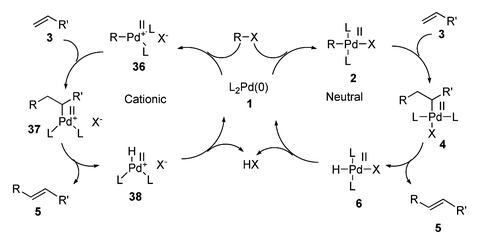 | ||
| Scheme 7 Comparison of cationic and neutral HM cycles. | ||
For a long time there has been considerable evidence that oxidative addition is not rate limiting in HM reactions. The observation of a reversal of the expected reactivity for aryl iodides92,159 is noteworthy, and the unexpectedly low reactivity of aryl chlorides in HM reactions, when compared to their rates of oxidative addition , suggests that even for these species, other steps may limit the reaction rate.160
It has been proposed that for the reaction of aryl iodides with acrylates using triphenylphosphine as a ligand, the rate determining step depends on the phosphine–palladium ratio, olefin coordination being rate limiting when the ratio is 2 : 1 or more whilst migratory insertion being rate limiting for ratios of 1 : 1.159 Strong effects of the phosphine–palladium ratio have also been observed in other systems,161,162 although mechanistic reasoning was not given.
6.1 Neutral monodentate intermediates
For monodentate ligands and ligand free systems, various suggestions for the rate determining step have been put forward. These include β-hydride elimination,163 coordination/insertion of the olefin164 and halide dissociation.165An interesting observation came in an investigation of a ligand free HM reaction. It was found that for certain combinations, addition of one olefin to a HM reaction accelerated the arylation of another olefin.166 This was rationalised by a modification of the β-hydride elimination step; the elimination of palladium hydride 41 being replaced by the transfer of the palladium hydride species 41 to another olefin 42 furnishing the product and unsubstituted palladium(II) alkyl species 44 (Scheme 8). This palladium hydride transfer was suggested to be the rate determining step,166 although a traditional β-hydride elimination step, followed by an elimination of HX, would still be required to generate an active catalyst. The study also proposed that both revised and traditional mechanisms could occur at the same time.166
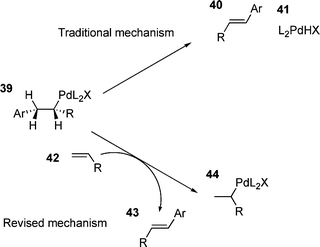 | ||
| Scheme 8 | ||
The olefin insertion has been shown to be irreversible in the ligand free palladium system in a study which also found β-hydride elimination to be rate determining.163 High pressure experiments have shown that olefin coordination or insertion is rate determining when triphenylphosphine is the ligand and also prove the presence of a polar transition state .164 Gas phase DFT studies have suggested that for carbene ligands, halide dissociation is required before the olefin can bind to the resulting cationic intermediate.165
Another interesting suggestion was that the aryl iodide species 45, resulting from the oxidative addition of PhI to Pd(PPh3)4, was unreactive to olefins, whilst the corresponding aryl acetate species 46 (arising from the proposed oxidative addition to palladium anions, vide supra) was the active species.109 However, the problem with this suggestion is that HM reactions of aryl iodides can be performed in the absence of acetate,1,167 showing that aryl acetate 46 is not the only possible reactive intermediate. This appears to be another case of stoichiometric reactions being unrepresentative of those taking place under catalytic conditions,120 and presumably can be explained by several factors, including isomerisation.121 Hence, it thus appears likely that the increase in reactivity observed on addition of, for example, acetate anions to aryl iodide 45 is due to an increased rate of isomerism to the reactive cis-form,121 or that the cis–trans equilibrium for aryl acetate 46 lies more on the side of cis-form than the equivalent equilibrium for aryl iodide 45.
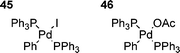
6.2 Cationic intermediates
Although one study has suggested that the reaction of cationic palladium aryl species with olefins is slow,109 this does not appear to be a general phenomenon since it has been shown that insertion reactions of olefins into palladium carbon σ-bonds is faster for cationic species.156 It has also been found that the addition of chloride to cationic systems, to form the neutral chloride species, can completely inhibit the reaction with olefins.131 Cationic palladium systems have also been used to great success in the asymmetric HM reaction.133 In these systems, reactions are accelerated by electron donating groups on the aryl moiety, which is the reverse order of reactivity observed for oxidative addition and this behaviour can be ascribed to rate limiting olefin coordination or carbometallation.126 The high enantiomeric excesses observed in the asymmetric HM reactions under cationic conditions are due to the olefin being able to bind to palladium without partial dissociation of the bidenatate phosphine.124,127 This observation has led to the proposal of some sort of dissociation from a four-coordinate palladium(II) complex, which is necessary for the olefin to coordinate.128 In cationic systems, the availability of a free site for an olefin to bind is due to the dissociation of an anion, whilst in neutral systems phosphine dissociation is necessary.126 Because dissociation of many chelated phosphines is slow,25 HM reactions using aryl halides and bidentate phosphines often do not occur.129,130 The olefin insertion reaction can be facile in cationic systems; in the intramolecular HM reaction shown in Scheme 9, this occurs rapidly at –40 °C.129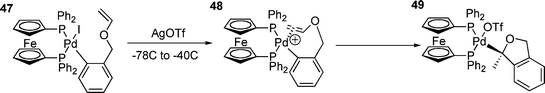 | ||
| Scheme 9 | ||
A great deal of work has been done on styrene-related systems, which has provided information on the olefin insertion step in the cationic HM reaction of PhOTf with a range of para-substituted styrenes.168 A negative Hammett parameter (ρ = –0.74) was obtained for α-substituted products only, and notably no correlation being found for β-substituted products. This observation was rationalised by the transition states 50 and 51. For α-substitution, the build-up of positive charge occurs at the α-carbon and is stabilised by electron donating groups on the styrene. For β-substitution, the build-up of positive charge occurs at the β-carbon and no stabilisation from groups on the styrene is possible.168
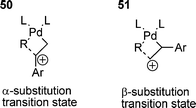
Carbene ligated palladium catalysts have also been used with some success under cationic conditions (AgBF4). These strong donor ligand systems presumably assist in stabilising the positive metal centre.54
The use of cationic catalysts has also been shown to have a pronounced effect on the regioselectivity of the HM reaction. Typically, in the neutral manifold, steric effects have a large impact upon regiocontrol and tend to favour β-substitution, however, by using a cationic catalyst, electronic effects can be made to dominate.128 For electron rich olefins, coordination to the cationic palladium atom favours migration to the α-carbon.126 Acrylates, however, always favour complete β-selectivity.128 Interestingly, it has been reported that it is possible to achieve highly selective α-substitution using a neutral catalyst.169 DFT studies including solvation have been used to generate a selectivity index for α/β-selectivities of a variety of olefins, in both neutral and cationic pathways, and the results appear to be quite accurate.170
6.3 Neutral bidentate intermediates
As previously noted, reactions involving this sort of species tend to be sluggish due to the reluctance of both chelated phosphines and halides to dissociate from palladium to give a co-ordinately unsaturated species.25 However, it appears that under certain circumstances, the olefin can displace the halide to give a reactive species. This was suggested as being the mechanism operating in the neutral asymmetric HM reaction shown in eqn (1).132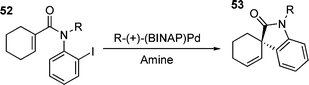 | (1) |
High e.e.s were obtained in the absence of added halide scavengers, indicating that phosphine dissociation was not occurring. The proposed mechanism involved associative displacement of halide by the olefin to give a reactive cationic species.132 In this case, the five-coordinate palladium transition state required contains two bidentate ligands which makes it less disfavoured than the five-coordinate palladium intermediates discussed previously. Another interesting observation to come from this study was the reversal of enantioselectivity upon the addition of AgOTf to the reaction,132 thus forming a cationic species upon oxidative addition .
More generally, however, small ring chelated palladium species seem to be poor catalysts of the HM reaction. Although the oxidative addition can occur, the resulting palladium species is unreactive towards olefins due to its coordination saturation and lack of labile groups.25,160 For some chelated species, the poor catalytic activity observed is due to disproportionation of the catalyst. For the reaction shown in Scheme 10, evolution of hydrogen occurs and the catalyst is oxidised to palladium(II) chloride in two catalytic cycles.94
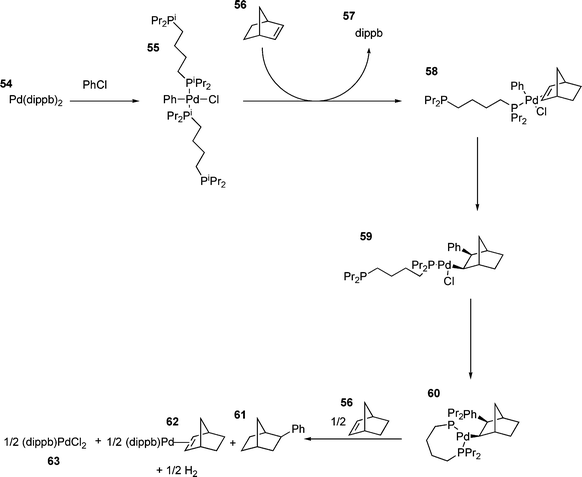 | ||
| Scheme 10 | ||
Whilst the small ring chelates from phosphines such as dippe and dippp generally give poor catalysts, increasing the size of the ring tends to increase activity.25,160 The use of dippb as a ligand allows the coupling of aryl chlorides;25 the increased ring size apparently increasing the lability of the phosphine and causing it to behave more like a monodentate ligand (Scheme 11).
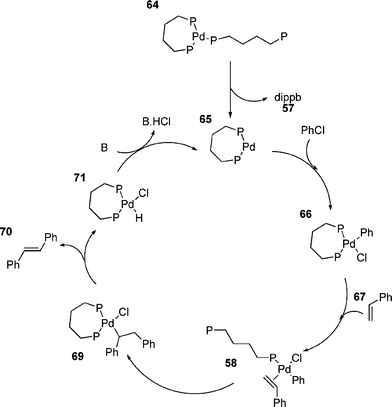 | ||
| Scheme 11 | ||
The kinetics of the HM reaction catalysed by chelated neutral species have been investigated.37 Use of a range of para-substituted iodides gave a Hammett parameter ρ = 1.39, and although the positive sign of this value is consistent with oxidative addition , its magnitude is not and rate limiting olefin insertion was proposed.37
Bidentate carbene–phosphine ligands have also been investigated by gas phase DFT studies and a mechanism involving rate limiting phosphine dissociation (i.e. chelate opening) has been proposed.165
7 Asymmetric HM (AHM) reactions
The Heck reaction discussed hereto is concerned with the generation of an sp2 hybridised centre and consequently no induction of chirality is possible. However, if the incoming group is added to a di- or tri-substituted carbon and a β’-proton is present, the competing β’-hydride elimination can give rise to a chiral centre (Scheme 12). In this case, the use of chiral ligands for palladium can give asymmetric induction .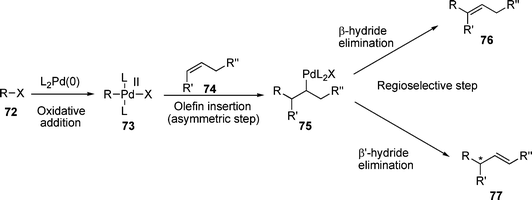 | ||
| Scheme 12 | ||
The discovery of this reaction came considerably later than that of the achiral variant, Shibasaki31 and Overman32 independently discovering the intramolecular version and Hayashi124,127 subsequently developing an intermolecular variant. The importance of this variant of the HM reaction is demonstrated by its extensive application to natural product total synthesis.171 Although the mechanism of this variant of the HM reaction is the same, the greater complexity introduced by the required enantioselective and regioselective steps merits further discussion.
Good control over both the enantioselectivity of the olefin insertion and the regioselectivity of the subsequent β-hydride elimination from 75 are essential to the AHM reaction. Even if great enantioselectivity can be achieved in the first step, the induced chirality will be lost if poor control of the β-hydride elimination step yields an achiral product. Additionally, the reinsertion of olefin 77 into the palladium hydride bond can lead to an equilibrium between the two products with consequent loss of chirality.172
7.1 Enantioselectivity
This requires a chiral palladium catalyst and hence the use of a chiral ligand. Bidentate ligands are required to achieve good asymmetric induction and a huge number of different ligands have been used. These include homochiral chelating P,P, P,N and N,N ligands.133Another important point relating to the enantioselectivity of the olefin insertion is whether the palladium(II) species is neutral or cationic. As a rule, the cationic pathway is found to give much greater asymmetric induction , this being ascribed to the requirement for ligand dissociation in the neutral pathway (vide supra). Whilst in the neutral pathway, partial dissociation of the bidentate chiral ligand reduces the influence of the ligand and thus e.e., in the cationic pathway the olefin can bind without ligand dissociation and thus high e.e. can be achieved.
Important exceptions to this rule have been observed in which high e.e.s have been found using the neutral pathway.173 Indeed, in some cases it has been found that the addition of silver additives to promote the cationic pathway can be detrimental to asymmetric induction .174 Also, the observed stereochemistry is generally reversed on moving from a cationic catalyst to a neutral catalyst.174 It has been shown that in these cases, phosphine dissociation does not occur and although halide dissociation to yield a cationic intermediate has been postulated, this would not explain either the change in configuration, the lack of selectivity for aryl triflates or the lack of solvent effects.132 Two other potential mechanisms which have been suggested, involve associative displacement of halide by the olefin and insertion from a five-coordinate palladium(II) intermediate.174 As noted earlier, the reluctance of palladium to form five-coordinate intermediates is documented,111,112 however, in this case with both the phosphine and the halide/olefin being bidentate, and thus having an enforced bite angle, such intermediates seem more plausible than in the case where all substituents are monodentate. Both mechanisms have the potential to explain the reversal in product configuration over the cationic pathway and thus without further studies it is not possible to say which occurs although the authors favour the associative displacement process.132
7.2 Regioselectivity
This requires a way to favour β’-hydride elimination over β-hydride elimination. The most obvious way to achieve this is through the generation of a quaternary chiral centre, in this way no β-hydride elimination is possible. Unfortunately, the formation of asymmetric quaternary centres is rather less well documented than for tertiary centres although it has been known to be possible for some time. Presumably, the tri-substituted olefins required for this tend to be less reactive due to steric hindrance and although possible, this reaction remains a challenge.A number of other, more common ways of favouring β’-hydride elimination include: i) use of intramolecular AMH reactions since when the product is an endocyclic alkene, the rotation required around the alkene σ-bond for β-hydride elimination to occur is not possible; ii) use of a themodynamic driving force for β’-elimination through choice of the group R″. For instance, R″ = OH gives an enol which tautomerises to the corresponding aldehyde or ketone, R″ = OR gives an enol ether or R″ = alkenyl gives a conjugated diene; iii) the use of an allylsilane as the olefin component has also allowed controlled β’-hydride elimination under AMH reaction conditions.175 Additionally, it may be expected that β’-hydride elimination would be favoured kinetically since the rotation around the alkene σ-bond necessary for β-hydride elimination is not necessary for β’-hydride elimination.
These points explain why the AMH reaction is most often used in intramolecular reactions to form endocyclic alkenes, another drive for β’-hydride elimination often also being present.172
7.3 Product isomerism
In order to prevent the loss of chirality associated with isomerism by reinsertion of products back into the palladium(II) hydride bond, it is often necessary to add chemicals to suppress this, these typically being thallium or silver salts.77,78,1768 Aryl–aryl exchange
The observation of the formation of unexpected products in HM reactions using triarylphosphine ligands has been known for some time.50,177 Typically, when triphenylphosphine is used, the alkenylated benzene product 81 is found as one of the products (eqn (2)).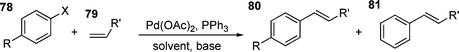 | (2) |
In addition, the phosphine resulting from exchange of one of the aryl groups of the phosphine with one of those from the halide can be observed.177 This problem tends to occur in systems which generally show poor reactivity, such as deactivated bromides and possibly because of the temperatures required to achieve coupling in such systems.160 It has been found that addition of stoichiometric Pd(PPh3)4 to vinyl triflates and aryl halides gives rise to vinylphosphonium123,178 and tetra-aryl phosphonium salts.179 This process can also be performed with catalytic palladium as a useful method to generate mixed aryl triarylphosphines.180
The mechanism for this scrambling involves the oxidative addition of the aryl halide to palladium(0) phosphine ligated catalyst 82 and subsequent reductive elimination to generate phosphonium salt 85 (Scheme 11).181,182 The eliminated phosphonium salt can then undergo oxidative addition to the palladium(0) species 84 generated in this process. However, the phosphorus–carbon bond undergoing the oxidative addition may not be the same as that which was formed in the reductive elimination , hence, aryl–aryl exchange can occur.181,182 It has been demonstrated that the selection of the phosphorus–carbon bonds that undergo oxidative addition is entirely random, allowing the statistical modelling of product distributions.181
Various attempts have been made to eliminate these reactions, mainly by varying the phosphine ligand employed,160,183 however, the only way to eliminate such side reactions is through the use of ortho-substituted phosphines, such as tri(o-tolyl)phosphine183 and tri(mesityl)phosphine,160 or by using trialkylphosphines.183 The reaction also appears to be promoted by electron donating substituents, either on the phosphine or the aryl group of the halide.181 This is presumably due to the promotion of the reductive elimination reaction by stabilisation of the positive charge on phosphorus.
Summary and conclusions
At best it can be said that the mechanism of the oxidative addition is strongly dependent on conditions, particularly depending on whether the reaction is saturated with halide. However, as the reaction proceeds, assuming aryl or vinyl halides are involved, the mechanism of the oxidative addition may well change as the halide generated saturates the reaction mixture. Clearly, not one proposed mechanism for the oxidative addition explains all the phenomena observed and it seems likely that several mechanisms may be in operation, either independently, depending on the reaction conditions, or in parallel.For subsequent steps, the mechanism is more poorly understood due to difficulties of investigations. It seems that even for aryl chlorides, the rate determining step may be after the oxidative addition step, since the rates of reaction of such species are lower than those seen for the oxidative addition reactions. This may be due to the electron rich, chelating nature of the phosphines required to activate aryl chlorides, which in turn disfavours carbometallation or dissociation. For iodides, and some other activated leaving groups, it seems to be generally accepted that the rate determining step comes after initial oxidative addition . However, the nature of this step is unclear and has a strong dependence on the nature of the species produced in the oxidative addition step.
Clearly, for a full understanding of the mechanism in operation, further studies are required, although it may well be the case that due to the reactive nature of the intermediates involved, a comprehensive understanding will be challenging to achieve.
References
- T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CAS.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- T. Hayashi, M. Konishi and M. Kumada, Tetrahedron Lett., 1979, 20, 1871–1874 CrossRef.
- A. Minato, K. Tamao, T. Hayashi, K. Suzuki and M. Kumada, Tetrahedron Lett., 1980, 21, 845–848 CrossRef CAS.
- E. Negishi and S. Baba, J. Chem. Soc., Chem. Commun., 1976, 596–597 Search PubMed.
- S. Baba and E. Negishi, J. Am. Chem. Soc., 1976, 98, 6729–6731 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636–3638 CrossRef.
- D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1979, 101, 4992–4998 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- D. Prim, J.-M. Campagne, D. Joseph and B. Andrioletti, Tetrahedron, 2002, 58, 2041–2075 CrossRef CAS.
- T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS.
- M. E. Logan and M. E. Oinen, Organometallics, 2006, 25, 1052–1054 CrossRef CAS.
- M. J. Schultz and M. S. Sigman, J. Am. Chem. Soc., 2006, 128, 1460–1461 CrossRef CAS.
- O. Gaertzen and S. L. Buchwald, J. Org. Chem., 2002, 67, 465–475 CrossRef CAS.
- E. M. Beck, N. P. Grimster, R. Hatley and M. J. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528–2529 CrossRef CAS.
- J.-L. Malleron, J.-C. Fiaud and J.-Y. Legros, Handbook of Palladium Catalysed Organic Reactions, Academic Press, San Diego, 1997 Search PubMed.
- Y. Hatanaka and T. Hayama, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS.
- R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5531–5534 CrossRef CAS.
- R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5535–5538 CrossRef CAS.
- R. F. Heck, J. Am. Chem. Soc., 1971, 93, 6896–6901 CrossRef CAS.
- H. A. Dieck and R. F. Heck, J. Am. Chem. Soc., 1974, 96, 1133–1136 CrossRef CAS.
- Y. Ben-David, M. Portnoy, M. Gozin and D. Milstein, Organometallics, 1992, 11, 1995–1996 CrossRef CAS.
- A. Jutand and A. Mosleh, Organometallics, 1995, 14, 1810–1817 CrossRef CAS.
- X. Fu, S. Zhang, J. Yin, T. L. McAllister, S. A. Jiang, C.-H. Tann, T. K. Thiruvengadam and F. Zhang, Tetrahedron Lett., 2002, 43, 573–576 CrossRef CAS.
- A. L. Hansen and T. Skrydstrup, Org. Lett., 2005, 7, 5585–5587 CrossRef CAS.
- K. Kikukawa and T. Matsuda, Chem. Lett., 1977, 159–162 CAS.
- T. Jeffery, J. Chem. Soc., Chem. Commun., 1984, 1287–1289 RSC.
- Y. Sato, M. Sodeoka and M. Shibasaki, J. Org. Chem., 1989, 54, 4738–4739 CrossRef CAS.
- N. E. Carpenter, D. J. Kucera and L. E. Overman, J. Org. Chem., 1989, 54, 5846–5848 CrossRef CAS.
- Y. Sato, M. Sodeoha and M. Shibasaki, Chem. Lett., 1990, 1953–1954 CAS.
- X. Du, M. Suguro, K. Hirabayashi and A. Mori, Org. Lett., 2001, 3, 3313–3316 CrossRef CAS.
- R. F. Heck, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS.
- B. L. Shaw, S. D. Perera and E. A. Staley, Chem. Commun., 1998, 1361–1362 RSC.
- M. Ohff, A. Ohff, M. E. van der Boom and D. Milstein, J. Am. Chem. Soc., 1997, 119, 11687–11688 CrossRef CAS.
- M. Beller and T. H. Riermeier, Eur. J. Inorg. Chem., 1998, 29–35 CrossRef CAS.
- V. P. W. Bohm and W. A. Herrmann, Chem.–Eur. J., 2001, 7, 4191–4197 CrossRef CAS.
- M. Rosol and A. Moyano, J. Organomet. Chem., 2005, 690, 2291–2296 CrossRef CAS.
- P. Nilsson and O. F. Wendt, J. Organomet. Chem., 2005, 690, 4197–4202 CrossRef CAS.
- F. d'Orlye and A. Jutand, Tetrahedron, 2005, 61, 9670–9678 CrossRef CAS.
- C. S. Consorti, G. Ebeling, F. R. Flores, F. Rominger and J. Dupont, Adv. Synth. Catal., 2004, 346, 617–624 CrossRef CAS.
- K. Yu, W. Sommer, M. Weck and C. W. Jones, J. Catal., 2004, 226, 101–110 CrossRef CAS.
- A. Sundermann, O. Uzan and J. M. L. Martin, Chem.–Eur. J., 2001, 7, 1703–1711 CrossRef CAS.
- G. P. F. van Strijdonck, M. D. K. Boele, P. C. J. Kamer, J. G. de Vries and P. W. N. M. Leewen, Eur. J. Inorg. Chem., 1999, 1073–1076 CrossRef CAS.
- G. T. Crisp, Chem. Soc. Rev., 1998, 27, 427–436 RSC.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS.
- L. Xu, W. Chen, J. Ross and J. Xiao, Org. Lett., 2001, 3, 295–297 CrossRef CAS.
- W. A. Herrmann, C. Brossmer, K. Ofele, M. Beller and H. Fischer, J. Organomet. Chem., 1995, 491, C1–C4 CrossRef CAS.
- M. Ohff, A. Ohff and D. Milstein, Chem. Commun., 1999, 357–358 RSC.
- R. A. Baber, S. Collard, M. Hooper, A. G. Orpen, P. G. Pringle, M. J. Wilkinson and R. L. Wingad, Dalton Trans., 2005, 1491–1498 RSC.
- D. E. Bergbreiter, P. L. Osburn and J. D. Frels, Adv. Synth. Catal., 2005, 347, 172–184 CrossRef CAS.
- D. S. McGuinness, K. J. Cavell, B. W. Skelton and A. H. White, Organometallics, 1999, 18, 1596–1605 CrossRef.
- A. Albinati, F. Lianza, H. Berger, P. S. Pregosin, H. Ruegger and R. W. Kunz, Inorg. Chem., 1993, 32, 478–486 CrossRef CAS.
- F. G. de la Torre, F. A. Jalon, A. M. Lopez-Agenjo, B. R. Manzano, A. Rodriguez, W. Weissensteiner, T. Sturm and M. Martinez-Ripoll, Organometallics, 1998, 17, 4634–4644 CrossRef CAS.
- K. Selvakumar, M. Valentini, M. Worle, P. S. Pregosin and A. Albinati, Organometallics, 1999, 18, 1207–1215 CrossRef CAS.
- F. A. Jalon, B. R. Manzano, F. G. de la Torre, A. M. Lopez-Agenjo, A. M. Rodriguez, W. Weissensteiner, T. Sturm, J. Mahia and M. Maestro, J. Chem. Soc., Dalton Trans., 2001, 2417–2424 RSC.
- P. Dotta, A. Magistrato, U. Rothlisberger, P. S. Pregosin and A. Albinati, Organometallics, 21, 3033–3041 Search PubMed.
- R. van Asselt and C. J. Elsevier, Organometallics, 1992, 11, 1999–2001 CrossRef CAS.
- R. van Asselt, K. Vrieze and C. J. Elsevier, J. Organomet. Chem., 1994, 480, 27–40 CrossRef CAS.
- R. van Asselt, C. J. Elsevier, W. J. J. Smeets and A. L. Spek, Inorg. Chem., 1994, 33, 1521–1531 CrossRef CAS.
- R. van Asselt and C. J. Elsevier, Organometallics, 1994, 13, 1972–1980 CrossRef CAS.
- R. van Asselt, C. J. Elsevier, C. Amatore and A. Jutand, Organometallics, 1997, 16, 317–328 CrossRef CAS.
- C. J. Elsevier, Coord. Chem. Rev., 1999, 185–186, 809–822 CrossRef CAS.
- J. Elguero, A. Guerrero, F. G. de la Torre, A. de la Hoz, F. A. Jalon, B. R. Manzano and A. Rodriguez, New J. Chem., 2001, 25, 1050–1060 RSC.
- M. W. van Laren, M. A. Duin, C. Klerk, M. Naglia, D. Rogolino, P. Pelagatti, A. Bacchi, C. Pelizzi and C. J. Elsevier, Organometallics, 2002, 21, 1546–1553 CrossRef CAS.
- M. C. Carrion, A. Guerrero, F. A. Jalon, B. R. Manzano, A. de la Hoz, R. M. Claramunt, V. Milata and J. Elguero, Inorg. Chem., 2003, 42, 885–895 CrossRef CAS.
- K. Boog-Wick, P. S. Pregosin and G. Trabesinger, Organometallics, 1998, 17, 3254–3264 CrossRef CAS.
- M. Tschoerner, G. Trabesinger, A. Albinati and P. S. Pregosin, Organometallics, 1997, 16, 3447–3453 CrossRef CAS.
- K. Selvakumar, M. Valentini, M. Worle, P. S. Pregosin and A. Albinati, Organometallics, 1999, 18, 4591–4597 CrossRef CAS.
- K. K. Hii, M. Thornton-Pett, A. Jutand and R. P. Tooze, Organometallics, 1999, 18, 1887–1896 CrossRef CAS.
- M. Qadir, T. Mochel and K. K. Hii, Tetrahedron, 2000, 56, 7975–7979 CrossRef CAS.
- B. R. Manzano, F. A. Jalon, F. G. de la Torre, A. M. Lopez-Agenjo, A. M. Rodriguez, K. Mereiter, W. Weissensteiner and T. Sturm, Organometallics, 2002, 21, 789–802 CrossRef CAS.
- T. Jeffery, Tetrahedron Lett., 1985, 26, 2667–2670 CrossRef CAS.
- T. Jeffery, Tetrahedron Lett., 1990, 31, 6641–6644 CrossRef CAS.
- K. Karabelas, C. Westerlund and A. Hallberg, J. Org. Chem., 1985, 50, 3896–3900 CrossRef CAS.
- R. Grigg, V. Loganathan, V. Santhakumar, V. Sridharan and A. Teasdale, Tetrahedron Lett., 1991, 32, 687–690 CrossRef CAS.
- J. Campora, P. Palma, D. del Rio, J. A. Lopez, E. Alvarez and N. G. Connelly, Organometallics, 2005, 24, 3624–3628 CrossRef CAS.
- L. Malatesta and M. Angoletta, J. Chem. Soc., 1957, 1186–1188 RSC.
- C. R. Coulson, Inorg. Synth., 1970, 13, 121–124.
- E. Negishi, T. Takahashi and K. Akiyoshi, J. Chem. Soc., Chem. Commun., 1986, 1338–1339 RSC.
- C. Amatore, M. Azzabi and A. Jutand, J. Organomet. Chem., 1989, 363, C41–C45 CrossRef CAS.
- C. Amatore, A. Jutand and M. A. M'Barki, Organometallics, 1992, 11, 3009–3013 CrossRef CAS.
- F. Ozawa, A. Kubo and T. Hayashi, Chem. Lett., 1992, 2177–2180 CAS.
- C. Amatore, A. Jutand, F. Lemaitre, J. L. Ricard, S. Kozuch and S. Shaik, J. Organomet. Chem., 2004, 689, 3728–3734 CrossRef CAS.
- M. Basato, B. Sesto, M. Zecca, G. Valle, S. Antonello and F. Maran, J. Organomet. Chem., 2000, 601, 201–210 CrossRef CAS.
- C. Amatore, E. Carre, A. Jutand and M. A. M'Barki, Organometallics, 1995, 14, 1818–1826 CrossRef CAS.
- J. Louie and J. F. Hartwig, Angew. Chem., Int. Ed. Engl., 1996, 35, 2359–2361 CrossRef CAS.
- T. Rosner, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 4621–4622 CrossRef CAS.
- A. F. Shmidt and V. V. Smirnov, Kinet. Catal., 2002, 43, 195–198 CAS.
- R. Benhaddou, S. Czernecki, G. Ville and A. Zegar, Organometallics, 1988, 7, 2435–2439 CrossRef CAS.
- G. Rothenberg, S. C. Cruz, G. P. F. van Strijdonck and H. C. J. Hoefsloot, Adv. Synth. Catal., 2004, 346, 467–473 CrossRef CAS.
- M. Portnoy, Y. Ben-David, I. Rousso and D. Milstein, Organometallics, 1994, 13, 3465–3479 CrossRef CAS.
- S. G. Fiddy, J. Evans, M. A. Newton, T. Neisius, R. P. Tooze and R. Oldman, Chem. Commun., 2003, 2682–2683 RSC.
- R. Ugo, A. Pasini, A. Fusi and S. Cenini, J. Am. Chem. Soc., 1972, 94, 7364–7370 CrossRef CAS.
- W. H. Thompson and C. T. Sears, Inorg. Chem., 1977, 16, 769–774 CrossRef CAS.
- R. J. Mureinik, M. Weitzberg and J. Blum, Inorg. Chem., 1979, 18, 915–918 CrossRef CAS.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS.
- P. K. Wong, K. S. Y. Lau and J. K. Stille, J. Am. Chem. Soc., 1974, 96, 5956–5957 CrossRef CAS.
- J.-F. Fauvarque, F. Pfluger and M. Troupel, J. Organomet. Chem., 1981, 208, 419–427 CrossRef CAS.
- C. A. Tolman, W. C. Seidel and D. H. Gerlach, J. Am. Chem. Soc., 1972, 94, 2669–2676 CrossRef CAS.
- W. Kuran and A. Musco, Inorg. Chim. Acta, 1975, 12, 187–193 CrossRef CAS.
- M. Ahlquist, P. Fristrup, D. Tanner and P.-O. Norrby, Organometallics, 2006, 25, 2066–2073 CrossRef CAS.
- C. Amatore and F. Pfluger, Organometallics, 1990, 9, 2276–2282 CrossRef CAS.
- M. Portnoy and D. Milstein, Organometallics, 1993, 12, 1665–1673 CrossRef CAS.
- A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 1533–1541 CrossRef CAS.
- C. Amatore, M. Azzabi and A. Jutand, J. Am. Chem. Soc., 1991, 113, 8375–8384 CrossRef CAS.
- C. Amatore, E. Carre, A. Jutand, M. A. M'Barki and G. Meyer, Organometallics, 1995, 14, 5605–5614 CrossRef CAS.
- C. Amatore, A. Jutand and A. Suarez, J. Am. Chem. Soc., 1993, 115, 9531–9541 CrossRef CAS.
- M. Broring and C. D. Brandt, Chem. Commun., 2003, 2156–2157 RSC.
- S. Hansson, P.-O. Norrby, M. P. T. Sjogren, B. Akermark, M. E. Cucciolito, F. Giordano and A. Vitagliano, Organometallics, 1993, 12, 4940–4948 CrossRef CAS.
- A. Jutand, Pure Appl. Chem., 2004, 76, 565–576 CrossRef CAS.
- L. J. Goossen, D. Koley, H. Hermann and W. Thiel, Chem. Commun., 2004, 2141–2143 RSC.
- L. J. Goossen, D. Koley, H. Hermann and W. Thiel, Organometallics, 2005, 24, 2398–2410 CrossRef CAS.
- S. Kozuch, C. Amatore, A. Jutand and S. Shaik, Organometallics, 2005, 24, 2319–2330 CrossRef CAS.
- S. Kozuch, S. Shaik, A. Jutand and C. Amatore, Chem.–Eur. J., 2004, 10, 3072–3080 CrossRef CAS.
- T. Rosner, J. Le Bars, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 1848–1855 CrossRef CAS.
- H. Urata, M. Tanaka and T. Fuchikami, Chem. Lett., 1987, 751–754 CrossRef CAS.
- J. F. Fauvarque and A. Jutand, J. Organomet. Chem., 1977, 132, C17–C19 CrossRef CAS.
- A. L. Casado and P. Espinet, Organometallics, 1998, 17, 954–959 CrossRef CAS.
- P. J. Stang, M. H. Kowalski, M. D. Schiavelli and D. Longford, J. Am. Chem. Soc., 1989, 111, 3347–3356 CrossRef CAS.
- R. J. Hinckle, P. J. Stang and M. H. Kowalski, J. Org. Chem., 1990, 55, 5033–5036 CrossRef CAS.
- F. Ozawa, A. Kubo and T. Hayashi, J. Am. Chem. Soc., 1991, 113, 1417–1419 CrossRef CAS.
- W. Cabri, I. Candiani, S. DeBernardinis, F. Francalanci, S. Penco and R. Santi, J. Org. Chem., 1991, 56, 5796–5800 CrossRef CAS.
- W. Cabri, I. Candiani, A. Bedeschi, S. Penco and R. Santi, J. Org. Chem., 1992, 57, 1481–1486 CrossRef CAS.
- T. Hayashi, A. Kubo and F. Ozawa, Pure Appl. Chem., 1992, 64, 421–427 CrossRef CAS.
- W. Cabri, I. Candiani, A. Bedeschi and R. Santi, J. Org. Chem., 1992, 57, 3558–3563 CrossRef CAS.
- J. M. Brown, J. J. Perez-Torrente, N. W. Alcock and H. J. Clase, Organometallics, 1995, 14, 207–213 CrossRef CAS.
- W. Cabri and I. Candiani, Acc. Chem. Res., 1995, 28, 2–7 CrossRef.
- S. Aoki, T. Fujimura, E. Nakamura and I. Kuwajima, J. Am. Chem. Soc., 1988, 110, 3296–3298 CrossRef CAS.
- A. Ashimori, B. Bachand, M. A. Calter, S. P. Govek, L. E. Overman and D. J. Poon, J. Am. Chem. Soc., 1998, 120, 6488–6499 CrossRef CAS.
- M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533–1552 CrossRef CAS.
- P. W. N. M. van Leeuwen, P. C. J. Kamer and J. N. H. Reek, Pure Appl. Chem., 1999, 71, 1443–1452.
- R. J. van Haaren, K. Goubitz, J. Fraanje, G. P. F. van Strijdonck, H. Oevering, B. Coussens, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Inorg. Chem., 2001, 40, 3363–3372 CrossRef CAS.
- C. Amatore, A. Jutand and A. Thuilliez, Organometallics, 2001, 20, 3241–3249 CrossRef CAS.
- C. Amatore, G. Broeker, A. Jutand and F. Khalil, J. Am. Chem. Soc., 1997, 119, 5176–5185 CrossRef CAS.
- M. Tschoerner, P. S. Pregosin and A. Albinati, Organometallics, 1999, 18, 670–678 CrossRef CAS.
- H. M. Senn and T. Ziegler, Organometallics, 2004, 23, 2980–2988 CrossRef CAS.
- A. Jutand, K. K. Hii, M. Thornton-Pett and J. M. Brown, Organometallics, 1999, 18, 5367–5374 CrossRef CAS.
- C. Amatore, A. Jutand, F. Khalil, M. A. M'Barki and L. Mottier, Organometallics, 1993, 12, 3168–3178 CrossRef CAS.
- Y. Mace, A. R. Kapdi, I. J. S. Fairlamb and A. Jutand, Organometallics, 2006, 25, 1795–1800 CrossRef CAS.
- U. K. Singh, E. R. Strieter, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14104–14114 CrossRef CAS.
- S. Shekhar, P. Ryberg and J. F. Hartwig, Org. Lett., 2006, 8, 851–854 CrossRef CAS.
- S. Shekhar, P. Ryberg, J. F. Hartwig, J. S. Mathew, D. G. Blackmond, E. R. Strieter and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 3584–3591 CrossRef CAS.
- L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, Organometallics, 2006, 25, 54–67 CrossRef CAS.
- F. Barrios-Landeros and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 6944–6945 CrossRef CAS.
- A. Beeby, S. Bettington, I. J. S. Fairlamb, A. E. Goeta, A. R. Kapdi, E. H. Niemela and A. L. Thosmpson, New J. Chem., 2004, 28, 600–605 RSC.
- M. Ahlquist, G. Fabrizi, S. Cacchi and P.-O. Norrby, Chem. Commun., 2005, 4196–4198 RSC.
- A. Jutand, S. Negri and J. G. de Vries, Eur. J. Inorg. Chem., 2002, 1711–1717 CrossRef CAS.
- (a) S. Brase and A. de Meijere, in Metal-catalysed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998 Search PubMed; (b) J. T. Link and L. E. Overman, in Metal-catalysed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998 Search PubMed; (c) R. F. Heck, Org. React., 1982, 27, 345–390 CAS.
- J. Halpern and T. Okamoto, Inorg. Chim. Acta, 1984, 89, L53–L54 CrossRef CAS.
- N. M. Doherty and J. E. Bercaw, J. Am. Chem. Soc., 1985, 107, 2670–2682 CrossRef CAS.
- B. J. Burger, B. D. Santarsiero, M. S. Trimmer and J. E. Bercaw, J. Am. Chem. Soc., 1988, 110, 3134–3146 CrossRef CAS.
- F. C. Rix, M. Brookhart and P. S. White, J. Am. Chem. Soc., 1996, 118, 2436–2448 CrossRef CAS.
- J. S. Brumbaugh, R. R. Whittle, M. Parvez and A. Sen, Organometallics, 1990, 9, 1735–1747 CrossRef CAS.
- E. G. Samsel and J. R. Norton, J. Am. Chem. Soc., 1984, 106, 5505–5512 CrossRef CAS.
- A. A. Sabino, A. H. L. Machado, C. R. D. Correia and M. N. Eberlin, Angew. Chem., Int. Ed., 2004, 43, 2514–2518 CrossRef CAS.
- M. Casey, J. Lawless and C. Shirran, Polyhedron, 2000, 19, 517–520 CrossRef CAS.
- W. A. Herrmann, C. Brossmer, K. Ofele, M. Beller and H. Fischer, J. Mol. Catal. A: Chem., 1995, 103, 133–146 CrossRef CAS.
- T. Mandai, T. Matsumoto and J. Tsuji, Tetrahedron Lett., 1993, 34, 2513–2516 CrossRef CAS.
- F. Zhao, B. M. Bhanage, M. Shirai and M. Arai, J. Mol. Catal. A: Chem., 1999, 142, 383–388 CrossRef CAS.
- A. F. Schmidt and V. V. Smirnov, Kinet. Catal., 2001, 42, 800–804.
- M. Buback, T. Perkovic, S. Redlich and A. de Mejere, Eur. J. Org. Chem., 2003, 2375–2382 CrossRef CAS.
- K. Albert, P. Gisdakis and N. Rosch, Organometallics, 1998, 17, 1608–1616 CrossRef CAS.
- A. F. Schmidt and V. V. Smirnov, Kinet. Catal., 2003, 44, 518–523 CAS.
- M. Feuerstein, H. Doucet and M. Santelli, J. Org. Chem., 2001, 66, 5923–5925 CrossRef CAS.
- P. Fristrup, S. Le Quement, D. Tanner and P.-O. Norrby, Organometallics, 2004, 23, 6160–6165 CrossRef CAS.
- L. Xu, W. Chen and J. Xiao, J. Mol. Catal. A: Chem., 2002, 187, 189–193 CrossRef CAS.
- R. J. Deeth, A. Smith and J. M. Brown, J. Am. Chem. Soc., 2004, 126, 7144–7151 CrossRef CAS.
- A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2963 CrossRef CAS.
- M. Shibasaki and E. M. Vogl, J. Organomet. Chem., 1999, 576, 1–15 CrossRef CAS.
- A. Ashimori, T. Matsuura, L. E. Overman and D. J. Poon, J. Org. Chem., 1993, 58, 6949–6951 CrossRef CAS.
- L. E. Overman and D. J. Poon, Angew. Chem., Int. Ed. Engl., 1997, 36, 518–521 CrossRef CAS.
- L. F. Tietze and R. Schimpf, Angew. Chem., Int. Ed. Engl., 1994, 33, 1089–1091 CrossRef.
- M. M. Abelman, T. Oh and L. E. Overman, J. Org. Chem., 1987, 52, 4133–4135 CrossRef CAS.
- K.-C. Kong and C.-H. Cheng, J. Am. Chem. Soc., 1991, 113, 6313–6315 CrossRef CAS.
- M. H. Kowalski, R. J. Hinckle and P. J. Stang, J. Org. Chem., 1989, 54, 2783–2784 CrossRef CAS.
- G. de la Torre, A. Gouloumis, P. Vazquez and T. Torres, Angew. Chem., Int. Ed., 2001, 40, 2895–2898 CrossRef CAS.
- F. Y. Kwong, C. W. Lai, Y. Tian and K. S. Chan, Tetrahedron Lett., 2000, 41, 10285–10289 CrossRef CAS.
- F. E. Goodson, T. I. Wallow and B. M. Novak, J. Am. Chem. Soc., 1997, 119, 12441–12453 CrossRef CAS.
- V. V. Grushin, Organometallics, 2000, 19, 1888–1900 CrossRef CAS.
- P. B. Hodgson and F. H. Salingue, Tetrahedron Lett., 2004, 45, 685–687 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |