Microcins, gene-encoded antibacterial peptides from enterobacteria
Sophie
Duquesne
,
Delphine
Destoumieux-Garzón
,
Jean
Peduzzi
and
Sylvie
Rebuffat
*
Laboratory of Chemistry and Biochemistry of Natural Substances, UMR 5154 CNRS, Department of Regulations, Development and Molecular Diversity, National Museum of Natural History, CP 54, 57 rue Cuvier, 75005, Paris, France
First published on 18th April 2007
Abstract
Covering 1982 to 2006
Microcins are gene-encoded antibacterial peptides , with molecular masses below 10 kDa, produced by enterobacteria. They are secreted under conditions of nutrient depletion and exert potent antibacterial activity against closely related species. Typical gene clusters encoding the microcin precursor, the self-immunity factor, the secretionproteins and frequently the post-translational modification enzymes are located either on plasmids or on the chromosome. In contrast to most of the antibiotics of microbial origin, which are non-ribosomally synthesized by multimodular enzymes termed peptide synthetases, microcins are ribosomally synthesized as precursors, which are further modified enzymatically. They form a restricted class of potent antibacterial peptides . Fourteen microcins have been reported so far, among which only seven have been isolated and characterized. Despite the low number of known representatives, microcins exhibit a diversity of structures and antibacterial mechanisms. This review provides an updated overview of microcin structures, antibacterial activities, genetic systems and biosyntheses, as well as of their mechanisms of action.
 Sophie Duquesne Sophie Duquesne | Sophie Duquesne learned chemistry and biochemistry at the National School of Chemistry, in Paris (France). In 2003, she joined the Rebuffat group in Paris as a PhD student in biochemistry. She is currently in the final stages of completing her PhD, which has focused on the enzymes involved in microcin biosynthesis. |
 Delphine Destoumieux-Garzón Delphine Destoumieux-Garzón | Delphine Destoumieux-Garzón studied biochemistry and microbiology at the National Institute for Applied Sciences and at the University Paul Sabatier in Toulouse (France). During her PhD, obtained from the University of Montpellier in 1998, she focused on antimicrobial peptides in invertebrate immunity. As a post-doctoral fellow in the Ganz lab, at the University of California, Los Angeles, she studied the role of defensins in skin immunity. In 2000, she obtained a permanent research position at the CNRS and joined the Rebuffat group in Paris. There, she studied the molecular basis of microbial competitions, with particular interest in microcin recognition and uptake pathways. In 2006, she returned to the University of Montpellier, where she is studying the resistance of Vibrio species to marine invertebrate antibacterials. |
 Jean Peduzzi Jean Peduzzi | Jean Peduzzi studied biochemistry, molecular and cellular pharmacology at the University Paris VI, Pierre and Marie Curie, from which he obtained his PhD in 1979. His PhD research focused on the isolation and inhibition of β-lactamases by clavulanic acid. He was hired by the CNRS in 1980 and obtained a permanent research position in 1983 after a post-doctorate in the laboratory of Professor J. Rosa, at the hospital Henri Mondor in Créteil (France), where he studied diphosphoglycerate mutase, a minor protein from red blood cells. Since 1985 he has been a member of the Laboratory of Chemistry and Biochemistry of Natural Substances at the National Museum of Natural History, in Paris (France). Together with Sylvie Rebuffat, he is currently heading the Biochemistry team, in which he is studying the structures, biosynthetic pathways and mechanisms of action of microcins. |
 Sylvie Rebuffat Sylvie Rebuffat | Sylvie Rebuffat studied chemistry and biochemistry at the University Paris VI, Pierre and Marie Curie, and then completed a PhD in chemistry and spectroscopy in 1982, carrying out her research at the National Museum of Natural History. Subsequently she got a Maitre de Conferences position at the same Institution. She was a CNRS fellow in 1987 at the Gesellshaft für Biotechnologische Forschung (GBF) in Germany in the group of Professor G. Höfle. She is now Professor in the Laboratory of Chemistry and Biochemistry of Natural Substances at the National Museum of Natural History. Her present research focuses on the molecular aspects of the adaptative processes and defence mechanisms developed by microorganisms in specific ecosystems, such as the intestinal microflora and the symbiotic bacteria associated with marine invertebrates. |
1 Introduction
Together with colicins, microcins are toxic peptides secreted by enterobacteria (mostly Escherichia coli) that belong to the large class of bacteriocins. The name microcin was introduced1 to distinguish this class of antibacterial peptides , with molecular masses below 10 kDa, from the higher molecular mass colicins.2–4 Microcins are generally hydrophobic and show a high stability to heat, extreme pH and proteases. Produced under conditions of stress, such as nutrient depletion, they have potent antibacterial activity against closely related bacteria, with minimum inhibitory concentrations (MICs) in the nanomolar range. They are therefore believed to be efficient weapons of the intestinal microbiota, contributing to the control of possible takeover by competing enterobacteria. The potent activity exerted by microcins, associated with a narrow spectrum of bacterial targets, make them particularly attractive tools for food preservation applications or for the replacement of conventional antibiotics.Whereas many antimicrobial peptides from microbial origin are produced by large multidomain enzyme complexes termed peptide synthetases, microcins are typically produced as ribosomally synthesized precursors, similar to the bacteriocins from Gram-positive bacteria (for reviews, see Jack et al.5 and Drider et al.6). Microcins are encoded by gene clusters carried either by plasmids or by the chromosome. Their gene clusters, which typically include open reading frames (ORFs) encoding the microcin precursor, self-immunity factors, secretionproteins and in general modification enzymes, give rise to an amazing diversity of microcin structures and mechanisms of action.
Microcins have been studied to a much lesser extent compared to other antibacterials such as colicins from Gram-negative bacteria, and bacteriocins from Gram-positive bacteria. However, numerous articles have been produced on the subject over the last few years. Among the fourteen microcins identified so far, only seven have been structurally characterized. Those are microcin B17 (MccB17), MccC7/C51, MccE492, MccJ25, MccL, MccM, and MccV (also known as ColV). Other microcins (MccH47, MccI47, Mcc24) had their structures predicted by genetic studies only. Finally, MccD93, Mcc140, Mcc15m and Mcc15n, which would be microcins of low molecular mass (below 1000 Da),7–10 were only evidenced by few partial biochemical studies, and will not be described further in this review article.
In contrast with the very large number of bacteriocins from Gram-positive bacteria and colicins (for reviews, see Sablon et al.11 and Braun et al.4), which have been assembled into classes according to common structural features and mechanisms of action, it appears to be more difficult to define sub-groups inside a family that is as restricted and diverse as the microcins. The first classification was attempted by Pons and collaborators,12,13 who proposed to define two classes of microcins according to the occurrence of post-translational modifications. However, our recent finding that MccE492, initially described as an unmodified 84 amino acid peptide , was also secreted in a modified form14 changed this vision. Therefore, we propose, in this review, a novel classification of microcins (Table 1) that agrees with most of the following criteria: (i) the presence, nature and localization of the post-translational modifications, (ii) the gene cluster organization, and (iii) the leader peptide sequences. In this classification, class I microcins are peptides with a molecular mass below 5 kDa, which are subject to extensive backbone post-translational modifications (MccB17, MccC7/C51, MccJ25). Class II includes higher molecular mass peptides (in the 5–10 kDa range). Class II is further subdivided into two subclasses: class IIa, some of which contain disulfide bonds but no further post-translational modification (MccL, MccV, Mcc24), and class IIb, which gathers together those linear microcins that may carry a C-terminal post-translational modification (MccE492, MccM and presumably MccH47 and MccI47).
| Microcin | Class | Precursor/Promicrocina | Leader peptide /Propeptidea | Microcin | |||||
|---|---|---|---|---|---|---|---|---|---|
| Size (number of residues) | Size (number of residues) | Size (number of residues) | Molecular mass (Da) | Gly content (%) | Ser content (%) | Cys content (number of residues) | Net charge | ||
| a Numbers in parentheses refer to the length of the precursor and leader peptide , assuming transcription starts at the second of the two neighboring AUG codons. b Values from the theoretical amino acid composition determined for the precursor after cleavage of the leader peptide and before modification of Gly-Cys and Gly-Ser sequences to thiazole and oxazole rings. c This microcin has not been isolated; the sizes of the precursor, of the leader peptide and of the mature microcin have been hypothesized from the amino acid sequence deduced from the nucleotide sequence of the structural gene and from leader peptide alignments. d The biochemical characteristics (molecular mass, Gly/Ser/Cys content, and net charge) have been calculated from the putative sequence. e MccE492 and u-MccE492 were formerly termed MccE492m and MccE492, respectively. f The three extra serines from the siderophore-type post-translational modification are not included. | |||||||||
| MccB17 | I | 69 | 26 | 43 | 3093 | 60.5b | 13.9b | 4b | 1+b |
| MccC7/C51 | I | 7 | 0 | 7 | 1177 | 0 | 0 | 0 | 0 |
| MccJ25 | I | 58 | 37 | 21 | 2107 | 28.6 | 4.8 | 0 | 0 |
| MccV | IIa | 103 | 15 | 88 | 8733 | 16.9 | 9.1 | 2 | 1+ |
| MccL | IIa | 105 | 15 | 90 | 8884 | 15.6 | 6.6 | 4 | 3– |
| Mcc24c,d | IIa | 90 (88) | 17 (15) | 73 | 7457 | 13.5 | 5.4 | 0 | 3+ |
| MccE492e | IIb | 103 (99) | 19 (15) | 84 | 8717 | 22.6 | 11.9 | 0 | 3– |
| u-MccE492e | IIb | 103 (99) | 19 (15) | 84 | 7886 | 22.6 | 11.9f | 0 | 3– |
| MccM | IIb | 92 | 15 | 77 | 7283 | 19.5 | 23.4 | 0 | 1– |
| MccH47c,d | IIb | 75 | 15 | 60 | 4865 | 26.6 | 15 | 0 | 0 |
| MccI47c,d | IIb | 77 | 15 | 62 | 6276 | 10.8 | 16.2 | 1 | 2– |
This review provides an updated overview of microcin structures and antibacterial activities, of their genetic systems and biosyntheses, as well as of their mechanisms of action.
2 Genetic system organization
The organization of microcin gene clusters is partially conserved and involves at least four clustered genes grouped in a single or several operons. The minimal structure is composed of (i) the structural gene encoding the microcin precursor, (ii) the self-immunity gene generally adjacent to the former, which encodes the self-immunity factor that protects the producing strain from its own antibacterial substance, and (iii) genes encoding the microcin export system necessary for the external secretion of the microcin. Additionally, genes encoding post-translational modification enzymes can be found. The content of microcin gene clusters and their overall organization are summarized in Table 2 and Fig. 1, respectively. The reader should be aware that the name given to each gene is not standardized throughout the different microcin gene clusters. For instance, genes encoding microcin precursors were often termed A (class I microcins as well as MccE492 and MccM), but some genes encoding microcin export proteins were also termed A (class IIa microcins). Moreover, with the exception of MccE492 genetic system, all genes termed I encode a self-immunity protein , but not all self-immunity proteins are encoded by a gene termed I. Two strategies have been used to identify the role of the different genes in microcin gene clusters. The first was based on genetics (mutagenesis, functional complementation, subcloning, gene fusion, etc.), and the second resulted from sequence homologies. The detailed roles of gene products are specified in Sections 4, 5 and 6.| Microcin | Structural gene | Self-immunity genes | Export genes | Post-translational modification genes | Genes of unknown function | EMBL database accession number | G + C content (%) |
|---|---|---|---|---|---|---|---|
| a Gene absent from MccC51 gene cluster. b Genes of unknown function that are necessary for microcin production. c Genes specific for MccI47. d Genes specific for MccM. e Genes absent from E. coli Nissle 1917 strain. | |||||||
| MccB17 | mcbA (210) | mcbE (726), mcbF (744), mcbG (564) | mcbE (726), mcbF (744) | mcbB (888), mcbC (819), mcbD (1191) | — | M24253, X07875 | 38.0 |
| MccC7/C51 | mccA (24) | mccC (1215), mccE (1566), mccFa (1035) | mccC (1215) | mccB (1053), mccD (804), mccE (1566) | — | X57583, AJ487788 | 34.4 |
| MccJ25 | mcjA (177) | mcjD (1743) | mcjD (1743) | mcjB (627), mcjC (1542) | — | AF061787, AM116873 | 33.1 |
| MccV | cvaC (312) | cvi (237) | cvaA (1242), cvaB (2097) | — | — | X57524, X57525 | 42.0 |
| MccL | mclC (318) | mclI (156) | mclA (1242), mclB (2097) | — | — | AY237108 | 40.1 |
| Mcc24 | mtfS (273) | mtfI (282) | mtfA (1245), mtfB (2124) | — | — | U47048 | 43.2 |
| MccE492 | mceA (312) | mceB (288) | mceH (1242), mceG (2097), mceF (540) | mceC (1113), mceD (1245), mceI (492) | mceE (345), mceJb (1575) | AF063590 | 40.0 |
| MccH47/MccI47 | mchB (228), mchS2c (234) | mchI (210), mchS3c (435) | mchE (1242), mchF (2097) | mchA (1119), mchS1 (1266), mchD (453) | mchX (120), mchCb (1551), mchS4c (246) | AJ009631 | 40.3 |
| MccM/MccH47 | mcmA d (279), mchB (228) | mcmI d (222), mchI (210) | mchE (1242), mchF (2097) | mcmL d , e (1119), mcmKd,e (1275), mchD (453) | mcmM d (687), mchX (120), mchC (1551) | AJ515251, AJ515252, AJ586887 | 40.3–41.4 |
 | ||
| Fig. 1 Genetic organization of microcin gene clusters. Genes are indicated by arrows whose direction refers to gene transcription. An overview of the gene functions is given in Table 2. Genes encoding microcin precursors are shown in yellow. Genes required for self-immunity, microcin export and post-translational modifications are shown in red, blue, and green, respectively. Direct repeats flanking the MccC7/C51 and MccH47 gene clusters are indicated by vertical lines. Promoters are indicated by flags. Sequences with the most significant homology to the fur (ferric uptake regulation) boxes are shown by diamonds. The name of genes is indicated below or above each gene. A colour code is used for genes specific for one microcin within the gene cluster. Thus, names in green, blue or red are specific for microcins whose names are labelled with the same colour. Class I microcins are shown in (A). Genes required for both immunity and export are shown in purple. The gene mccE, whose product is involved both in post-translational modification (N-terminal region) and immunity (C-terminal region) towards MccC7/C51 is shown in green and red gradations. Class IIa and class IIb microcins are shown in (B) and (C), respectively. For class IIb microcins the name of the E. coli strain is indicated in parentheses. Genes encoding proteins of unknown function are indicated in grey. Genes encoding homologous or identical proteins in different clusters are coloured by different shades of the same colour. The genes tra5, insC, and insE, coloured in brown, encode transposases for insertion sequences IS2 and IS3. Truncated genes in MccL, MccH47/I47 and MccM/H47 (Nissle 1917) gene clusters are crossed through. | ||
2.1 Class I microcins: MccB17, MccC7/C51 and MccJ25
Class I microcins are encoded by gene clusters in which the self-immunity gene is not located near to the microcin structural gene. Two or three genes involved in post-translational modifications of the amino acid backbone are located adjacent to the structural gene. Furthermore, at least one gene is involved in both self-immunity and export.MccB17 is produced by various E. coli strains harbouring the 70-kb single-copy, conjugative pMccB17 plasmid (formerly pRYC17).15,16 The MccB17 gene cluster is composed of seven genes17–19 (Fig. 1A). The gene mcbA encodes the 69-aa MccB17 precursor,20 while mcbB, mcbC and mcbD encode the three components of the MccB17 synthetase19 involved in post-translational modifications of McbA.21 The genes mcbE and mcbF encode two proteins mainly involved in MccB17 secretion, which also contribute to self-immunity towards MccB17.18 Full self-immunity requires the product of a last gene, mcbG.18 The mcb genes probably form a single transcriptional unit22,23 under the control of a stationary-phase promoter Pmcb,19,24 located upstream of mcbA. Two additional promoters were identified within the MccB17 gene cluster:19 P2, located within mcbC, can direct a weak transcription of mcbD, whereas the role of P3, located within mcbD, and which directs transcription in the opposite direction, remains unclear.
MccC7/C51 is the smallest microcin hitherto characterized. MccC7 was first isolated by Moreno and collaborators from culture supernatants of E. coli strains harbouring the 43-kb single-copy pMccC7 plasmid (formerly pRYC7).25,26 The authors termed it MccC7 after the name of the plasmid. Later, the same molecule was isolated by Khmel and collaborators from E. coli strains harbouring the 38-kb low-copy number pMccC51 plasmid (formerly pC51),27 and the peptide was termed MccC51. A 6.5-kb and a 5.7-kb DNA fragment containing the microcin gene clusters were cloned from pMccC7 and pMccC51, respectively.27–29 The nucleotide sequences of the two microcin gene clusters (Fig. 1A) display 98–100% sequence identity for mccA to mccE.29 The 24 bp structural gene, mccA has been described as the smallest known gene.30 It encodes the 7-aa precursor of MccC7/C51, MccA. The genes mccB, mccD and mccE are involved in the post-translational modifications of MccA, whereas mccC and mccE are required for self-immunity towards MccC7/C51. MccC, which exhibits similarity to multidrug efflux transporters, is also probably involved in the MccC7/C51 export. The product of the last gene on the cluster, mccF, which is transcribed from the opposite strand, contributes weakly to the self-immunity towards MccC728 but not to self-immunity towards MccC51, since only a truncated mccF gene is present on MccC51 genetic system.29 One promoter was identified in the MccC7/C51 gene clusters.30,31 Located upstream of mccA, Pmcc directs transcription from mccA to mccE. Therefore, this region most probably forms an operon.23
MccJ25 is encoded by the 60-kb low-copy number pTUC100 plasmid, found in the E. coli AY25 faecal strain.32 A 4.8-kb DNA fragment of pTUC100 plasmid, containing all the MccJ25 determinants, has been cloned and sequenced.33,34 Four genes, arranged in two divergent operons, are required for MccJ25 production, export and self-immunity (Fig. 1A). The gene mcjA encodes the 58-aa MccJ25 precursor, while mcjB and mcjC encode proteins probably involved in the post-translational modifications of McjA. The sequence of mcjC, which was recently reinvestigated, is 213 bp longer than that previously described (Duquesne et al., unpublished work, GenBank, accession no. AM116873). The last gene, mcjD, is required for both MccJ25 secretion and self-immunity towards MccJ25. Similar to an ABC (ATP-binding cassette) transporter, McjD is responsible for the secretion of endogenous MccJ25 outside the cell,34 but also for the export of exogenous MccJ25 that may enter in the producing bacteria.33 Two promoters were found in the mcjA–mcjBintergenic region. The first one, PmcjA, directs the transcription of the structural gene, whereas PmcjB directs the transcription of the three other genes, in the opposite direction.23,35
2.2 Class II microcins
In class II microcin gene clusters, at least two genes are involved in export. This set of genes, which are homologous among class II microcins, requires the chromosomally located tolC to be functional.4,36–39
2.2.1 Class IIa microcins: MccV, MccL and Mcc24. Class IIa microcin gene clusters are composed of only four plasmid-borne genes, which are organized in a similar fashion.
MccV was the first antibiotic substance reported to be produced by E. coli.40 This antibacterial agent was initially named colicin V (ColV).41 However, on account of several characteristics (low molecular mass, non-inducible production, and dedicated export system), it became obvious that ColV should be classified within the microcins.36,42 In this review, we therefore propose to name it MccV, but the reader should keep in mind that most of the literature on this microcin uses the ColV terminology. MccV is secreted by various E. coli strains harbouring large (>80 kb), low-copy number pColV plasmids.43 A 4.2-kb DNA fragment from the 144-kb pColV-K30 plasmid is required for MccV production, export and self-immunity. Four genes distributed in two converging operons have been identified (Fig. 1B).36,44,45 The structural gene cvaC, encoding the 103-aa MccV precursor, and the self-immunity gene cvi form the first operon. The dedicated export system of MccV has been well characterized46–48 and involves two genes that form the second operon.36 The gene cvaA encodes a protein anchored at the inner membrane with a C-terminal region extending into the perisplamic space.49 The gene cvaB encodes an inner membrane ABC transporter. Two promoters were identified upstream of cvaA and downstream of cvi.50 The nucleotide sequence analysis of 12 MccV-producing plasmids isolated from natural E. coli strains revealed a low level of polymorphism in the 683 bp cvaC–cvi region,51 which suggests a strong stability of the MccV gene cluster.
MccL is produced by the E. coli LR05 strain isolated from poultry intestine.12 This isolate also expresses MccB17, MccD93 and MccJ25.52 Sequencing of pL102, which results from the cloning of the DNA conjugative plasmids of E. coli LR05, showed that the MccL gene cluster (Fig. 1B) consists of four genes encoding the 105-aa MccL precursor (mclC), the microcin self-immunity protein (mclI), and the microcin export proteins (mclA and mclB). The genes mclA and mclB are highly homologous (99% and 96% identity) to cvaA and cvaB encoding the MccV export system. The concomitant expression of MccV self-immunity and precursor genes inferred that the two genes are grouped in an operon.39 Furthermore, mclA and mclB are translated from the opposite strand and probably form a second operon. Downstream of mclI, two ORFs were identified. Surprisingly, the first encodes a 27-aa peptide whose first 15 amino acids are identical to MccV leader peptide . The second exhibits 98% identity with the MccV self-immunity gene cvi, and makes the MccL-producing strain resistant to MccV.52
Mcc24 (formerly colicin 24) is secreted by the uropathogenic E. coli strain 2424, and its genetic determinants are located on the 43.5-kb conjugative plasmid p24-2.53 A 5.3-kb DNA fragment from the pGOB18 recombinant plasmid was sequenced (O'Brien and Mahanty, 1996, unpublished work). Analysis of the nucleotide sequence (EMBL database accession no U47048) revealed that Mcc24 gene cluster (Fig. 1B) contains the following genes: mtfS, which encodes the probable 90-aa Mcc24 precursor; mtfI, which encodes the self-immunity protein ; mtfA and mtfB, which encode proteins similar to the MccV export proteins , CvaA and CvaB. In contrast with MccV and MccL, the four genes apparently form a single operon.
2.2.2 Class IIb microcins: MccE492, MccH47, MccI47 and MccM. Unlike the previously described microcins, which are all plasmid-encoded, the class IIb microcins are chromosomally encoded. In addition, their gene clusters show a complex transcriptional organization.
MccE492 is secreted by Klebsiella pneumoniae RYC492, a strain isolated from human faeces.54 A 13-kb DNA fragment containing the entire MccE492 gene cluster was cloned to raise the pJAM434 recombinant plasmid.55 Ten genes (mceA to mceJ) are necessary for MccE492 production, export and self-immunity (Fig. 1C).38 The structural gene mceA encodes the 103-aa MccE492 precursor, and mceB is involved in the self-immunity towards MccE492.56 The genes mceC, mceD and mceI, which encode proteins homologous to a glycosyltransferase, a ferric enterobactin esterase, and an acyltransferase, respectively, are required for MccE492 post-translational modifications.57 The gene mceJ would also be involved in the maturation process but its exact role remains to be elucidated.38,58 Two other genes, mceG and mceH, are required for the export of MccE492. They encode an ABC transporter and an accessory protein , respectively. The gene mceF would also be involved in export.38 The role of the last gene, mceE, remains unknown. The ten genes are organized in at least six transcriptional units, which is unusual for a bacterial gene cluster.38 The gene mceA is transcribed with the self-immunity gene mceB, while mceC, mceD, mceE, and mceF are all transcribed as monocistronic single units. Finally, mceGHIJ are organized in a polycistronic operon, but mceGH may also be transcribed as a bicistronic unit.38 Amazingly, the orientation of mceGHIJ on the chromosome of K. pneumoniae RYC492 is opposite to that of the homologous mchCDEF from MccH47 and MccM gene clusters (see below). MccE492 is also produced by E. coli harbouring the pJAM229 recombinant plasmid. This plasmid differs from pJAM434 by an inverted orientation of the 6.9-kb XhoI fragment that contains mceGHIJ. However, both plasmids are reported to express MccE492.55
Since MccH47, MccI47 and MccM gene clusters are closely interwoven, they are described simultaneously. MccH47 was initially detected in culture supernatants of E. coli H47 strain, isolated from human faeces.59 MccM is secreted by the nonpathogenic E. coli Nissle 1917 (DSM 6601) isolated from human faeces. This strain, also named Mutaflor,60 is used as a probiotic agent for the treatment of various intestinal diseases.61–63 Initially described as colicin X,64 the antibacterial substance was recently renamed MccM after the name Mutaflor.65 MccM and MccH47 are both secreted by E. coli CA46 and CA58 strains,65 which were initially described as producers of colicins G and H.66,67
The genetic determinants required for MccH47 production, export and self-immunity are all located within a 10.5-kb DNA fragment on E. coli H47 chromosome59 (Fig. 1C), and eight genes were first identified. These include mchA–mchF, mchI, and mchS1, which is located in a 3-kb silent region neither involved in MccH47 production nor in self-immunity.37 The gene mchB encodes the 75-aa MccH47 precursor68 and mchI confers the self-immunity towards MccH47.69 The genes mchE and mchF, which exhibit high homology (96.3% and 90% identity) to cvaA and cvaB, respectively, are probably involved in MccH47 secretion.37,70 The genes mchA, mchS1 and mchD, which are homologous to mceC, mceD and mceI, respectively, are presumably involved in MccH47 post-translational modifications. The gene mchC, which is homologous to mceJ, is necessary for the activity of the microcin, but its precise function is unknown. Upstream of mchI, mchX was later identified. It encodes a 39-aa peptide that may be involved in the regulation of mchI and mchB expression.69 More recently, three other genes were identified in the silent region.71 The gene mchS2 encodes the 77-aa precursor of a new microcin termed MccI47, and mchS3 confers the specific self-immunity towards MccI47.72 Additionally, mchS4, which was found to be responsible for the overproduction of the catecholate siderophore enterobactin,71 encodes an 81-aa protein whose role is unclear. Downstream of mchF, three truncated genes (mcmI, mcmA, mcmM) were also found (see below).4,72
Analysis of microcin gene clusters on genomic island I from E. coli Nissle 191773 and E. coli CA46 and CA58 genomes4,65 showed that these gene clusters direct the synthesis of both MccM and MccH47. The three gene clusters share a common organization except for the 5′ region located upstream of mchX (Fig. 1C). Three MccM-specific genes were identified in the 3′ region. The gene mcmI encodes the MccM self-immunity protein , mcmA (formerly mcmC),4 encodes the 92-aa MccM precursor, whereas mcmM, which is transcribed in the opposite direction, encodes a protein similar to MceF (62% identity over 176 residues).65 The MccM secretion does not involve specific genes and is probably carried out by mchE and mchF gene products. The minimal region necessary for the MccM production65 is carried by mchDEF and mcmIA. In E. coli CA46 and CA58, two additional genes, mcmL/mcmK, were identified upstream of mchX4 (Fig. 1C). They are homologous to mchA/mchS1 and mceC/mceD, and are probably involved, together with mchD, in MccM post-translational modifications. Moreover, in the MccM/MccH47 gene cluster from E. coli CA46 (Fig. 1C), three genes, mchS2S3S4, are present between mcmK and mchX. The gene mchS2 encodes a 77-aa protein that differs from the MccI47 precursor by only one amino acid substitution (glycine for alanine in position 22). Thus, the E. coli CA46 strain could secrete a third microcin. Similar features are observed in the MccM/MccH47 gene cluster from E. coli CA58 (Fig. 1C). Downstream of mcmK, a putative mchS2 is also found when the undetermined nucleotide N, located at position 3452, is deleted. Nevertheless, an additional 1.3-kb DNA fragment, composed of genes encoding transposase and insertion sequences,4 is inserted upstream of mchS3. The MccM/MccH47 gene cluster from E. coli Nissle 1917 differs from those of E. coli CA46 and CA58 mostly by the absence of mcmL and mcmK65 (Fig. 1C). A MccM/MccH47 gene cluster identical to that of E. coli Nissle 1917 is also encountered with 100% nucleotide sequence identity in the serX pathogenicity island of the uropathogenic E. coli strain CFT073.74 Moreover, a partial MccM gene cluster including the 3′ region of mchF and mcmIAM is also encountered in the pathogenicity island II from E. coli strain 478775 and in the pathogenicity island III from E. coli strain 536.76 The presence of genes encoding transposase and insertion sequences, which are known to be involved in genetic recombination, strongly supports the hypothesis of a MccM/MccH47 gene cluster exchange between bacteria.
The G+C contents of all Mcc gene clusters, which range from 33.1% to 43.2% (Table 2), are lower than those of their bacterial host genomes (about 51% for various E. coli and 57.5% for K. pneumoniae MGH78578 strain). Thus, these bacteria would appear not to be the original hosts of microcin gene clusters. Moreover, partial or complete MccM gene clusters are encountered in genomic or pathogenicity islands. Such structures represent a large group of mobile elements that contribute to microbial evolution (for reviews, see Hacker et al.77 and Dobrindt et al.78). Altogether, the identification of short direct repeats flanking the MccC51 and MccH47/I47 gene clusters29,72 (Fig. 1), the location of some gene cluster in genomic islands, and the G+C content strongly suggest the possibility of a horizontal transfer of genes responsible for the microcin biosynthesis.
3 Purification, structures and antibacterial activity
Elucidation of microcin structures, which include in many cases complex and unusual post-translational modifications, requires the optimization of culture conditions and purification protocols in order to isolate substantial amounts of highly purified microcins. These time-consuming and difficult (but necessary) steps have often hampered the elucidation of many microcin structures and the reliable determination of their antibacterial activities. Indeed, while all microcins show a potent antibacterial activity specifically directed against enterobacteria, the literature often reports the activity of the producing strains instead of the purified microcins. Since a number of microcinogenic strains were described as producing several microcins52,65,72 or other antibacterials such as colicins, old published data should be interpreted with the greatest care. Because all microcins have not been purified to homogeneity nor accurately quantified prior to antimicrobial assays, quantitative measurements leading to MICs and minimal bactericidal concentrations (MBCs) are rarely available. Finally, MICs and MBCs are rarely comparable due to differences in the experimental protocols used.3.1 Class I microcins: MccB17, MccC7/C51, MccJ25
Class I microcins have the lowest molecular masses, ranging from 1 to 3 kDa, and display extensive post-translational modifications of their peptide backbone. Thus, MccB17 is a 43-residue peptide characterized by the presence of thiazole and oxazole rings, MccC7/C51 is a nucleotide –heptapeptide, and MccJ25 is a 21-residue cyclic peptide that adopts a particular lasso three-dimensional structure.MccB17, as purified for structure determination, was produced by E. coli BM21 cells harbouring the pMM39 plasmid, and grown in tryptone–yeast extract medium. The purification protocol included an initial hot acid extraction step of the harvested cells at pH 2.9 and 100 °C for about 10 min to ensure complete extraction from the cells.79 The following steps consisted of size-exclusion chromatography and reversed-phase high performance liquid chromatography (RP-HPLC). MccB17 was reported to display potent bactericidal activity against a wide range of Gram-negative bacteria including Escherichia, Citrobacter, Klebsiella, Salmonella, Shigella and Pseudomonas.1,80 MccB17 was shown to derive from a 69-aa precursor, McbA (Fig. 2), endowed with an atypical 26-residue leader peptide . Mature MccB17 carries four oxazole and four thiazole rings that derive from the unusual post-translational modification of six glycines, four serines, and four cysteines spanning residues 39–66 of the MccB17 precursor. Those rings are formed by the reaction of serine and cysteine side-chains with the carbonyl groups of the preceding glycine in the peptide chain79,81 (Fig. 2). This complex structure was elucidated by Jung and collaborators in 1995, through the combined use of UV spectroscopy, mass spectrometry (MS), amino acid analysis, Edman sequencing and, above all, multi-dimensional nuclear magnetic resonance (NMR) applied to unlabelled and stable isotope-labelled samples.79 The complete structure of the microcin was indeed deduced from a detailed analysis of the data arising from homo- and heteronuclear multi-dimensional NMR and triple resonance experiments performed on the 13C/15N doubly labelled MccB17. Two isolated thiazole and oxazole rings and two adjacent bis-heterocyclic systems consisting of directly linked oxazole–thiazole and thiazole–oxazole entities were characterized. They were found to result from two Gly-Cys and two Gly-Ser dipeptides on the one hand, and from Gly-Ser-Cys and Gly-Cys-Ser tripeptides, respectively, on the other hand. This unique structure was fully confirmed through the total synthesis of MccB17.82 The three-dimensional structure of MccB17 has never been described until now.
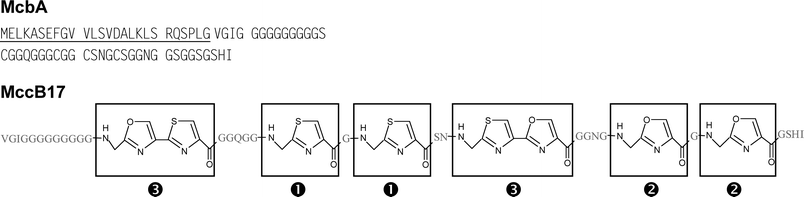 | ||
| Fig. 2 Sequence of the MccB17 precursor (McbA) and structure of mature MccB17. The 26-residue leader peptide is underlined in McbA. The amino acids are figured in grey for mature MccB17. In MccB17, thiazoles (1), oxazoles (2), and bis-heterocycles (3) are boxed. | ||
MccC7 was isolated from E. coli MC4100 harbouring the pMM550 plasmid and cultivated in M63 medium, using a protocol associating size-exclusion and RP chromatographies.83 MccC51 was purified from culture supernatants of E. coli TG1 harbouring either the pUHAB or pBM43 plasmid and grown in M63 minimal medium. The purification protocol involved solid-phase extraction and subsequent RP-HPLC.84,85 The spectrum of activity of MccC7 and MccC51 was reported to cover several genera of enterobacteria, such as Escherichia, Enterobacter, Klebsiella, Salmonella, Shigella, Proteus and Yersinia.27,86,87 MccC7 and MccC51 were shown to have an identical structure (see below). They were characterized as an N-formylated heptapeptide, which also contains a modified adenosine monophosphate (AMP) covalently attached to the C-terminal Asp through a phosphoramide bond. Conversely to mccA, which encodes an Asn as the seventh amino acid, MccC7/C51 heptapeptide ends with an Asp (Fig. 3). The phosphoramidate group, substituted by an n-aminopropanol chain, subsequently contains a chiral phosphorus atom. This is the only microcin known to carry a nucleotide as post-translational modification. The structure was identified for MccC7 by Delepierre and collaborators in 1995.83 The same year, the structure of MccC51 was published.88 The structures of MccC7 and MccC51, based on NMR studies, differed in both the linkage between the peptide and nucleotide parts and the nucleotide structure itself. MccC51 was described as a nebularin 5′-monophosphate C-terminal entity linked to the Asp7 side-chain through three methylene bonds. In 2000, the structure of MccC51 was re-investigated in our group. By a combined hetero- and homonuclear NMR study, we determined that the structure of MccC51 was actually identical to that of MccC7.29,84 In particular, the presence of a phosphoramide bond acting as a linker between the heptapeptide and the nucleotide and the location of the n-aminopropanol chain, which were the two critical points of the structure, were unambiguously assigned in MccC51 through typical cross-peaks in two-dimensional 1H–31P NMR heteronuclear single quantum coherence spectra.84 Therefore, MccC7 and MccC51, which arise from two distinct E. coli strains, share a common nucleotide –peptide structure (Fig. 3), disclosing the first observation of two closely related microcins. At the present time, the three-dimensional structure of MccC7 and MccC51 remains unknown. It is worth noting that because of their common structure, MccC7 and MccC51 have been occasionally termed MccC.23,85 However, this terminology is also used for the mccC gene product. We therefore prefer to use the MccC7/C51 terminology, which avoids this confusion. Interestingly, the secreted microcin undergoes activation by proteolytic cleavage inside the susceptible bacteria (Fig. 3).85 To distinguish these two forms of microcin, we propose to term MccC7/C51* the intracellularly processed MccC7/C51.
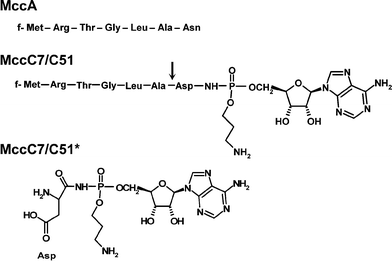 | ||
| Fig. 3 Sequence of MccA and structures of MccC7/C51 and MccC7/C51*. Formylation of Met1 is indicated by an ‘f’ in the sequences. MccC7/C51 is the antibacterial peptide secreted by the producer, whereas MccC7/C51* is the translation inhibitor generated by cleavage of MccC7/C51 within susceptible cells. The arrow indicates the cleavage site of MccC7/C51. | ||
MccJ25 was efficiently purified from culture supernatants of E. coli MC4100 harbouring the pTUC202 plasmid, and grown in M9 or M63 minimal medium. Thus, as MccC7/C51, MccJ25 was isolated by solid-phase extraction and further RP-HPLC.89 The spectrum of MccJ25 antibacterial activity was found to be restricted to few genera of enterobacteria, mainly Escherichia and Salmonella, with MICs in the 2–5 nM range.89,90 MccJ25 was shown to be bactericidal against strains of E. coli and S. enterica, serovars Enteritidis and Paratyphi.90 MccJ25 is generated from a 58-aa precursor, McjA (Fig. 4A). Mature MccJ25 is a 21-residue hydrophobic peptide that displays a three-dimensional lasso-type structure (Fig. 4B). MccJ25 cyclization results from the linkage between the N-terminal Gly1 amino group and the Glu8 side-chain carboxylate, leading to a small ring (Fig. 4B). The resulting 13-residue linear C-terminal tail is entrapped into this ring through (i) non-covalent interactions, and (ii) steric hindrance by the two bulky aromatic side-chains from Phe19 and Tyr20, straddling each side of the ring (Fig. 4B). The tail can only be released by cleavage of the ring. This structure was identified simultaneously by three groups using combined MS and NMR studies.91–93 However, similar to MccC7/C51, the structure of MccJ25 has been a subject of debate in the literature.94 MccJ25 was first isolated in 1992 by Salomón et al.,32 and characterized as a 20-residue hydrophobic peptide with a blocked N-terminal end. It was further shown in our group to be a 21-residue head-to-tail macrocyclic peptide .89,95 Re-investigation of the structure in 2003 showed that the cycle actually engaged Glu8 side-chain carboxylate instead of Gly21 one.91–93 The MccJ25 lasso structure was shown to be required for optimal antibacterial activity96 and to be responsible for MccJ25 high stability. Indeed, MccJ25 retained both its three-dimensional structure and its antibacterial activity at 165 °C, as well as up to 95 °C in the presence of potent denaturing agents.96 MccJ25 structure is also resistant to proteolysis. The loop can be enzymatically opened by thermolysin at the Phe10–Val11 amide bond (Fig. 5), or can be targeted by a strong acidic medium (Fig. 5).96,97 However, the initial lasso structure is not destroyed during these processes and the resultant entities are two-chain peptides , the tail (or a shortened tail) remaining firmly anchored to the ring, both in solution and in gas phase, as shown by NMR and MS studies.97 In fact, ring cleavage can only be accomplished by partial hydrolysis in basic medium.93 Such an original structure had never been identified previously among antibacterial peptides . Nevertheless, either additionally stabilized or not by disulfide bond(s), similar lasso-type structures had already been encountered among enzyme inhibitors synthesized by Streptomyces species.94,98–100 Recently, such a structure was also found in lariatins from Rhodococcus sp.101
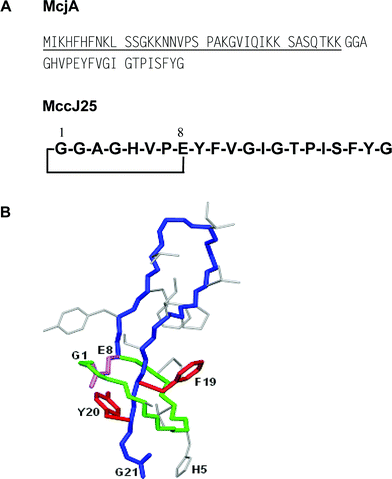 | ||
| Fig. 4 (A) Sequence of the MccJ25 precursor (McjA) and structure of mature MccJ25. The 37-residue leader peptide is underlined. (B) Three-dimensional structure of MccJ25. Note the steric hindrance imposed by Phe19 and Tyr20, which strongly contribute to the blocking of the C-terminal tail into the ring. | ||
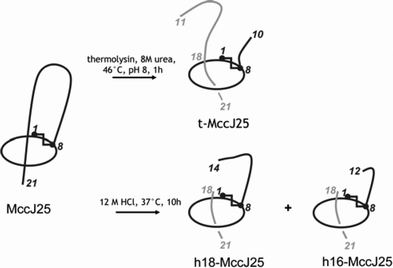 | ||
| Fig. 5 Structures of the two-chain peptides generated from MccJ25. t-MccJ25 is obtained by thermolysin cleavage of MccJ25, which breaks the lasso structure between Phe10 and Val11. h18-MccJ25 and h16-MccJ25, which contain 18 and 16 amino acids, respectively, are obtained by hydrochloric acid cleavage of MccJ25. The cleaved fragments (in grey) remain tightly attached to the main peptide chain (in black), which contains the cycle. | ||
3.2 Class II microcins
Class II microcins are higher molecular mass microcins (4.9 to 8.9 kDa). Their peptide backbones do not undergo extensive modifications. Besides disulfide bonds, they may carry a siderophore-type post-translational modification.
3.2.1 Class IIa microcins: MccV, MccL and Mcc24. MccV was isolated, purified and characterized in 1994 by Kolter and collaborators102 simultaneously to Håvarstein and collaborators.103 From these studies, MccV can be isolated from culture supernatants of E. coli MC4100 harbouring the pHK11 or pHK22 plasmid. Since the MccV gene expression is repressed by excess iron, the strains were grown on tryptone broth or Luria broth containing the iron chelator 2,2′-dipyridyl. MccV purification used a four-step procedure involving trichloroacetic acid or ammonium sulfate precipitation, amberlite XAD16 absorption, cation exchange chromatography and RP-HPLC. MccV showed an antibacterial activity directed against related Gram-negative bacteria with an MIC of about 0.1 nM against E. coli.103 Expressed as a 103-aa precursor, the mature MccV is an 88-aa peptide (Table 1), without post-translational modification, which possesses a single disulfide bond connecting Cys76 to Cys87 (Fig. 6A).103
 | ||
| Fig. 6 (A) Sequences of the MccV precursor (CvaC) and of mature MccV. (B) Sequences of the MccL precursor (MclC) and of mature MccL. The 15-residue leader peptides are underlined. Disulfide bonds are shown as black lines. | ||
MccL was isolated from the supernatant of the wild-type producer E. coli LR05 grown in M63 medium, and its purification mainly used RP-chromatography.12 MccL was reported to be active against Shigella sp., several E. coli including diarrheagenic strains, Pseudomonas sp. and several Salmonella enterica strains, including serovars Enteritidis and Typhimurium, with MICs in the nanomolar range.39 MccL, which is generated from a 105-aa precursor, is composed of 90 unmodified amino acids.39 It is a glycine-rich, anionic, and highly hydrophobic peptide (46.7% non-polar amino acids) (Table 1, Fig. 6B). MccL shares an identical 13-aa C-terminal sequence with MccV, which is believed to be folded by a disulfide bond connecting Cys78 and Cys89 in MccL.39 However, MccL possesses an additional disulfide bond connecting Cys29 to Cys33. Homology searches show a strong similarity between the 32 C-terminal amino acids of MccL and MccV (87.5% identity). Moreover, in the region surrounding the first disulfide bond (Ile20–Ala38), MccL exhibits significant similarity (43–52% identity) with lafA subunit from lactacin F104 and gassericin T,105 two non-lantibiotic bacteriocins from Lactobacillus.
Mcc24 has neither been isolated nor characterized. Although its precursor sequence makes it undoubtedly a class II microcin (Table 1, Fig. 7), it is difficult to classify it within class IIa/IIb. Indeed, it does not contain any cysteine and lacks the C-terminal sequence found in other class IIa microcins (Fig. 6). Similarly, the Mcc24 precursor displays major homologies (50% identity) with MccE492 precursor (Section 5.1.1), but lacks the 10-aa C-terminal sequence typical of class IIb microcins. Therefore, Mcc24 appears atypical among class II microcins. Because the structure of Mcc24 does not allow its classification, we have considered that its gene cluster, which contains four genes only, makes it belong to class IIa.
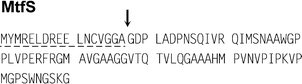 | ||
| Fig. 7 Sequence of the Mcc24 precursor (MtfS). The putative leader peptide is underlined with a dashed line. The putative cleavage site of Mcc24 precursor, whose location is based on multiple alignment of class II microcin precursors deduced from their DNA sequences (Section 5.1.1; Fig. 10), is indicated by an arrow. | ||
3.2.2 Class IIb microcins: MccE492, MccM, MccH47 and MccI47. Class IIb microcins are devoid of disulfide bonds. All of them have a conserved serine-rich C-terminal region and they may carry a siderophore-type post-translational modification.
MccE492 was purified from culture supernatants of E. coli strains harbouring the recombinant pJAM229 plasmid.55 Culture conditions were found to be critical to obtain fully mature MccE492.14 Basically, MccE492 should be expressed under iron-poor conditions, M63 minimal medium being appropriate, and in the absence of free aromatic amino acids (unpublished work). Indeed, the use of casamino acids should be prevented, since it led to unmodified MccE492 (termed here u-MccE492), which we have recently shown to be an incompletely processed microcin (unpublished work). Efficient purification of both MccE492 and u-MccE492 can be achieved by solid-phase extraction followed by RP-HPLC.14,106 Both MccE492 and u-MccE492 were found to be bactericidal, mainly against E. coli strains. However, upon complete maturation, MccE492 activity increased by 4–8 fold (MICs in the 40–80 nM range) and its spectrum of activity extended to K. pneumoniae and Enterobacter cloacae.14 MccE492 is generated from a 103-aa precursor, MceA, by elimination of a 19-aa leader peptide .56 This cleavage results in the release of u-MccE492, the unmodified form initially characterized by Pons et al.107 (Table 1, Fig. 8). Fully mature MccE492 (formerly termed MccE492m) is a siderophore-peptide that carries a linear trimer of N-(2,3-dihydroxybenzoyl)-L-serine (DHBS) anchored at the C-terminus (Ser84) through a β-D-glucose.14 This structure was determined in our group by subjecting the 11-residue C-terminal fragment of MccE492 to ion trap MS and high field two-dimensional 1H–13C NMR. The β-D-glucose was shown to be linked to the Ser84 carboxylate through an O-glycosidic bond at C6, and to the first DHBS entity through a C-glycosidic bond at C1 (Fig. 8).14 The amino acids composing MccE492 are mainly uncharged and hydrophobic, with the exception of one histidine, three aspartic acids and one glutamic acid (Table 1) that give an anionic character to this microcin. MccE492 post-translational modification mimics siderophores (i.e. molecules designed by bacteria to chelate ferric iron, enabling its uptake across the bacterial outer membranevia specific receptors (Section 7.1.1), and particularly salmochelins.108 Indeed, we showed by MS that mature MccE492 selectively binds ferric iron through its catecholate moieties. This makes MccE492 the first natural siderophore-peptide to be described.14
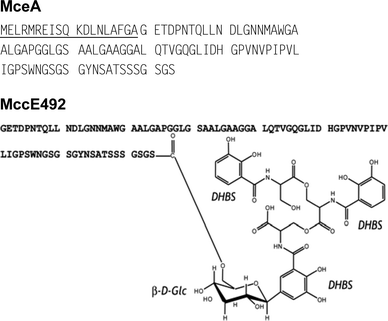 | ||
| Fig. 8 Sequence of the MccE492 precursor (MceA) and structure of mature MccE492 carrying the siderophore post-translational modification. The leader peptide is underlined. Glc and DHBS stand for glucose and N-(2,3-dihydroxybenzoyl)-L-serine, respectively. | ||
E. coli Nissle 1917 was shown to produce two bactericidal activities attributed to MccH47 and MccM,65 but none of these microcins had been isolated until now. Very recently, MccM was isolated and purified in our group (unpublished work) from E. coli MC4100 transformed with the pMM75 plasmid (Moreno and collaborators, unpublished work). MccM was produced in M63 medium and purified by a protocol similar to that used for MccE492 purification.14 No spectrum of activity is available at this time. MccM is a 77-aa peptide generated from a 92-aa precursor, McmA (Table 1, Fig. 9A). This was recently shown in our group (unpublished work) by MS, Edman sequencing and analysis of MccM trypsin digest. Matrix-assisted laser desorption/ionization time-of-flight MS data also suggested that MccM secreted by E. coli harbouring the pMM75 plasmid carries a post-translational modification similar to that characterized for MccE492. Interestingly, although MccM appears to belong to class IIb microcins, it exhibits 34% identity with MccV.
 | ||
| Fig. 9 (A) Sequence of the MccM precursor (McmA). The leader peptide is underlined. Similar to mature MccE492, MccM can be modified by a siderophore moiety linked to the C-terminal serine. (B) Sequences of the precursors of MccH47 (MchB) and MccI47 (MchS2). The putative leader peptides are underlined with dashed lines. The putative cleavage sites of MccH47 and MccI47 precursors, whose location is based on multiple alignment of class II microcin precursors (Section 5.1.1; Fig. 10), are indicated by an arrow. | ||
Difficulties in isolating MccH47 have hampered the characterization of its primary structure. The recently discovered MccI47 has never been isolated either. The structures of both microcins were predicted by the genetic studies of Laviña and collaborators.37,59,72 On the basis of their deduced amino acid sequences (Fig. 9B), both microcins are believed to display the highly conserved C-terminus found in MccE492 and MccM (Fig. 10, Section 5.1.1). Because MccE492, and probably MccM (see above), carry a catechol-type siderophore on this conserved C-terminal region, a similar post-translational modification could occur in MccH47 and MccI47. As discussed earlier,57 this hypothesis is reinforced by (i) highly conserved genes believed to encode modification enzymes in MccE492, MccM/MccH47, and MccH47/MccI47 gene clusters (Fig. 1C, Section 2.2.2), and (ii) the requirement of all four microcins (MccE492, MccM, MccH47, MccI47) for catechol-type siderophore receptors at the outer membrane of E. coli (Section 7.1.1).
 | ||
| Fig. 10 Multiple alignment of N- and C-terminal regions of class II microcin precursors. The sequences of the central regions are not aligned. Their lengths in amino acids (aa) are given in italics. Alignments were performed with Multalin122 and improved manually. Dashes indicate gaps. All the amino acid sequences are from the Swiss-Prot Database. CvaC (accession no P22522), MclC (accession no Q841V4), MtfS (accession no Q46971), MceA (accession no Q9Z4N4), MchB (accession no P62530), McmA (accession no Q83TS1) and MchS2 (accession no Q712Q0) correspond to MccV, MccL, Mcc24, MccE492, McH47, MccM and MccI47 precursors, respectively. The arrow indicates the known or putative cleavage site of microcin precursors. The extra 4 and 2 residues in the N-terminal region of MceA and MtfS, respectively, are in italics (see Section 5.1.1). | ||
It thus appears that mature microcins share common structural features, such as high hydrophobicity and high content in glycine and serine (Table 1). Most of the isolated microcins are devoid of cysteine residues, except the class IIa microcins MccV and MccL, which show 1 and 2 disulfide bonds, respectively. All 4 cysteine residues present in MccB17 precursor are modified to heterocyclic rings in the mature microcin. Most microcins are anionic peptides , except MccB17, MccV and putatively Mcc24, which are slightly cationic. Within class I microcins, no similarity can be highlighted, but low molecular mass and extensive backbone modification. Alignment of class II microcins (Section 5.1.1, Fig. 10) shows strong similarities in the C-terminal region of the class IIa MccL and MccV on the one hand, and of the class IIb MccE492, MccM, MccH47 and MccI47 on the other hand. However, similarities between members of class IIa and IIb are also underlined.
4 Export machinery
The machinery in charge of microcin secretion into the extracellular medium was identified either by genetic or homology analysis. In many cases, this machinery appears to be associated either to self-immunity or to proteolytic cleavage of the promicrocin.There is no standard export machinery for class I microcins. MccB17 export has been shown to be driven by McbF and McbE,18 which are related to an ABC transporter and its accessory protein , respectively. McbF is predicted to contain a nucleotide -binding domain and McbE to span the inner membrane. For this reason, McbF and McbE could serve as a pump to export MccB17 to the perisplasmic space.18 However, the presumed ABC transporter involved in MccB17 export does not show any proteolytic domain, as class II microcins ABC transporters do. Moreover, the outer membrane component required for MccB17 export across the outer membrane remains unidentified. The mechanism of MccC7/C51 secretion is unclear. It involves MccC, a hydrophobic protein that shows significant similarity to multidrug efflux transporters belonging to the major facilitator superfamily (MFS).28,29 MFS transporters export small solutes only, such as sugars and secondary metabolites (for a review, see Pao et al.109). This efflux mechanism would be consistent with the low molecular mass of MccC7/C51. To date, three proteins involved in MccJ25 export have been identified. McjD, whose sequence contains a putative transmembrane domain and a C-terminal domain similar to nucleotide binding domain, was classified as an ABC transporter.34 It would be responsible for MccJ25 efflux, a mechanism by which it would also confer self-immunity to the producing strain. McjD would work together with the chromosomally encoded TolC.110 This outer membraneprotein forms a trimeric channel in the outer membrane, extending from the extracellular side of the outer membrane, through most of the periplasmic space, to finally end close to the periplasmic side of the inner membrane.111 Another chromosomally encoded protein of E. coli, YojI, which is also similar to ABC transporters, was found to protect TolC-expressing bacteria against MccJ25.112 McjD and YojI display the same ABC transporter features, and could play the same role in MccJ25 export, i.e. form a complex with TolC, with or without a supplementary accessory protein .
Class II microcin export machinery displays a canonical structure. It consists of three components, two of which, the ABC transporter and the accessory protein , are encoded by the microcin gene cluster. Table 3 summarizes the percentage identities of class II microcin export machineries. The CvaB protein from MccV export machinery and the highly homologous MclB, MtfB, MceG and MchF proteins responsible for the export of MccL, Mcc24, MccE492 and MccH47/I47/M, respectively, are all similar to ABC transporters (for reviews, see Jones and George113 and Holland et al.114) responsible for the export of other antibacterials such as class II bacteriocins from Gram-positive bacteria115 and RTX toxins .116 These ABC transporters possess three domains. Besides a central, poorly conserved, transmembrane domain, they contain (i) an N-terminal domain (about 130 amino acids), which has a protease activity and would be located in the cytoplasm of bacteria,117,118 and (ii) a C-terminal domain, which contains a highly conserved nucleotide -binding cassette required for ATP binding.119 A model in which binding of the microcin promotes the transition of the ABC transporter from an inactive dimer bound to nucleotide diphosphate to an active high-energy dimer bound to nucleotide triphosphate has been proposed for MccV.48 This energized state would enable the transmembrane domain of the protein to form a channel in the inner membrane. The second component of the export system is referred to as the accessory protein . It is predicted to be a periplasmic protein anchored at the inner membrane by an N-terminal transmembrane helix.49,120 In class II microcins, accessory proteins are CvaA from the MccV export machinery, as well as MclA, MtfA, MceH and MchE for MccL, Mcc24, MccE492 and MccH47/I47/M, respectively. As ABC transporters, the accessory proteins are also highly conserved (Table 3). Although their role in secretion is still unclear, they may serve as connectors to the outer membraneprotein TolC, which is the third component of the class II microcin export machinery.36–39 TolC is believed to enable the secretion of the microcins by forming a continuous channel from the cytoplasm to the extracellular medium. The MccE492 export machinery seems to require another protein encoded by the microcin gene cluster, MceF. This putative inner membrane protein could interact with MceGH for processing or export.38 Similarly, McmM which is homologous to MceF (61.6% identity), would be involved in MccM processing or export.
5 From genes to structures: biosynthesis and regulation
5.1 Microcin precursors, the promicrocins
Similar to bacteriocins from Gram-positive bacteria,5,121 microcins generally derive from a precursor, the promicrocin (Table 1). This latter consists of (i) a C-terminal structural region and (ii) an N-terminal leader peptide comprising 15 to 37 residues.22 As previously mentioned, MccC7/C51 is the only one to be secreted by the producing strain without previous cleavage of a longer precursor.25,27
5.1.1 Biochemical characteristics and conserved domains. Class I microcin precursors do not display common features. MccB17 and MccJ25 have long leader peptides (26 and 37 amino acids) relative to the size of their precursor (69 and 58 amino acids, respectively). The processing site of both microcins was accurately determined by isolation of the mature peptide.20,89 As with several other microcins from class II (see below), processing of the MccB17 precursor occurs C-terminal to a glycine (Fig. 2). However, this residue is not involved in a double-glycine or glycine–alanine motif, and the leader has not the typical sequence of double-glycine-type leader peptides , as do class II microcin precursors (see below). In MccJ25, cleavage occurs C-terminal to a lysine and N-terminal to a double-glycine motif (Fig. 4A).
Class II microcins are generated from large precursors carrying small conserved leader peptides . Indeed, alignment of class II microcin precursors using the Multalin program122 reveals that these microcins exhibit highly conserved leader peptides (Fig. 10). All seven microcin precursors, whose sizes range from 75 to 105 residues, have a 15–19-residue leader peptide . Because Mcc24 and MccE492 precursors derive from genes with two neighboring AUG codons, transcription may actually begin at a second AUG encoding Met3 and Met5 in Mcc24 and MccE492, respectively. Mcc24 and Mcc492 precursors would then be lacking the extra 2 and 4 residues in the N-terminal position, respectively. It is therefore likely that all class II microcin leader peptides , including those of Mcc24 and Mcc492, are 15 residues long. Leader peptides from class II microcins contain an M-R-X-[I/L]-X9-G-[A/G] (X denotes any amino acid) conserved sequence (Fig. 10), with a typical double-glycine motif (MccV, MccM and MccI47) or a glycine–alanine motif (MccL, Mcc24, MccE492, and MccH47), found as an alternative to the double-glycine motif in proteins exported through ABC transporters.115,123 Based on the N-terminal sequences of mature MccV,102 MccL,12 MccE492,107 and MccM (unpublished work), it is likely that Mcc24, MccI47, and MccH47 precursors are also processed after the double-glycine or the glycine–alanine motif.
5.1.2 Role of the leader peptide. A wide variety of functions have been proposed for N-terminal leader peptides from antimicrobial peptide precursors. The leader peptide could alternatively (i) ensure stabilization of the antibacterial peptide by preventing intracellular degradation of the produced peptide or its encoding DNA/RNA ,124 (ii) act as a chaperone folding the molecule so that it is recognized by the maturation machinery,125 or (iii) serve as a recognition sequence for the maturation and/or export machineries.126 Microcin leader peptides appear to achieve function(s) that differ from one microcin to another.
Several class I microcins do not require their leader peptide for export. Instead, it can be used for recognition by post-translational modification enzymes,127 as shown for MccB17. Indeed, following the formation of the oxazole and thiazole rings of various fusion peptides , Kolter and collaborators demonstrated that MccB17 leader peptide is essential for the post-translational modifications of MccB17, and serves as a prime determinant for the recognition and recruitment of the precursor by MccB17 synthetase.21,127 In addition, because exogenous MccB17 (lacking the leader peptide ) is pumped out by McbEF-expressing strains,18 it was proposed that the MccB17 leader peptide was involved in MccB17 recognition by the post-translational modification enzymes rather than by the export machinery. Although the MccJ25 precursor has been poorly studied until now, it is possible that the MccJ25 leader peptide would serve an identical function. Indeed, the McjD export protein confers resistance to exogenous MccJ25.33 This indicates that the MccJ25 leader peptide is not required for recognition by the export machinery.
Based on MccV studies, leader peptides from class II microcins could be involved in export. Indeed, the MccV leader peptide was shown to be an N-terminal export signal. Thus, while the 29 N-terminal residues of the MccV precursor are sufficient to promote translocation across the inner membrane, amino acids 30–38 contain the information affecting the efficiency of recognition/export of the protein .36 Conversely, deletion of 21 residues at MccV C-terminal end does not modify its secretion.102 Because of the sequence similarities described above, leader peptides of class II microcins, which do not resemble the typical N-terminal signal sequence-specific for Sec-dependent translocation,128,129 could nonetheless exhibit a common export recognition signal.
5.1.3 Antibacterial activity of promicrocins. Due to their self-immunity and export genes, microcin-producing bacteria are protected from the toxicity of their own microcin. However, several microcin precursors do not display antibacterial activity until processed. This is the case for class I microcin precursors.
Studies on the MccB17 precursor (McbA) revealed that compounds with six out of the eight heterocycles found in mature MccB17 are active, in contrast to McbA.130 This indicates that at least partial modification is required for antibacterial activity. Similarly, the synthetic heptapeptide moieties of both MccC7/C51 and MccA lack antibacterial activity.83 Moreover, it was recently shown that the secreted nucleotide –peptide remains inactive until proteolysis inside target cells.85 As with MccB17 and MccC7/C51, MccJ25 precursor (McjA) is devoid of antibacterial activity. This was recently demonstrated using an E. coli expressing recombinant McjA (Duquesne et al., unpublished work). Interestingly, the chemically synthesized 21-residue linear MccJ25 was not active either.96 This suggests that in the case of MccJ25, the antibacterial activity depends on the acquisition of the three-dimensional structure, rather than on the elimination of the leader peptide .
In contrast, class II microcin precursors could display some antibacterial activity. Indeed, bacteria only harbouring MccV structural (cvaC) and self-immunity (cvi) genes showed antibacterial activity in the lysates,44,131 suggesting that CvaC, the MccV precursor, possesses antibacterial activity. Interestingly, antibacterial activity was completely abolished upon alanine replacement of the two cysteines involved in the MccV disulfide bond.46 This indicates that, as with MccJ25, folding of MccV is required for antibacterial activity. Similar to MccV, lysates from bacteria only harbouring MccL structural (mclL) and self-immunity genes (mclI) were found to possess antibacterial activity,39 suggesting that unprocessed MccL is active. However, in contrast to MccV, the folding imposed by the two disulfide bridges seems not to be involved in MccL activity, since addition of high amounts of dithiothreitol does not abolish the antibacterial activity.39 Disulfide bonds were therefore proposed to be exclusively responsible for the high stability of mature MccL.39 Since production of the MccH47 precursor (MchB) was deleterious to the producing strain, MchB was proposed to display antibacterial activity.68 However, lysates from bacteria only harbouring the structural (mchB) and self-immunity (mchI) genes were inactive against MccH47-susceptible cells. Lack of activity was also observed in lysates of bacteria that do not express mchACD,68 three genes putatively involved in MccH47 production. This might be due to a dramatic decrease in MccH47 production, as also observed with u-MccE492 when mceC (homologous to mchA) is disrupted (unpublished work). The same lowered microcin production could explain why no antibiotic activity was detected for bacteria harbouring mchS2 (MccI47 structural gene) but disrupted mchA, mchC or mchD, the genes thought to be responsible for MccI47 production.72 The unprocessed MccE492 precursor (MceA) has never been isolated to date. However, the presence of the MccE492 siderophore post-translational modification increased the peptide potency against all tested E. coli and Salmonella strains and broadened its spectrum of activity.14
Altogether, these studies conclude that the class I microcin precursors are devoid of antibacterial activity. To gain their activity, extensive backbone post-translational modifications are required. In contrast, class II microcin precursors are thought to have some antibacterial activity. The gain of activity associated with the processing step was not directly measured. However, for MccE492 and MccV, antibacterial activity is significantly improved by subsequent post-translational modifications and/or folding.
5.2 Maturation of microcins
Maturation of microcins requires proteolytic enzymes that cleave the leader peptides from the promicrocins, and enzymes that ensure post-translational modifications.
5.2.1 Proteolytic cleavage of promicrocins. Although all promicrocins except MccC7/C51 require proteolytic removal of a leader peptide before secretion, little is known about the proteases involved. Nevertheless, it seems that proteolytic cleavage of class II microcin leader peptides occurs during export, and that it would result from the action of the export machinery. However, this does not apply to class I microcins, for which precursor processing appears to rely on a broader variety of mechanisms.
The best-studied class I microcin is MccB17. Its processing is carried out by chromosomally encoded protease(s). Indeed, mcbA–lacZ fusions expressed in mcbBCDEF-deficient strains were found to be cleaved from their 26-residue N-terminal leader peptide .132 A chromosomal gene called pmbA (or tldE) was shown to be involved in MccB17 maturation133 (Fig. 11). Subsequently, studies of tldE and tldD mutant strains harbouring mcbABCDEF demonstrated that modified McbA accumulated in those bacteria, indicating that MccB17 export machinery was not responsible for the cleavage of the leader peptide .134 MccC7/C51 is secreted as an uncleaved nucleotide –heptapeptide. However, it was recently shown to be cleaved after entry into the target bacteria to generate MccC7/C51*, a modified aspartyl adenylate (Fig. 3), which is the actual intracellularly active form of the microcin85 (Section 7.2.2). As for MccB17, cleavage of the last peptide bond in MccC7/C51 does not depend on any of the genes encountered on the microcin gene cluster. Indeed, in this case, the processing is performed in susceptible bacteria, and amazingly, it can also be carried out by peptidases from eukaryotic extracts.85 MccJ25 processing enzymes remain unidentified to date. Nevertheless, bacteria harbouring disrupted mcjB or mcjC genes are impaired in the production of mature MccJ25 (Duquesne et al., unpublished work). Therefore, and although neither McjB nor McjC are similar to known proteases, they are believed to be involved in MccJ25 processing rather than the self-immunity/export protein McjD or any other chromosomally encoded enzyme.34 However, which of these two proteins displays a proteolytic activity, and at what stage of the maturation this step happens, remain to be elucidated.
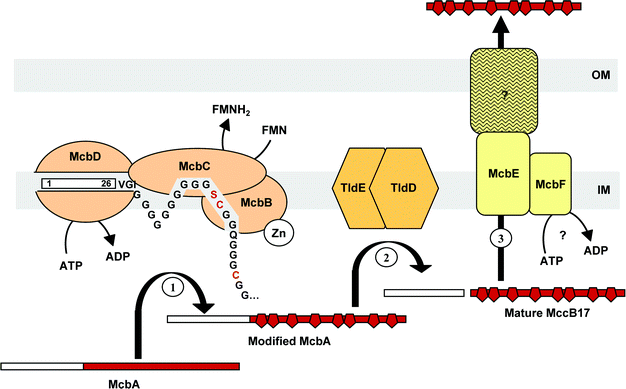 | ||
| Fig. 11 Biosynthesis of MccB17. (1) McbA is modified by the MccB17 synthetase, consisting of McbB, McbC and McbD. The ATP-dependent McbD subunit first binds the McbA leader peptide , the polyglycine linker enabling the correct positioning of the substrate. The zinc-dependent cyclodehydratase McbB subunit then cyclizes Cys and Ser residue side chains at the upstream peptide carbonyl groups. The resulting oxazolines and thiazolines are finally desaturated by the dehydrogenase flavine-dependent McbC subunit. (2) The leader peptide is cleaved off from modified McbA by the chromosomally encoded TldE and TldD. (3) Mature MccB17 is secreted by the export machinery, consisting of McbE/McbF located in the inner membrane (IM), and an unknown component of the outer membrane (OM). | ||
Maturation of class II microcins is concomitant with export. Indeed, processing of MccV precursor involves the CvaA/CvaB/TolC export machinery described above.135 Because MccV processing could be inhibited by N-ethylmaleimide and antipain, the protease was proposed to be a cysteine protease.135 Since CvaA is devoid of cysteine residues, and the N-terminal cytoplasmic domain of CvaB contains a proteolytic domain, the protease activity may be accomplished by CvaB. Similar to MccV, MccH47 precursor is believed to be cleaved from its leader peptide during export.70 This is consistent with the identification of a glycine–alanine motif in MchB,68 while the alternative double-glycine motif is found in MccV precursor, CvaC (Fig. 10). MchF, which is homologous to CvaB, would then be responsible for the cleavage of MccH47 leader peptide . Consistently, MccV export system was actually shown to be competent for recognizing and exporting mature MccH47 into the extracellular medium.70 Moreover, since MccM and MccI47 (i) share the conserved leader sequence displayed by MccH47 and MccV (Fig. 10), and (ii) are encoded on the same gene cluster as MccH47 in E. coli CA46/CA58 and E. coli H47, respectively (Fig. 1), they are likely to share the processing/export machinery of MccH47. As both the leader peptides of MccE492, MccL and Mcc24 precursors (Fig. 10) and their export machineries are highly similar to those of MccV (Table 3), processing is likely to be carried out by the same mechanism involving the ABC transporter of their dedicated export machinery, namely MceG, MclB and MtfB for MccE492, MccL, and Mcc24, respectively.
5.2.2 Post-translational modifications. Biosynthesis of post-translationally modified microcins involves enzymes encoded on the microcin gene clusters. They are believed to be responsible for the large panel of post-translational modifications displayed by microcins. However, only a few reports on in vitro synthesis of microcins have been published.
Consistent with the heterogeneity of structures they display, class I microcins use various enzyme machineries to achieve post-translational modifications. MccB17 post-translational modification has undoubtedly been the most extensively studied, and at least three gene products are required for MccB17 cyclization process. In 1996, Walsh and collaborators reported the first in vitro reconstitution of MccB17 biosynthesis. The purified synthetase, consisting of McbB, McbC, and McbD, was used to synthesize oxazole and thiazole rings within a recombinant His-tagged McbA.21 The three-step model proposed at that time could be further verified.136,137 Thus, the zinc-dependent McbB performs the initial cyclodehydration step, leading to oxazoline and thiazoline rings, which are further desaturated by the flavine-dependent dehydrogenase, McbC. Photo-labelling of McbA showed that within the complex, the ATPase McbD is responsible for the initial recognition and interaction with McbA137 (Fig. 11). The enzymes responsible for MccB17 biosynthesis were shown to be chemoselective, processing cysteine residues faster than serine ones, and regioselective, only one ring being made before nascent product is released. In addition, the post-translational modification process was shown to be carried out directionally, ring formation taking place from the N- to the C-terminal extremity.138 NMR analysis of MccB17 leader peptide showed it consists of an amphipathic α-helix spanning residues 5–21.139 Ser6, Ser13 and Ser20 on one face of the helix form a polar stretch, while the side-chains of Phe8, Leu12 and in a lesser extent Val11 form a hydrophobic patch. Mutagenesis analysis demonstrated the stringent role of Phe8 and Leu12 in the recognition events by the MccB17 synthetase.139 Moreover, the polyglycine linker (Gly30 to Gly39), whose length influences the synthethase turnover, was proposed to act as a spacer allowing the correct positioning of the heterocyclization site (Fig. 11).140 The moderately polar face of the helical leader peptide including the serine array would interact with the inner membrane in order to target the modified McbA for cleavage of the leader peptide , and subsequent export. Finally, substitution of the glycine located immediately upstream of the cyclizable sequence, as well as substitution of cysteine and serine residues involved in the cyclization process, inhibited ring formation.140
MccC7/C51 post-translational modification would be carried out by MccB, MccD and MccE.28,29 MccB exhibits similarity to proteins from the ThiF/MoeB/HesA family. These proteins catalyze the C-terminal adenylation of the ThiS and MoaD subunits from the thiamine and molybdopterin synthase, respectively.141,142 MccB, as all of these proteins , contains a nucleotide -binding domain and a repeated cysteine metal-binding motif. The latter motif may be important for the activity of MccB, since a Tn5insertion in this repeat abolishes MccC7/C51 production.28 The MccD sequence was found to be similar to proteins of the methyltransferase family.29 Finally MccE possesses two putative domains that consist of an N-terminal region resembling pyridoxal phosphate-dependent amino acid decarboxylase and a C-terminal region displaying homologies with proteins catalysing the acetylation of ribosomal proteins .29 This would account for a dual role of MccE in post-translational modification and self-immunity towards MccC7/C51. According to these similarities, three steps could be suggested for MccC7/C51 maturation. The inner membrane anchored MccB would adenylate the C-terminal aspartate of MccA. Moreover, MccE might be responsible for the formation of the n-aminopropanol through homoserine decarboxylation,29 whereas MccD would be involved in the transfer of the n-aminopropanol to the AMP group.28,29 This role was supported by synthesis of a MccC7/C51 related product missing one amino group when MccC51 gene cluster was mutated on mccD.29
MccJ25 post-translational modification consists in the formation of a β-lactam bond between Gly1 and Glu8 side chain of the C-terminal 21-residue peptide , resulting in a lasso structure.91–93 Although MccJ25 genetic system is one of the smallest among microcins, little is known about the genes involved in its post-translational modification. As discussed above, this process is thought to involve McjB and McjC. Whereas McjB does not share homologies with other known enzymes, McjC contains an ATP-binding motif and could thus be responsible for the activation of the glutamic acid, before β-lactam bond formation. Future studies should help identifying the functions of McjB and McjC, and whether or not they are sufficient to convert McjA into MccJ25. Whether maturation of MccJ25 occurs in one or two steps is unknown. Nevertheless, given the MccJ25 lasso structure (Fig. 4C), the ring closure involving Gly1 and Glu8 should occur after acquisition of the spatial structure of the molecule and an almost correct positioning of both Glu8 carboxylate and Gly1 amino group on the one hand, and Phe19 and Tyr20 aromatic side-chains on the other hand. Two hypotheses have thus to be considered: (i) initial cleavage from the leader sequence followed by cyclization by McjB and McjC separately, or (ii) concomitant cleavage from the leader sequence and subsequent cyclization by a McjBC complex.
Because class IIa microcins display little to no post-translational modification, their maturation process, limited to proteolytic cleavage and cysteine oxidation, has been little-studied. Conversely, an increasing interest is devoted to class IIb microcin post-translational modification enzymes. The recent finding in our group (i) that MccE492 is synthesized in a post-translationally modified form14 and (ii) that the siderophore–peptide is the mature microcin (unpublished work), enabled the attribution of putative functions to some enzymes encoded by the MccE492 gene cluster. Recent studies on salmochelin biosynthesis143–145 were very helpful to understand the MccE492 biosynthetic pathway. Indeed, MccE492 post-translational modification resembles salmochelins. On the basis of sequence similarity with iroB and iroD, two genes originally identified in Salmonella,146 an enzymatic activity was postulated for mceC and mceD gene products.38,57 Indeed, MceC is highly similar to IroB (76% identity), a glycosyl transferase involved in C-glycosylation of enterobactin,143 while MceD resembles the enterobactin esterase IroD (57% identity).144,145 It is therefore likely that MceC performs C-glycosylation of enterobactin, while MceD could break down the glycosylated enterobactin into its linear form. Because MceI shares homologies with acyltranferases involved in activation of RTX toxins such as hemolysin,38,57 it could catalyse the acylation of β-D-glucose by the C-terminal serine residue of MccE492 precursor. This process is likely to also involve mceJ, which is co-transcribed with mceI, and is required for the detectable production/secretion of MccE492.58 As discussed previously, the structures of class IIb microcins other than MccE492 remain either uncharacterized or partially characterized. However, depending on the producer strain, genes homologous to mceC, mceD, mceI and mceJ found in the MccE492 gene cluster can be encountered in these microcin gene clusters (Fig. 1C; Table 4). Moreover, heterologous complementation of mceAB by the MccH47 genetic system, which carries the iroB and D orthologues mchA and mchS1, produces MccE492.72 It was therefore proposed that, similar to MccE492,14 (i) other class IIb microcins can be modified by addition of a catechol-type siderophore (Glc-DHBS3),57 and (ii) that the conserved C-terminal sequence present in MccE492, MccH47, MccI47, and MccM (Fig. 10) serves as a recognition signal for modification enzymes.72 Altogether, these data indicate that class IIb microcins are most likely post-translationally modified by similar enzymes with rather related substrate specificity, one system being able to complement the other.
5.3 Regulation of microcin biosynthesis
The regulatory mechanisms underlying the production of microcins have been studied in detail for class I microcins, with much less attention being paid to class II microcins.Like most of the antibiotics, toxins and secondary metabolites, it was claimed that microcins were produced when bacteria enter the stationary phase.25,32,147 Gene expression studies, such as those performed with MccE492,58 showed this statement is not general to all microcins. However, the need for survival within a microbial community when stress conditions appear may stimulate most microcin production. Thus, nutrient depletion, which occurs at the approach of the stationary phase, provides a first level of regulation for most microcins. Another stress that was considered to regulate the production of microcins is oxygen starvation. Nevertheless, not all microcins are controlled by identical stress stimuli.
Expression of mcjA, which encodes the MccJ25 precursor, was shown to be controlled neither by the pH variation that occurs in exponential-stationary phase transition, nor by cell density.35 Because mcjA expression was immediately induced when cultures in mid-exponential phase were exposed to spent medium,35 its induction at the onset of the stationary phase was supposed to rely on nutrient depletion. In agreement with this hypothesis, most microcins were shown to be overproduced in minimal medium compared to rich medium,35,59,148 MccC7/C51 being the only exception reported to date.31 As many conventional antibiotics, class I microcins production is repressed by glucose.23,35,149 As glucose is a fast-used carbon source, it favours a high bacterial growth rate. Such culture conditions could then be related to a lowered production of stationary phase-produced microcins. Conversely, neither glucose depletion nor substitution of glucose by glycerol enhanced MccE492 production,150 in agreement with an exponential phase production of MccE492. The same difference was observed when examining the influence of the nitrogen source. Indeed, nitrogen starvation induced MccB17 production,24 whereas de Lorenzo showed that the easier the utilization of the nitrogen source, the higher the amount of secreted MccE492.150 As with glucose, nitrogen assimilation rate, on which the bacterial growth rate depends, was related to production of microcins. Mild air limitation, as well as shear stress could enhance MccB17 production.149,151 In contrast, MccJ25 production decreased under anaerobic conditions.35
The subtle role of growth conditions in microcin production was further related to the expression of transcriptional regulators. Given that the expression of most of the regulators also depends on growth conditions, nutrients cited above might have both a direct and an indirect role in the control of microcin gene transcription. Several regulators of the growth-phase-dependent transcription of class I microcin structural genes were shown to be involved in complex and interplaying mechanisms. Transcription of Pmcb regulated genes from MccB17 gene cluster (Fig. 1A) was shown to increase as cells enter the stationary phase of growth.147 This activation was dependent on the OmpR transcriptional factor, which is known to positively regulate the production of the outer membraneporins believed to be involved in the import of nutrients from nutritionally poor media (see Section 7.1.2). On the other hand, MccB17 production was found independent on RpoS,152 the RNA polymerase sigma S factor (σS), which controls transcription of several stationary-phase-induced genes. Instead, the alternative sigma 70 (σ70) factor seemed to be involved in this increased expression. In contrast, RpoS appeared to be involved in regulation of mcjA and mccA expression. Indeed, whereas the typical growth phase induction of mcjA was still observed in strains mutated on rpoS, these strains were reported to produce lower amounts of MccJ25 than wild-type strains.35 The same phenomenon was noticed for mccA, whose basal expression decreased in both exponential and stationary phase of growth upon inactivation of rpoS, but was still stimulated during transition to the stationary phase.31,153 Moreover, expression of mcjA was found positively regulated by a complex network at least consisting of the leucine responsive protein , the integration host factor and two unusual nucleotides (guanosine tetraphosphate or pentaphosphate, also termed (p)ppGpp).35 Taking into account that these regulators are themselves growth-phase-responsive, and that induction of mcjA is practically abolished in strains deficient in any of them, their concerted action would stimulate the expression of mcjA at the onset of stationary phase. Several regulators that would negatively control microcin gene expression in the exponential phase were also described. These include the histone-like protein H-NS, which acts as a repressor of the genes encoding MccC7/C51 or MccB17.23,31 Another repressor likely to affect transcription of mcb genes during exponential phase is encoded by the microcin production regulator gene mprA, which was further assimilated to the first gene of the emrRABoperon, and renamed emrR accordingly.154 This gene was also found to repress MccC7/C51 and MccV production when a high-copy number of this gene was expressed.155
Another factor controlling the production of some microcins is the iron availability in the culture media. Iron-regulated gene expression in E. coli is largely mediated by Fur (ferric uptake regulation), a ferrous iron-bindingprotein that binds to the so-called fur boxes (the 19-bp 5′-GATAATGATAATCATTATC-3′ inverted repeat consensus sequence) and blocks iron-regulated promoters in a metal-dependent fashion (for a review, see Hantke156). Gene clusters from class I microcins are devoid of fur boxes (Fig. 1A). Consistently, iron availability was reported not to significantly affect MccB17 and MccC7/C51 production.157 However, MccJ25 production dropped by 95% when iron was added to the culture medium. Accordingly, the use of chelating agents restored MccJ25 production. However, the iron-control of MccJ25 production is Fur-independent.157 Some significant fur boxes are found in all class II microcin gene clusters (Fig. 1B and 1C). However, iron regulation of microcin production has not been shown for all class II microcins. The production of MccV was shown to be induced under iron-limiting conditions. For this reason, the iron chelator 2,2′-dipyridyl is used in culture media to increase the production of MccV.36 Consistent with the presence of fur boxes ahead of cvi and cvaA (Fig. 1B), cvaC expression could be de-repressed upon mutation of the fur gene.45 Contradictory results have been published about iron-regulation of MccE492 production.45,157,158 This discrepancy is mostly due to the use of antibacterial assays for quantifying MccE492 production, since both MccE492 and an antagonist 158 are found in culture supernatants of MccE492-producing strains. No fur box could be found in an extensive region (240 bp) upstream of mceB56 (Fig. 1C). Therefore, mceB is likely to be expressed independently of the iron concentration. Conversely, MccE492 post-translational modification is repressed by high iron concentration (unpublished work). Despite the presence of a fur box ahead of mchX (Fig. 1C), MccH47 production was reported not to be regulated by iron either.71 However, synthesis of both MccE492 and MccH47 have been shown to be dependent on enterobactin synthesis.14,71 Therefore, if MccH47 and MccM are secreted as siderophore–peptides similarly to MccE492, their putative post-translational modification is likely to be iron-dependent.
6 Self-immunity of the producing strains
Bacteria producing antimicrobial compounds must protect themselves from their toxic products. In contrast to colicin- and bacteriocin-producing strains, the mechanisms by which microcinogenic bacteria acquire self-immunity towards their own microcin remain largely unexplained. Nevertheless, the characterization of microcin gene clusters showed that at least one resistance-conferring gene is associated with the production of a given microcin.As previously mentioned (Sections 2.1 and 4), self-immunity and export of class I microcins are tightly associated, at least one gene being involved in both mechanisms. Self-immunity towards MccB17 involves three genes from the microcin gene cluster. Indeed, bacteria harbouring either mcbEF or mcbG showed partial self-immunity, whereas those harbouring all three genes were fully resistant to MccB17.18 McbE and McbF presumably mediate the export of mature MccB17 from the cytoplasm to the periplasmic space, but the exact role of McbG is still unknown. Besides McbE, McbF and McbG, Baquero et al. suggested that during the stationary phase, SbmC may contribute to bacterial cell protection by binding and sequestering MccB17, thus preventing the microcin interaction with its intracellular target, the DNA gyrase.159 Self-immunity towards MccC7/C51 seems to involve at least MccC and MccE. MccC would function as an efflux pump, decreasing the intracellular microcin concentration. The C-terminal region of MccE is similar to RimL and RimJ, two bacterial enzymes that catalyze the acetylation of the N-terminal residue of ribosomal proteins .28,29 Thus, it was originally proposed that MccE could modify the putative intracellular target of the microcin.28 However, it was recently shown85 that secreted MccC7/C51 requires activation by an unknown peptidase to generate the intracellularly active MccC7/C51* (Section 7.2.2). Thus, MccE could prevent MccC7/C51 activation by either downregulating the expression of the gene encoding the peptidase or by interacting with the peptidase or its substrate, MccC7/C51, in the producing strain. A third protein , MccF, seems to contribute, although to a lesser extent, to self-immunity towards MccC7.28 However, mccF is not functional in the MccC51 genetic system.29 Because of its similarity to numerous hypothetical proteins from non-microcinogenic bacteria, MccF is thought to have a widely distributed uncharacterized function.29 Uniquely, MccJ25 gene cluster (Fig. 1A) possesses a single gene, mcjD, putatively involved in self-immunity. On the basis of sequence homologies and mutational assays, McjD was proposed to be responsible for both the export and self-immunity towards MccJ25.33 The mechanism of protection would then be an efflux of the antibiotic that enables keeping intracellular MccJ25 below the inhibitory concentration.
In contrast to class I microcins, all class II microcins are characterized by a dedicated self-immunity protein . This is also true when several microcins are encoded by a same gene cluster (e.g. E. coli H47, CA46, CA58 and Nissle 1917). Thus, MccH47, MccI47 and MccM, which have a common export system consisting of MchE and MchF, have specific self-immunity proteins . Self-immunity proteins of class II microcins likely range in size from 51 to 144 amino acids. Except for MchS3, the MccI47 self-immunity protein , class II microcin self-immunity proteins are probably membrane-bound, with two or three transmembrane helices. Cloning of MccL self-immunity and precursor genes only, led to an intracellular antibacterial activity and to full immunity of the bacteria to exogenous MccL.39 This suggests that the self-immunity protein MclI protects the bacteria from the MccL precursor as well as from MccL itself. As previously mentioned (Sections 5.1.1 and 5.2.1), the leader peptides of class II microcins are highly similar and are cleaved by the export machinery. Moreover, it was proposed that the similar C-terminal region of class IIb microcins could be involved in receptor recognition (Section 7.1.1). Thus, the N-terminal region of mature class II microcins, which is highly variable, could be involved in a specific interaction with the self-immunity protein . No significant similarity is observed between self-immunity proteins of class II microcins, except for MceB and MtfI, which are involved in MccE492 and Mcc24 self-immunity, respectively, and exhibit 38.7% identity. Such a similarity is also found between MccE492 and Mcc24 precursors (Section 3.2.1) and between their export proteins (Section 4). Therefore, it is tempting to speculate that either the short Mcc24 gene cluster derives from that of MccE492, or that both microcin gene clusters have a common ancestor.
The data available show that self-immunity towards microcins may arise from different mechanisms that confer either partial or full immunity to the producers. Further studies will be required to elucidate the delineated mechanisms in greater details.
7 Mechanisms of action
The broad variety of microcin structures correlates with diverse mechanisms of action, such as inhibition of vital enzymatic functions and damages to the inner membrane. While microcins display a broad diversity of cellular targets, the initial recognition/uptake pathways may be common to several microcins.7.1 Recognition/uptake: role of the stress response-regulated machineries
Evocative of receptor-mediated mechanisms of action, mature microcins were shown to display narrow spectra of activity, limited to few genera of enterobacteria, and low MICs (often below 0.1 µM).14,90 The existence of microcin receptors in enterobacteria was also strongly supported by the isolation of microcin-resistant mutants impaired in outer membraneproteins normally involved in nutrient uptake.160–164 As these proteins are also exploited by bacteriophages, antibiotics, and bacterial toxins for cell entry, they constitute an “Achilles’ heel” for the bacterium. Their utilization by microcins is illustrated below.
7.1.1 Role of TonB-dependent iron-uptake machineries. Over recent years, it has become obvious that the iron uptake machineries and their associated energy-transduction system, the TonB/ExbB/ExbD inner membrane complex, are required for recognition of various microcins. Iron is imported into enterobacteria through three pathways that involve dedicated outer membrane receptors: (i) FhuA, which binds hydroxamate siderophores (e.g. ferrichrome), (ii) FepA, Cir and Fiu, which bind catecholate siderophores (e.g. enterobactin), and (iii) FecA, which is involved in the uptake of hydroxycarboxylates (e.g.citrate) (for reviews, see Ferguson and Deisenhofer165 and Letellier and Santamaria166). FepA and FhuA show similar three-dimensional structures.167–169 Both receptors are composed of a β-barrel embedded in the outer membrane with an N-terminal globular domain, either called the plug or the cork domain, folded inside the barrel. This domain spans most of the interior of the barrel and occludes it. It is connected to the β-barrel and to the external hydrophilic loops by numerous hydrogen bonds and salt bridges. The external loops contain the binding sites for iron–siderophore complexes.170,171 Interestingly, FhuA and FepA are multifunctional proteins which, besides their physiological function, also transport antibiotics and serve as receptors for colicins and bacteriophages (for a review, see Letellier and Santamaria166), which bind to diverse external loops on the receptors.171 As iron–siderophore complexes, most of these ligands require the TonB/ExbB/ExbD complex to be anchored at the inner membrane for uptake. This complex is responsible for the transduction of the proton-motive force energy from the inner membrane, where it is generated, to the outer membrane (for a review, see Postle and Kadner172).
The ferrichrome receptor FhuA. Early studies showed that the iron–siderophore receptor FhuA, as well as the TonB and SbmA inner membrane proteins , were most likely involved in MccJ25 uptake.162,163 The MccJ25 requirement for both FhuA and the TonB/ExbB/ExbD complex (Fig. 12A) was ascertained by homologous complementation assays in E. coli strains impaired in one of these proteins .90 Heterologous complementation in Salmonella species, whose FhuA genes were sequenced,173 indicated that resistance of S. enterica serovars such as Typhimurium is due to variations in the FhuA sequence.174 Besides genetic evidence, the role of FhuA in MccJ25 recognition was demonstrated functionally. Indeed, MccJ25 was shown to inhibit phage T5 adhesion to its receptor FhuA both in vivo and in vitro.90 Moreover, MccJ25/FhuA interaction was demonstrated by size-exclusion chromatography and isothermal titration calorimetry. MccJ25 binds to FhuA with a 2 : 1 stoichiometry and a Kd of 1.2 µM. Both differential scanning calorimetry and antibacterial assays showed that MccJ25 binding involves FhuA external loops. By using the thermolysin-cleaved variant of MccJ25 (Fig. 5; Section 3.1), it was also demonstrated that the MccJ25 Val11-Pro16 β-hairpin region, which is disrupted upon thermolysin cleavage, is required for microcin recognition by FhuA.90
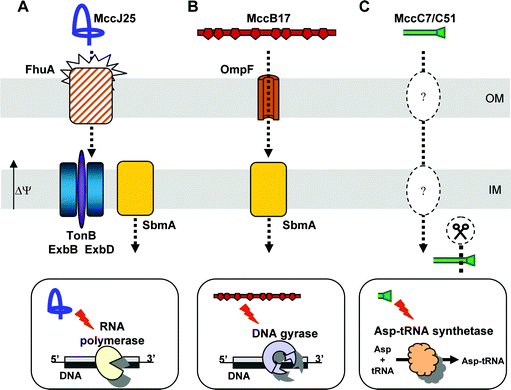 | ||
| Fig. 12 Uptake and mechanism of action of class I microcins. (A) MccJ25 is recognized by the high affinity receptor FhuA at the outer membrane (OM). The recognized structural motif is the MccJ25 β-hairpin region. Translocation of MccJ25 across the OM requires the inner membrane (IM) potential Δψ and needs the TonB/ExbB/ExbD complex as well as the SbmA protein at the IM. Once into the cytoplasm, MccJ25 inhibits transcription by obstructing the RNA polymerase secondary channel. The ring-tail part of MccJ25 is proposed to be involved in this obstruction. (B) MccB17 passes across the OM through the OmpF porin. The IM protein SbmA is involved in MccB17 further uptake into the cytoplasm, whereupon it inhibits DNA supercoiling by the DNA gyrase. The binding site of MccB17 is likely to be the C-terminal domain of GyrB. (C) The components enabling the translocation of MccC7/C51 across bacterial membranes are unknown. MccC7/C51*, which is generated by proteolytic cleavage of MccC7/C51 after uptake, is a modified aspartyl-adenylate that inhibits translation by targeting the aspartyl-tRNA synthetase. | ||
The catecholate siderophore receptors FepA, Cir and Fiu. MccE492 was shown to require the catecholate siderophore receptors for recognition at the outer membrane (Fig. 13). Indeed, MccE492, which inhibited the growth of E. coli H1443 at 40 nM, was inactive against the isogenic fepA cir fiu triple mutant (MIC >10 µM).14 Besides the need for catecholate siderophore receptors, MccE492 was found to be dependent on both energy and TonB for antibacterial activity and translocation across the outer membrane14,57 (Fig. 13). The need for FepA, Cir and Fiu as well as for TonB was also demonstrated for u-MccE492, which lacks the siderophore post-translational modification.57,90 As with MccE492, MccM- and MccH47-producing strains failed to inhibit the growth of a tonB mutant and a fepA cir fiu triple mutant, where both strains were derived from a susceptible E. coli with an identical genetic background.65 This suggests that not only MccE492, but also MccM and MccH47, require the catecholate siderophore receptors and the associated TonB for antibacterial activity. Moreover, it was recently shown that the C-terminal region, which is conserved among class IIb microcins (Fig. 10; Section 5.1.1), is essential for the activity of extracellular but not intracellular MccE492.175 This strongly suggests that the C-terminal sequence from class IIb microcins is required for receptor recognition and/or translocation accross the outer membrane. As discussed previously,57 MccM and MccH47, as well as the recently discovered MccI47, are able to carry a catecholate siderophore as a post-translational modification on their C-terminal serine (Section 3.2.2). We showed that the modification increases the antibacterial activity,14 probably by providing the microcin with a higher affinity for its receptors.57 Thus, as with MccE492, it is tempting to speculate that all class IIb microcins use structural mimicry (i.e. a siderophore post-translational modification) to improve their recognition by the catecholate siderophore receptors. Furthermore, while the C-terminal region is thought to be required for optimal uptake, the remaining part of the protein is likely to endow the specificity of the mechanism of action, which greatly differs among these microcins (see below). While the three receptors FepA, Cir and Fiu would be needed for class IIb microcin antibacterial activity, genetic evidence strongly supports the idea that Cir alone is involved in MccV recognition at the outer membrane. Indeed, contrary to what was shown for the former microcins, mutations in cir were found to be sufficient to confer resistance to MccV.45 Evocative of an energy-dependent uptake similar to that of MccE492, MccV was also shown to require tonB and exbB for antibacterial activity.45 As for MccV, the sole receptor Cir and the TonB/ExbB/ExbD complex were also shown to be involved in MccL recognition/translocation (Sable et al., personal communication).
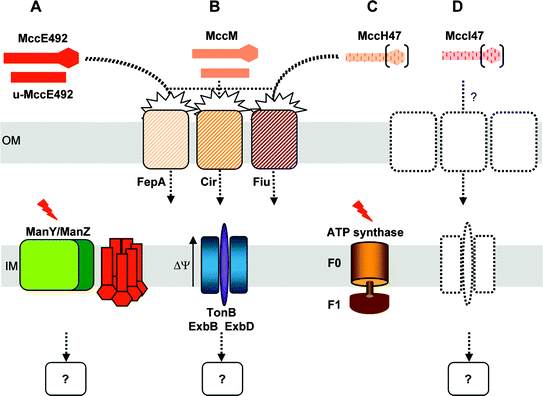 | ||
| Fig. 13 Uptake and mechanism of action of class IIb microcins. (A) MccE492 and its incompletely processed form, u-MccE492, are recognized by the high affinity receptors FepA, and to a lesser extent Cir and Fiu, at the outer membrane (OM) of E. coli. MccE492 is then translocated across the OM via an uncharacterized but TonB- and energy-dependent mechanism. Once into the periplasmic space, MccE492 inserts into the inner membrane (IM), whereupon it induces proton leakage and subsequent drop of the IM potential (Δψ). Membrane insertion and antibacterial activity are dependent on ManY and ManZ, two membrane components of the mannose permease. The C-terminal sequence of MccE492 is not required for interaction with the mannose permease. The orientation of MccE492 monomers in the IM remains hypothetical. The occurrence of a cytoplasmic target remains unknown. (B) MccM in both modified and unmodified forms is also recognized by FepA, Cir and Fiu, and further translocated into the periplasmic space by a TonB-dependent process. Its mechanism of action remains unknown. (C) MccH47 has never been isolated to date but it is hypothesized to bear a C-terminal siderophore modification similar to MccE492 and MccM. It utilizes the same receptors for recognition and is translocated in a TonB-dependent process. It then inhibits the membrane component F0 of the ATP synthase in the IM. (D) MccI47 has not been purified either, and its uptake and mechanism of action have not been investigated. Nevertheless, MccI47 bears significant C-terminal sequence homologies to other class IIb microcins, which could account for the same uptake mechanism. Siderophore-type post-translational modifications are depicted as hexagons. | ||
In a recent study, by screening a total of 49 Gram-negative clinical isolates from urine, Laviña and collaborators demonstrated that 71% of the strains generating an antibacterial activity were inactive against a tonB-deficient mutant and that 37% were both inactive against the tonB-deficient mutant and a fepA cir fiu triple mutant.72 Among these, only 2 strains produced an activity attributable to MccE492 or MccH47. This strongly suggests that urinary tract bacteria synthesize other microcins, colicins, or other bacterial toxins that use the TonB-dependent catecholate siderophore uptake pathway.
7.1.2 Role of the outer membraneprotein OmpF. Early studies on MccB17 mode of action led to the isolation of MccB17-insensitive mutants, most of which were uptake-deficient.161 They contained mutations in ompF and ompR, two genes encoding outer membraneproteins (Fig. 12B). It was shown that OmpF, which serves as a passive diffusion pore across the outer membrane, is also required for the uptake of various group A colicins in association with the Tol/Pal translocation system (for a review, see Cao and Klebba176). It was proposed that OmpF is important for efficient nutrient uptake from nutritionally poor media.177 The transcriptional upregulation of ompF expression under conditions of nutrient depletion is reminiscent of the upregulation of fhuA, fepA, cir and fiu under iron-poor conditions.178,179 Since MccB17, MccJ25, MccE492, MccH47 and MccM are known and/or believed to use these porins or high affinity receptors for recognition, it is likely that nutritionally poor media enhance susceptibility to microcins23,45,157 (Section 5.3).
7.1.3 Role of the inner membrane proteins SdaC and SbmA. We showed above that inner membrane proteins, such as TonB, and in some cases ExbB and ExbD, are required for the activity of MccJ25 and class IIb microcins. MccV bactericidal activity was shown to be dependent on another inner membrane protein, SdaC, also termed DcrA, which is involved in serine uptake.180 SdaC was previously known as being required for infection by bacteriophages together with the outer membrane receptor FhuA (phage C6) or BtuB (phage C1).181,182 It was recently proposed that SdaC also serves as a specific inner membrane receptor for MccV, thus helping it locate the inner membrane, a step required for channel formation and disruption of membrane potential180 (Section 7.2.1).
Another inner membrane protein , SbmA, was proposed to be required for the activity of the class I microcins MccB17 and MccJ25 (Fig. 12A and B). Indeed, E. coli achieved high and specific resistance to MccB17 upon inactivation of sbmA.161 Given that SbmA is also involved in the uptake of the antitumoral antibiotic bleomycin, whose backbone displays thiazole rings, it was proposed that heterocycles could be a structural feature necessary for the recognition by SbmA, and further uptake into the cytoplasm.183 The finding that SbmA is required for MccJ25 antibacterial activity163 indicates that structural features different from the heterocycles may also be recognized by SbmA. Recently, de Cristóbal et al. showed that osmotic shock-treated bacteria, in which the FhuA-dependent outer membrane recognition step is bypassed, were resistant to His5 mutants of MccJ25 but not to wild-type MccJ25. Overexpression of SbmA sensitized these strains equally to both MccJ25 and its His5 mutant.184 It was therefore inferred that MccJ25 interacts with SbmA, and that the interaction may involve the His5 located in the ring-tail part of MccJ25.
7.2 Cellular targets: from inner membrane to cytoplasmic targets
7.2.1 Inner membrane targets. While most of the gene-encoded antimicrobial peptides are believed to inhibit the growth of microorganisms by targeting the cell phospholipid bilayers,185–187 such a mechanism of action was only reported for few microcins.
Modification of membrane permeability was shown to be induced by three microcins. Thus, MccV was reported to abolish E. coli membrane potential in vivo. However, pore formation could not be observed with liposomes,42 and difficulties in isolating sufficient amounts of the peptide have hampered further studies on the MccV mechanism of action. MccJ25 was also reported to disrupt membrane integrity in S. enterica Newport,188 in liposomes189 and uncharged phospholipid monolayers.190 However, these properties were reported to be specific to S. enterica serovars and were observed at concentrations much higher than the MICs. The best-studied microcin with regard to membrane-permeabilization properties is undoubtedly MccE492. Most of the work has been done prior to the elucidation of the structure of mature MccE492, by using either pre-purified culture supernatants (containing both MccE492 and u-MccE492) or u-MccE492 homogenous preparations. In vitro, the microcin was able to form ion-channels in planar lipid bilayers.191 The u-MccE492 pore-forming activity could be observed at concentrations as low as 2 × 10–10 M, and the insertion was shown to be voltage-independent.106In vivo, the microcin depolarized the inner membrane of E. coli192 in an energy- and TonB-dependent manner106 and made the inner membrane permeable to chromogenic substrates, such as o-nitrophenyl-β-D-galactopyranoside.106 Studies in our group also showed that mature MccE492 damages the inner membrane57 (Fig. 13). Interestingly, we found (i) that membrane integrity (as observed by electron microscopy ) was preserved in MccE492-killed bacteria, indicating that the observed leakage does not result from membrane lysis, and (ii) that interference with the inner membrane is not responsible by itself for the lethal effect of either u-MccE492 or MccE492.57,106 It was therefore proposed that the inner membrane is not the sole target of the microcin.
In a recent study, Bieler et al. have further investigated the MccE492 mechanism of action. The authors have evidenced that, as MccH47, MccE492 targets inner membrane proteins . These belong to the mannose permease.175 Indeed, by Tn10transposoninsertionmutagenesis, manY and manZ were identified as critical for MccE492 antibacterial activity against E. coli175 (Fig. 13). ManYZ is an inner membrane complex that functions together with the cytoplasmic ManX to form the mannose permease involved in the uptake of mannose and related hexoses.193,194 It was shown that all the manYZ mutants resistant to MccE492 were unable to metabolize mannose. In addition, they became insensitive to the inner membrane depolarization mediated by periplasmic MccE492.175 At this stage, the molecular basis of MccE492 bactericidal activity thus remains to be established. As Mcc24 is similar to MccE492, Bieler et al. proposed that Mcc24 antibacterial activity could also require ManYZ at the inner membrane.175
The MccH47 mechanism of action has been fairly well documented by Laviña and collaborators, although the microcin has never been purified. In their study of MccH47 precursor (MchB), the authors found that strains expressing mchB only were not viable, but that the mutants exhibiting an Atp– phenotype resisted.68 Confirming this first observation, the authors characterized mutants resistant to MccH47, obtained by Tn5transposoninsertionmutagenesis, as impaired in the atpoperon. It was therefore proposed that F0F1ATP synthase was necessary for MccH47 antibacterial activity164 (Fig. 13). Since then, the authors have shown that all Tn5 insertions mapped to genetic determinants encoding the F0 membrane component of ATP synthase.195 In the same study, they used a complementation approach to confirm that the minimal structure of ATP synthase needed for MccH47 antibacterial activity was the F0 proton channel, while the F1 catalytic unit was dispensable.
7.2.2 Cytoplasmic targets. Class I microcins were shown to target intracellular enzymes responsible for DNA/RNA structure or synthesis.
MccB17 is a DNA gyrase inhibitor. A significant number of studies have been devoted to the elucidation of the mechanism of action of MccB17 over the past 20 years. MccB17 was shown to induce the SOS response and to block DNA replication.196 Consistent with this last finding, besides uptake-deficient mutants, bacteria resistant to MccB17 displayed mutations on DNA gyrase,161,197 a bacterial type II topoisomerase involved in DNA topology and essential in DNA replication. Point mutations in the DNA gyrase B subunit (encoded by gyrB) were actually sufficient to lower or abolish susceptibility to MccB17.198 By studying the effect of semipurified MccB17 on gyrB mutants and on replicative cell-free extracts prepared from these cells, MccB17 was shown to induce the irreversible trapping of DNA–DNA gyrase complexes, leading to the accumulation of double-stranded DNA breaks and replication inhibition197 (Fig. 12B). Confirming earlier finding that bisheterocycles are necessary for MccB17 antibacterial activity,199 Zamble et al. showed the activity of MccB17 on DNA gyrase extends to supercoiling inhibition.200 MccB17 was actually shown to slow down but not to completely inhibit the supercoiling and relaxation reactions of DNA gyrase and to stabilize the cleavage complex.201 Later, by establishing the proteolytic signature of the gyrase in the presence of MccB17, the same authors showed that the binding site of MccB17 was likely to be the C-terminal domain of GyrB.202 They also demonstrated that DNA strand passage was involved in the MccB17 mechanism of action. Based on this knowledge, it was inferred that MccB17 traps a transient intermediate state of gyrase reaction only present during DNA passage. At this stage, the MccB17 mechanism of action remains incompletely elucidated. In particular, the molecular details of the inhibition are not characterized.
MccC7/C51 targets the aspartyl-tRNA synthetase. First studies showed that MccC7/C51, used at MIC, blocked the in vivo incorporation of radiolabelled leucine to proteins , while transcription remained unchanged.86 This was the first indication that MccC7/C51 is a translation inhibitor. Afterwards, MccC7/C51 was shown to inhibit protein synthesis in a cell-free coupled transcription–translationassay . Based on the compared activities of MccC7/C51 and the synthetic heptapeptide part, it was suggested that the peptide backbone was required for translation inhibition, while the C-terminal post-translational modification was needed for recognition/uptake.83 Nevertheless, the exactly opposite conclusions were recently found, since highly pure MccC7/C51 was unable to inhibit translation.85 Actually, MccC7/C51 is processed by an uncharacterized intracellularpeptidase present in crude bacterial extracts, and the resulting product, MccC7/C51* (Fig. 3B; Section 3.1), strongly inhibits translation. The mechanism underlying this process was found to be an inhibition of aminoacylated tRNAAsp synthesis by aspartyl tRNA-synthetase (Fig. 12C). Since MccC7/C51* is devoid of antibacterial activity, the authors proposed that the peptide backbone is required for recognition, and that following uptake, MccC7/C51 is the subject of cleavage, which renders it active in translation inhibition. Thus MccC7/C51 develops a clever “Trojan horse” strategy that involves the cleavage of the microcin inside the target bacteria to generate a potent inhibitor of bacterial cell growth, MccC7/C51*. Such cleavage of the promicrocin in the target cell rather than in the producer is unique among microcins.
MccJ25 targets RNA polymerase. While most of the MccJ25-resistant mutants were found to carry mutations in genes encoding membrane transporters, one mutant was isolated that carried a point mutation in rpoC.203 Consistent with rpoC encoding the RNA polymerase β′ subunit, MccJ25 was able to inhibit the RNA polymerase of Gram-negative bacteria in vitro203,204 (Fig. 12A), with an apparent Ki ranging from 1.2 to 20 µM.205,206 Crosslinking, as well as fluorescence resonance energy transfer experiments, clearly showed that MccJ25 binds within the secondary channel of E. coliRNA polymerase.205,206 The MccJ25/RNA polymerase interaction was characterized by a Kd of 0.5 µM.206 Since MccJ25 was unable to bind RNA polymerases carrying mutations on the secondary channel, it is very likely that resistance to MccJ25 is conferred by inhibition of MccJ25 binding.205 Therefore, it is believed that MccJ25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel, whereupon it prevents incoming nucleoside triphosphates from trafficking through the channel.205,206 Structure–activity analyses were also performed. The thermolysin-cleaved variant of MccJ25, lacking the Val11–Pro16 β-hairpin region (Fig. 5; Section 3.1), was as efficient as the uncleaved microcin in inhibiting transcription.207 This implies that the β-hairpin, which is involved in FhuA receptor-recognition,90 is not involved in RNA polymerase binding.207 It was therefore proposed that the ring-tail part of MccJ25, which is preserved in all variants, is responsible for the interaction with the RNA polymerase,207 as well as with SbmA (Section 7.1.3), while the Val11-Pro16 β-hairpin region is required for recognition by FhuA (Section 7.1.1).
8 Comparison with other gene-encoded antibacterial peptides from bacteria
As mentioned in introduction, besides microcins, gene-encoded antibacterials from bacteria include colicins and bacteriocins secreted by Gram-negative (enterobacteria) and Gram-positive bacteria (lactic acid bacteria, LAB), respectively. Colicins have higher molecular masses (30–90 kDa) than microcins, while bacteriocins have molecular masses below 10 kDa, similar to microcins. In this section, microcins are compared to colicins and bacteriocins.8.1 Colicins from Gram-negative bacteria
Microcins and colicins are both encoded by dedicated gene clusters that contain most of the information required for their production, export and self-immunity. To date, all colicins have been found to be plasmid-encoded, in contrast to microcins, among which class IIb microcins are chromosome-encoded. Colicin gene clusters appear to be highly conserved, but amazingly simple compared to microcin gene clusters (Section 2). Indeed, they include two to three genes only, the minimum requisite being a structural gene and a self-immunity gene. A third gene encoding a lysis protein is required for colicin secretion (for a review, see Van der Wal et al.208). In contrast to microcins, the production of colicins is mainly induced via the DNA repair network, called the SOS response (for a review, see Janion209). It can be activated by an environmental stress, such as UV irradiation, exposure to DNA-damaging agents, or starvation of cells.209–211One of the major differences between microcins and colicins, besides an evident molecular mass difference, is their structure. Indeed, whereas most microcins were shown or proposed to bear post-translational modifications (Sections 3.1 and 3.2.2), the simple and conserved organization of colicin gene clusters leads to non-post-translationally modified proteins . Colicins are organized in three functional domains including a central receptor binding domain, an N-terminal translocation domain and a C-terminal catalytic domain. These domains, which are common to all colicins, ensure every common step of the colicin mechanisms of action, i.e. (i) recognition by a specific receptor at the outer membrane, (ii) translocation across the outer membrane and (iii) lethal interaction with a specific cellular target.212 Another noticeable difference between microcins and colicins is their mechanism of export. Contrary to microcins, which utilize ABC transporters or efflux pumps (Section 4), the release of colicin results from the sole presence of lysis factors.213,214 These small and highly similar lipoproteins, predominantly located in the outer membrane of colicinogenic strains, are first synthesized as precursor polypeptides . While the stable signal peptide would accumulate in the inner membrane, the mature lysis protein would activate the phospholipase A, both phenomena being responsible for the loss of membrane integrity and cell lysis.2,208,215
Both microcins and colicins have a narrow spectrum of activity, being active against bacterial strains phylogenetically related to the producer. This specificity relies in part on the use of outer membrane receptors specifically expressed by enterobacteria. Similar to microcins, most colicins parasitize multi-protein systems involved in important biological functions to enter bacteria, i.e. the vitamin B12 receptor BtuB, the siderophore receptors FepA, Cir, Fiu or FhuA, or the nucleoside receptor Tsx (Section 7.1).
We have shown in this review that all studied microcins have in common the use of outer membraneproteins for recognition/translocation, but differ in their cellular targets. Colicins are also distinguished by their bacterial killing mechanisms. Indeed, some possess nuclease-activity (for a review, see James et al.216), pore-forming activity (for reviews, see Cramer et al.217 and Duche218), or in rare cases, they target the peptidoglycan . This is evocative of microcin mechanisms of action. Nevertheless, whereas the mechanism of action of nuclease–colicins consists in hydrolysing DNA or RNA strands, microcin cytoplasmic targets are the enzymes responsible for the DNA/RNA structure or synthesis. Moreover, the third kind of activity, inhibition of peptidoglycan synthesis, which was reported for pesticin219 and colicin M,220 has never been described among microcins.
8.2 Bacteriocins from Gram-positive bacteria
As microcins and colicins, bacteriocins from Gram-positive bacteria are encoded by dedicated gene clusters, where the structural gene is accompanied by a self-immunity gene. Similar to microcins, bacteriocins from Gram-positive bacteria display a high stability to temperature, pH and most often proteases. They are assembled in four main classes according to their structural characteristics (for reviews, see Garneau et al.221 and Drider et al.6). Surprisingly, microcins, which form a much more restricted group, have more diverse structures. Indeed, most of the bacteriocins from Gram-positive bacteria, except the highly modified lantibiotics (for a review, see Jack and Sahl222), also referred to as class I bacteriocins, are unmodified peptides . The latter contain unusual amino acids such as lanthionine and dehydrated amino acids (for reviews, see Patton and van der Donk223 and Chatterjee et al.121). Precursors of bacteriocins from Gram-positive bacteria and microcins are similarly processed through elimination of a leader peptide . They also display similarities in their leader peptide sequence. In addition, bacteriocins from Gram-positive bacteria and microcins have in common the involvement of ABC transporters for their export into the extracellular medium.Compared to microcins, bacteriocins from Gram-positive bacteria may exhibit a broader spectrum of antibacterial activity. This is the case for nisin, a lantibiotic that uses lipid II (the crucial precursor of peptidoglycan biosynthesis) as a docking molecule,224,225 inhibits peptidoglycan synthesis and forms heteromolecular pores in bacterial membranes. Such a dual mode of action is responsible for the potent antibacterial activity of nisin. Nevertheless, bacteriocins from Gram-positive bacteria may also have mechanisms of action that are similar to those of microcins. Indeed, membrane permeabilization has been reported for nisin and for class II bacteriocins, such as leucocin A or mesentericin Y105 (for a review, see Fimland et al.226), which induce disruption of the proton-motive force at the inner membrane. This is reminiscent of the MccV and MccE492 mechanisms of action (see Section 7.2.1). Interestingly, similar to MccE492, class IIa bacteriocins target both the inner membrane and require the ManYZ components of mannose permease.175,227,228 However, other mechanisms, such as the inhibition of cell wall synthesis described for nisin,224,225 have not been described for microcins.
This rapid comparison with bacteriocins from Gram-positive bacteria and colicins illustrates how microcins combine features and strategies exemplified by these two classes of antibacterials. Indeed, they assemble the typical leader peptides , self-immunity, and maturation mechanisms of bacteriocins from Gram-positive bacteria with the uptake strategy of colicins. Such an efficient combination, which would be either at the root or at the top of evolution, constitutes an amazing model for the design and engineering of new antibacterials.
9 Current challenges in microcin research
We have shown in the previous sections that great progress has been made on microcin research over the last years, especially with the identification of several structures and mechanisms of action. However, some fundamental questions remain unresolved.9.1 Unresolved questions
Self-immunity of microcinogenic strains. The most poorly studied topic in microcin research is certainly self-immunity. As with colicins,229,230 multidisciplinary studies should aim at isolating and collecting structural information on microcin self-immunity proteins . This would clarify the basis of the self-immunity specificity, an essential point to determine how producers protect themselves from their own toxic substances.Pheromone activity of microcins. Bacterial communication such as that involved in quorum sensing, which regulates many bacterial behaviours including symbiosis or virulence,231 uses signalling molecules (for reviews, see Miller and Bassler232 and Reading and Sperandio233). In Gram-positive bacteria, these are peptide pheromones, which display similarities with microcins. Indeed, they are concomitantly cleaved for maturation and exported via ABC transporters. Since bacteriocins from Gram-positive bacteria were reported to play the role of inducing agents,234,235 one may speculate that microcins contribute to cell-to-cell signalling in Gram-negative bacteria. Future research should help determining whether, similar to eukaryotic defensins, prokaryotic antibacterial peptides are multifunctional, being involved in chemotaxis, signalling, and antibacterial defence.
Ecological role of microcins. Several surveys on enteric bacteria reveal that an average 10–50% of the strains sampled produce antibacterials including bacteriocins from Gram-positive bacteria, colicins and microcins.236 Early studies on the ecological role of microcins were often contradictory or inconclusive.8,237–239 However, recent studies emphasized the role of colicins and microcins as regulators of bacterial populations.236,240–242 Indeed, the resulting rock–paper–scissors model243 provides evidence that colicins, and potentially other bacteriocins, may promote rather than eliminate microbial diversity in their ecosystems.243,244 Further studies should firmly assess the specific roles of microcins as antibacterial weapons or signalling molecules.
9.2 Miscellaneous applications of microcins
Based on our current knowledge of microcin biological activities, several applications may be considered for these peptides as well as for their associated modification enzymes. Here we list the properties that may be valuable in terms of potential developments.Probiotic agents. Based on their potential ecological role, microcinogenic strains were tested as probiotics. On the one hand, the role of microcins in preventing Salmonella invasion in humans by E. coli Nissle 1917, a microcinogenic probiotic commercially available under the name Mutaflor®, could not be demonstrated clearly.245 Long term colonization and transmission of this strain, as well as the presence of microcins, was reported in swine herds.246 On the other hand, inhibition of Shigella flexneri by an E. coli H22 strain was shown to be mediated by the production of MccC7/C51.247 MccJ25-producing strains were observed to be widely distributed in poultry intestinal habitats.248 Moreover, an Mcc24-producing E. coli was shown to inhibit the growth of pathogenic Salmonella and Escherichia O157:H7 in the intestinal tract of chickens.238 Further studies should help defining how far microcins contribute to the prevention of intestinal infections.
Antitumoral agents. MccE492 was shown to induce biochemical and morphological changes typical of apoptosis in human cell lines,249 opening a new research field for potential applications of microcins as antitumoral agents. Indeed, inducers of apoptosis are of great interest in the search for novel antitumoral agents, since apoptosis was observed as a response of eukaryotic cells to infection by a wide range of pathogens and is mediated by an array of pathogen-encoded virulence determinants (for a review, see Weinrauch and Zychlinsky250). Some microcins are encountered on genomic islands together with virulence factors including siderophores,77 and could therefore promote virulence/pathogenicity.
Antimicrobials for health and food preservation. The worldwide emergence of antibiotic-resistant pathogens has led to an increasing demand for new antimicrobial agents. Since microcins differ from conventional antibiotics by their diverse mechanisms of action and a highly potent activity on a restricted bacterial spectrum, they may help in the design of novel drugs or substitutes. Colicin-engineered antibiotics obtained by fusing channel-forming colicins and pheromones from Gram-positive bacteria proved to be efficient and specific against pathogenic Gram-positive strains, without toxicity in mammal cells.251,252 Inspired by the lantibiotic nisin, which is commonly used as a preservative against food spoilage, and because bacteriocins from LAB do not kill Gram-negative bacteria, heterologous production of MccV in LAB was performed. This engineered microcin, which has a LAB peptide leader and displays MccV activity,253 should find applications to prevent food poisoning by Gram-negative bacteria. The design of chimeric peptides active against specific bacterial infections could thus be efficiently applied to microcins.
Enzymes for the design of more stable/specific antimicrobials. The large panel of microcin structures results from post-translational modifications. Some of the enzymes involved show interesting activity in terms of possible development. Thus, enzymes responsible for the lasso structure of MccJ25 would be of great biotechnological interest given the increased potency and stability of cyclic antimicrobial peptides . Similarly, several years before the isolation of the first natural siderophore–peptide ,14 conjugation with hydroxamate or catecholate siderophores provided vancomycin254 or cephalosporins 255 with enhanced antibacterial activity compared to unsubstituted antibiotics. Thus, the enzymes involved in MccE492 siderophore modification would also be worth isolating to improve antibacterial activities.
Taking into account these different aspects and the potential applications, there is no doubt that for many years, microcins will continue to be an active area of fundamental and applied research.
10 References
- C. Asensio, J. C. Perez-Díaz, M. C. Martínez and F. Baquero, Biochem. Biophys. Res. Commun., 1976, 69, 7–14 CrossRef CAS.
- A. P. Pugsley, Microbiol. Sci., 1984, 1, 168–175 Search PubMed.
- A. P. Pugsley, Microbiol. Sci., 1984, 1, 203–205 Search PubMed.
- V. Braun, S. I. Patzer and K. Hantke, Biochimie, 2002, 84, 365–380 CrossRef CAS.
- R. W. Jack, J. R. Tagg and B. Ray, Microbiol. Rev., 1995, 59, 171–200 CAS.
- D. Drider, G. Fimland, Y. Hechard, L. M. McMullen and H. Prevost, Microbiol. Mol. Biol. Rev., 2006, 70, 564–582 CrossRef CAS.
- A. F. Duro, R. Serrano and C. Asensio, Biochem. Biophys. Res. Commun., 1979, 88, 297–304 CrossRef CAS.
- A. Aguilar, J. C. Perez-Díaz, F. Baquero and C. Asensio, Antimicrob. Agents Chemother., 1982, 21, 381–386 CAS.
- A. Aguilar, F. Baquero, J. L. Martínez and C. Asensio, J. Antibiot., 1983, 36, 325–327 CAS.
- J. L. Martínez and J. C. Perez-Díaz, Antimicrob. Agents Chemother., 1986, 29, 456–460 CAS.
- E. Sablon, B. Contreras and E. Vandamme, Adv. Biochem. Eng. Biotechnol., 2000, 68, 21–60 Search PubMed.
- S. Gaillard-Gendron, D. Vignon, G. Cottenceau, M. Graber, N. Zorn, A. Van Dorsselaer and A. M. Pons, FEMS Microbiol. Lett., 2000, 193, 95–98 CrossRef CAS.
- A. M. Pons, I. Lanneluc, G. Cottenceau and S. Sablé, Biochimie, 2002, 84, 531–537 CrossRef CAS.
- X. Thomas, D. Destoumieux-Garzón, J. Péduzzi, C. Afonso, A. Blond, N. Birlirakis, C. Goulard, L. Dubost, R. Thai, J. C. Tabet and S. Rebuffat, J. Biol. Chem., 2004, 279, 28233–28242 CrossRef CAS.
- F. Baquero, D. Bouanchaud, M. C. Martínez-Perez and C. Fernandez, J. Bacteriol., 1978, 135, 342–347 CAS.
- J. L. San Millán, C. Hernández-Chico, P. Pereda and F. Moreno, J. Bacteriol., 1985, 163, 275–281 CAS.
- J. L. San Millán, R. Kolter and F. Moreno, J. Bacteriol., 1985, 163, 1016–1020 CAS.
- M. C. Garrido, M. Herrero, R. Kolter and F. Moreno, EMBO J., 1988, 7, 1853–1862 CAS.
- O. Genilloud, F. Moreno and R. Kolter, J. Bacteriol., 1989, 171, 1126–1135 CAS.
- J. Davagnino, M. Herrero, D. Furlong, F. Moreno and R. Kolter, Proteins, 1986, 1, 230–238 CAS.
- Y. M. Li, J. C. Milne, L. L. Madison, R. Kolter and C. T. Walsh, Science, 1996, 274, 1188–1193 CrossRef CAS.
- R. Kolter and F. Moreno, Annu. Rev. Microbiol., 1992, 46, 141–163 CrossRef CAS.
- F. Moreno, J. E. Gónzalez-Pastor, M. R. Baquero and D. Bravo, Biochimie, 2002, 84, 521–529 CrossRef CAS.
- N. Connell, Z. Han, F. Moreno and R. Kolter, Mol. Microbiol., 1987, 1, 195–201 CrossRef CAS.
- J. F. García-Bustos, N. Pezzi and C. Asensio, Biochem. Biophys. Res. Commun., 1984, 119, 779–785 CrossRef CAS.
- M. A. Novoa, L. Díaz-Guerra, J. L. San Millán and F. Moreno, J. Bacteriol., 1986, 168, 1384–1391 CAS.
- N. E. Kurepina, E. I. Basyuk, A. Z. Metlitskaya, D. A. Zaitsev and I. A. Khmel, Mol. Gen. Genet., 1993, 241, 700–706 CrossRef CAS.
- J. E. Gónzalez-Pastor, J. L. San Millán, M. A. Castilla and F. Moreno, J. Bacteriol., 1995, 177, 7131–7140 CAS.
- D. E. Fomenko, A. Z. Metlitskaya, J. Peduzzi, C. Goulard, G. S. Katrukha, L. V. Gening, S. Rebuffat and I. A. Khmel, Antimicrob. Agents Chemother., 2003, 47, 2868–2874 CrossRef CAS.
- J. E. Gónzalez-Pastor, J. L. San Millán and F. Moreno, Nature, 1994, 369, 281 CrossRef CAS.
- D. Fomenko, A. Veselovskii and I. Khmel, Res. Microbiol., 2001, 152, 469–479 CrossRef CAS.
- R. A. Salomón and R. N. Farías, J. Bacteriol., 1992, 174, 7428–7435 CAS.
- J. O. Solbiati, M. Ciaccio, R. N. Farías and R. A. Salomón, J. Bacteriol., 1996, 178, 3661–3663 CAS.
- J. O. Solbiati, M. Ciaccio, R. N. Farías, J. E. Gónzalez-Pastor, F. Moreno and R. A. Salomón, J. Bacteriol., 1999, 181, 2659–2662 CAS.
- M. J. Chiuchiolo, M. A. Delgado, R. N. Farías and R. A. Salomón, J. Bacteriol., 2001, 183, 1755–1764 CrossRef CAS.
- L. Gilson, H. K. Mahanty and R. Kolter, EMBO J., 1990, 9, 3875–3884 CAS.
- C. Gaggero, F. Moreno and M. Laviña, J. Bacteriol., 1993, 175, 5420–5427 CAS.
- R. Lagos, M. Baeza, G. Corsini, C. Hetz, E. Strahsburger, J. A. Castillo, C. Vergara and O. Monasterio, Mol. Microbiol., 2001, 42, 229–243 CrossRef CAS.
- A. M. Pons, F. Delalande, M. Duarte, S. Benoit, I. Lanneluc, S. Sablé, A. Van Dorsselaer and G. Cottenceau, Antimicrob. Agents Chemother., 2004, 48, 505–513 CrossRef CAS.
- A. Gratia, C. R. Soc. Biol., 1925, 93, 1041–1042 Search PubMed.
- P. Fredericq, E. Joiris, M. Betz-Barreau and A. Gratia, CR Soc. Biol., 1949, 143, 556–559 Search PubMed.
- C. C. Yang and J. Konisky, J. Bacteriol., 1984, 158, 757–759 CAS.
- V. L. Waters and J. H. Crosa, Microbiol. Rev., 1991, 55, 437–450 CAS.
- L. Gilson, H. K. Mahanty and R. Kolter, J. Bacteriol., 1987, 169, 2466–2470 CAS.
- H. Chehade and V. Braun, FEMS Microbiol. Lett., 1988, 52, 177–182 CrossRef CAS.
- L. H. Zhang, M. J. Fath, H. K. Mahanty, P. C. Tai and R. Kolter, Genetics, 1995, 141, 25–32 Search PubMed.
- J. Hwang, X. Zhong and P. C. Tai, J. Bacteriol., 1997, 179, 6264–6270 CAS.
- X. Guo, R. W. Harrison and P. C. Tai, J. Bacteriol., 2006, 188, 2383–2391 CrossRef CAS.
- R. C. Skvirsky, S. Reginald and X. Shen, J. Bacteriol., 1995, 177, 6153–6159 CAS.
- A. E. Boyer and P. C. Tai, J. Bacteriol., 1998, 180, 1662–1672 CAS.
- T. Pinou and M. A. Riley, Plasmid, 2001, 46, 1–9 CrossRef CAS.
- S. Sablé, M. Duarte, D. Bravo, I. Lanneluc, A. M. Pons, G. Cottenceau and F. Moreno, Can. J. Microbiol., 2003, 49, 357–361 CrossRef CAS.
- G. J. O'Brien and H. K. Mahanty, Plasmid, 1994, 31, 288–296 CrossRef CAS.
- V. de Lorenzo, Arch. Microbiol., 1984, 139, 72–75 CrossRef CAS.
- M. Wilkens, J. E. Villanueva, J. Cofre, J. Chnaiderman and R. Lagos, J. Bacteriol., 1997, 179, 4789–4794 CAS.
- R. Lagos, J. E. Villanueva and O. Monasterio, J. Bacteriol., 1999, 181, 212–217 CAS.
- D. Destoumieux-Garzón, J. Peduzzi, X. Thomas, C. Djediat and S. Rebuffat, Biometals, 2006, 19, 181–191 Search PubMed.
- G. Corsini, M. Baeza, O. Monasterio and R. Lagos, Biochimie, 2002, 84, 539–544 CrossRef CAS.
- M. Laviña, C. Gaggero and F. Moreno, J. Bacteriol., 1990, 172, 6585–6588 CAS.
- A. Nissle, Dtsch. Med. Wochenschr., 1925, 44, 1809–1813 CrossRef.
- G. Blum, R. Marre and J. Hacker, Infection, 1995, 23, 234–236 CrossRef CAS.
- B. J. Rembacken, A. M. Snelling, P. M. Hawkey, D. M. Chalmers and A. T. Axon, Lancet, 1999, 354, 635–639 CrossRef CAS.
- W. Kruis, Aliment. Pharmacol. Ther., 2004, 20(suppl. 4), 75–78 Search PubMed.
- J. Papavassiliou, Nature, 1959, 184(suppl. 17), 1339–1340 CrossRef.
- S. I. Patzer, M. R. Baquero, D. Bravo, F. Moreno and K. Hantke, Microbiology, 2003, 149, 2557–2570 CrossRef CAS.
- P. Fredericq, Rev. Belg. Pathol. Med. Exp., 1948, 19(suppl. 4), 1–107 Search PubMed.
- D. E. Bradley, Can. J. Microbiol., 1991, 37, 751–757 CAS.
- E. Rodríguez, C. Gaggero and M. Laviña, Antimicrob. Agents Chemother., 1999, 43, 2176–2182 CAS.
- E. Rodríguez and M. Laviña, Can. J. Microbiol., 1998, 44, 692–697 CrossRef CAS.
- M. F. Azpiroz, E. Rodríguez and M. Laviña, Antimicrob. Agents Chemother., 2001, 45, 969–972 CrossRef CAS.
- M. F. Azpiroz and M. Laviña, Antimicrob. Agents Chemother., 2004, 48, 1235–1241 CrossRef CAS.
- M. E. Poey, M. F. Azpiroz and M. Laviña, Antimicrob. Agents Chemother., 2006, 50, 1411–1418 CrossRef CAS.
- L. Grozdanov, C. Raasch, J. Schulze, U. Sonnenborn, G. Gottschalk, J. Hacker and U. Dobrindt, J. Bacteriol., 2004, 186, 5432–5441 CrossRef CAS.
- R. A. Welch, V. Burland, G. Plunkett, 3rd, P. Redford, P. Roesch, D. Rasko, E. L. Buckles, S. R. Liou, A. Boutin, J. Hackett, D. Stroud, G. F. Mayhew, D. J. Rose, S. Zhou, D. C. Schwartz, N. T. Perna, H. L. Mobley, M. S. Donnenberg and F. R. Blattner, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 17020–17024 CrossRef CAS.
- H. Dezfulian, D. Tremblay and J. Harel, FEMS Microbiol. Lett., 2004, 238, 321–332 CAS.
- U. Dobrindt, G. Blum-Oehler, T. Hartsch, G. Gottschalk, E. Z. Ron, R. Funfstuck and J. Hacker, Infect. Immun., 2001, 69, 4248–4256 CrossRef CAS.
- J. Hacker, B. Hochhut, B. Middendorf, G. Schneider, C. Buchrieser, G. Gottschalk and U. Dobrindt, Int J. Med. Microbiol., 2004, 293, 453–461 Search PubMed.
- U. Dobrindt, B. Hochhut, U. Hentschel and J. Hacker, Nat. Rev. Microbiol., 2004, 2, 414–424 Search PubMed.
- A. Bayer, S. Freund and G. Jung, Eur. J. Biochem., 1995, 234, 414–426 CrossRef CAS.
- F. Baquero and F. Moreno, FEMS Microbiol. Lett., 1984, 23, 117–124 CrossRef CAS.
- A. Bayer, S. Freund, G. Nicholson and G. Jung, Angew. Chem., Int. Ed. Engl., 1993, 32, 1336–1339 CrossRef.
- G. I. Videnov, D. Kaiser, M. Brooks and G. Jung, Angew. Chem., Int. Ed. Engl., 1996, 35, 1506–1508 CrossRef CAS.
- J. I. Guijarro, J. E. Gónzalez-Pastor, F. Baleux, J. L. San Millán, M. A. Castilla, M. Rico, F. Moreno and M. Delepierre, J. Biol. Chem., 1995, 270, 23520–23532 CrossRef CAS.
- A. Blond, C. Goulard, D. E. Fomenko, A. Z. Metlitskaya, J. Peduzzi, M. Barthélémy, G. Katrukha, I. Khmel and S. Rebuffat, in Peptides 2000. Proceedings of the 26th European Peptide Symposium, ed. J. Martínez and J. A. Fehrentz, EDK, Paris, France, 2000, pp. 601–602 Search PubMed.
- A. Metlitskaya, T. Kazakov, A. Kommer, O. Pavlova, M. Praetorius-Ibba, M. Ibba, I. Krasheninnikov, V. Kolb, I. Khmel and K. Severinov, J. Biol. Chem., 2006, 281, 18033–18042 CrossRef CAS.
- J. F. García-Bustos, N. Pezzi and E. Mendez, Antimicrob. Agents Chemother., 1985, 27, 791–797 CAS.
- I. A. Khmel, V. M. Bondarenko, I. M. Manokhina, E. I. Basyuk, A. Z. Metlitskaya, V. A. Lipasova and Y. M. Romanova, FEMS Microbiol. Lett., 1993, 111, 269–274 CrossRef CAS.
- A. Z. Metlitskaya, G. S. Katrukha, A. S. Shashkov, D. A. Zaitsev, T. A. Egorov and I. A. Khmel, FEBS Lett., 1995, 357, 235–238 CrossRef CAS.
- A. Blond, J. Peduzzi, C. Goulard, M. J. Chiuchiolo, M. Barthélémy, Y. Prigent, R. A. Salomón, R. N. Farías, F. Moreno and S. Rebuffat, Eur. J. Biochem., 1999, 259, 747–755 CrossRef CAS.
- D. Destoumieux-Garzón, S. Duquesne, J. Péduzzi, C. Goulard, M. Desmadril, L. Letellier, S. Rebuffat and P. Boulanger, Biochem. J., 2005, 389, 869–876 CrossRef CAS.
- M. J. Bayro, J. Mukhopadhyay, G. V. Swapna, J. Y. Huang, L. C. Ma, E. Sineva, P. E. Dawson, G. T. Montelione and R. H. Ebright, J. Am. Chem. Soc., 2003, 125, 12382–12383 CrossRef CAS.
- K. J. Rosengren, R. J. Clark, N. L. Daly, U. Goransson, A. Jones and D. J. Craik, J. Am. Chem. Soc., 2003, 125, 12464–12474 CrossRef CAS.
- K. A. Wilson, M. Kalkum, J. Ottesen, J. Yuzenkova, B. T. Chait, R. Landick, T. Muir, K. Severinov and S. A. Darst, J. Am. Chem. Soc., 2003, 125, 12475–12483 CrossRef CAS.
- S. Rebuffat, A. Blond, D. Destoumieux-Garzón, C. Goulard and J. Peduzzi, Curr. Protein Pept. Sci., 2004, 5, 383–391 Search PubMed.
- A. Blond, M. Cheminant, I. Ségalas-Milazzo, J. Peduzzi, M. Barthélémy, C. Goulard, R. Salomón, F. Moreno, R. Farías and S. Rebuffat, Eur. J. Biochem., 2001, 268, 2124–2133 CrossRef CAS.
- A. Blond, M. Cheminant, D. Destoumieux-Garzón, I. Ségalas-Milazzo, J. Peduzzi, C. Goulard and S. Rebuffat, Eur. J. Biochem., 2002, 269, 6212–6222 CrossRef CAS.
- K. J. Rosengren, A. Blond, C. Afonso, J. C. Tabet, S. Rebuffat and D. J. Craik, Biochemistry, 2004, 43, 4696–4702 CrossRef CAS.
- D. Frechet, J. D. Guitton, F. Herman, D. Faucher, G. Helynck, B. Monegier duSorbier, J. P. Ridoux, E. James-Surcouf and M. Vuilhorgne, Biochemistry, 1994, 33, 42–50 CrossRef CAS.
- R. Katahira, K. Shibata, M. Yamasaki, Y. Matsuda and M. Yoshida, Bioorg. Med. Chem., 1995, 3, 1273–1280 CrossRef CAS.
- R. Katahira, M. Yamasaki, Y. Matsuda and M. Yoshida, Bioorg. Med. Chem., 1996, 4, 121–129 CrossRef CAS.
- M. Iwatsuki, H. Tomoda, R. Uchida, H. Gouda, S. Hirono and S. Omura, J. Am. Chem. Soc., 2006, 128, 7486–7491 CrossRef CAS.
- M. J. Fath, L. H. Zhang, J. Rush and R. Kolter, Biochemistry, 1994, 33, 6911–6917 CrossRef CAS.
- L. S. Håvarstein, H. Holo and I. F. Nes, Microbiology, 1994, 140, 2383–2389 CrossRef CAS.
- P. M. Muriana and T. R. Klaenhammer, J. Bacteriol., 1991, 173, 1779–1788 CAS.
- Y. Kawai, B. Saitoh, O. Takahashi, H. Kitazawa, T. Saito, H. Nakajima and T. Itoh, Biosci., Biotechnol., Biochem., 2000, 64, 2201–2208 CrossRef CAS.
- D. Destoumieux-Garzón, X. Thomas, M. Santamaria, C. Goulard, M. Barthélémy, B. Boscher, Y. Bessin, G. Molle, A. M. Pons, L. Letellier, J. Péduzzi and S. Rebuffat, Mol. Microbiol., 2003, 49, 1031–1041 CrossRef CAS.
- A. M. Pons, N. Zorn, D. Vignon, F. Delalande, A. Van Dorsselaer and G. Cottenceau, Antimicrob. Agents Chemother., 2002, 46, 229–230 CrossRef CAS.
- K. Hantke, G. Nicholson, W. Rabsch and G. Winkelmann, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3677–3682 CrossRef CAS.
- S. S. Pao, I. T. Paulsen and M. H. Saier, Jr., Microbiol. Mol. Biol. Rev., 1998, 62, 1–34 CAS.
- M. A. Delgado, J. O. Solbiati, M. J. Chiuchiolo, R. N. Farías and R. A. Salomón, J. Bacteriol., 1999, 181, 1968–1970 CAS.
- S. K. Buchanan, Trends Biochem. Sci., 2001, 26, 3–6 CrossRef CAS.
- M. A. Delgado and R. A. Salomón, Plasmid, 2005, 53, 258–262 CrossRef CAS.
- P. M. Jones and A. M. George, Cell. Mol. Life Sci., 2004, 61, 682–699 CrossRef CAS.
- I. B. Holland, L. Schmitt and J. Young, Mol. Membr. Biol., 2005, 22, 29–39 CAS.
- L. S. Håvarstein, D. B. Diep and I. F. Nes, Mol. Microbiol., 1995, 16, 229–240 CAS.
- M. A. Blight, C. Chervaux and I. B. Holland, Curr. Opin. Biotechnol., 1994, 5, 468–474 CrossRef CAS.
- C. M. Franke, J. Tiemersma, G. Venema and J. Kok, J. Biol. Chem., 1999, 274, 8484–8490 CrossRef CAS.
- T. K. Wu, C. Y. Huang, C. Y. Ko, C. H. Chang, Y. J. Chen and H. K. Liao, Arch. Biochem. Biophys., 2004, 421, 42–53 CrossRef CAS.
- H. Benabdelhak, S. Kiontke, C. Horn, R. Ernst, M. A. Blight, I. B. Holland and L. Schmitt, J. Mol. Biol., 2003, 327, 1169–1179 CrossRef CAS.
- C. M. Franke, K. J. Leenhouts, A. J. Haandrikman, J. Kok, G. Venema and K. Venema, J. Bacteriol., 1996, 178, 1766–1769 CAS.
- C. Chatterjee, M. Paul, L. Xie and W. A. Van Der Donk, Chem. Rev., 2005, 105, 633–684 CrossRef CAS.
- F. Corpet, Nucleic Acids Res., 1988, 16, 10881–10890 CAS.
- M. J. van Belkum, R. W. Worobo and M. E. Stiles, Mol. Microbiol., 1997, 23, 1293–1301 CrossRef CAS.
- Z. J. Xu, D. B. Moffett, T. R. Peters, L. D. Smith, B. P. Perry, J. Whitmer, S. A. Stokke and M. Teintze, J. Biol. Chem., 1995, 270, 24858–24863 CrossRef CAS.
- B. R. Hubbard, M. Jacobs, M. M. Ulrich, C. Walsh, B. Furie and B. C. Furie, J. Biol. Chem., 1989, 264, 14145–14150 CAS.
- P. Huber, T. Schmitz, J. Griffin, M. Jacobs, C. Walsh, B. Furie and B. C. Furie, J. Biol. Chem., 1990, 265, 12467–12473 CAS.
- L. L. Madison, E. I. Vivas, Y. M. Li, C. T. Walsh and R. Kolter, Mol. Microbiol., 1997, 23, 161–168 CAS.
- C. K. Murphy and J. Beckwith, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2557–2561 CrossRef CAS.
- T. den Blaauwen, P. Fekkes, J. G. de Wit, W. Kuiper and A. J. Driessen, Biochemistry, 1996, 35, 11994–12004 CrossRef CAS.
- R. S. Roy, O. Allen and C. T. Walsh, Chem. Biol., 1999, 6, 789–799 CrossRef CAS.
- K. K. Frick, R. L. Quackenbush and J. Konisky, J. Bacteriol., 1981, 148, 498–507 CAS.
- P. Yorgey, J. Davagnino and R. Kolter, Mol. Microbiol., 1993, 9, 897–905 CAS.
- M. C. Rodríguez-Sainz, C. Hernández-Chico and F. Moreno, Mol. Microbiol., 1990, 4, 1921–1932 CrossRef CAS.
- N. Allali, H. Afif, M. Couturier and L. Van Melderen, J. Bacteriol., 2002, 184, 3224–3231 CrossRef CAS.
- X. Zhong, R. Kolter and P. C. Tai, J. Biol. Chem., 1996, 271, 28057–28063 CrossRef CAS.
- J. C. Milne, A. C. Eliot, N. L. Kelleher and C. T. Walsh, Biochemistry, 1998, 37, 13250–13261 CrossRef CAS.
- J. C. Milne, R. S. Roy, A. C. Eliot, N. L. Kelleher, A. Wokhlu, B. Nickels and C. T. Walsh, Biochemistry, 1999, 38, 4768–4781 CrossRef CAS.
- N. L. Kelleher, C. L. Hendrickson and C. T. Walsh, Biochemistry, 1999, 38, 15623–15630 CrossRef CAS.
- R. S. Roy, S. Kim, J. D. Baleja and C. T. Walsh, Chem. Biol., 1998, 5, 217–228 CAS.
- R. S. Roy, P. J. Belshaw and C. T. Walsh, Biochemistry, 1998, 37, 4125–4136 CrossRef CAS.
- S. V. Taylor, N. L. Kelleher, C. Kinsland, H. J. Chiu, C. A. Costello, A. D. Backstrom, F. W. McLafferty and T. P. Begley, J. Biol. Chem., 1998, 273, 16555–16560 CrossRef CAS.
- S. Leimkühler, M. M. Wuebbens and K. V. Rajagopalan, J. Biol. Chem., 2001, 276, 34695–34701 CrossRef CAS.
- M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 571–576 CrossRef CAS.
- H. Lin, M. A. Fischbach, D. R. Liu and C. T. Walsh, J. Am. Chem. Soc., 2005, 127, 11075–11084 CrossRef CAS.
- M. Zhu, M. Valdebenito, G. Winkelmann and K. Hantke, Microbiology, 2005, 151, 2363–2372 CrossRef CAS.
- A. J. Bäumler, T. L. Norris, T. Lasco, W. Voight, R. Reissbrodt, W. Rabsch and F. Heffron, J. Bacteriol., 1998, 180, 1446–1453 CAS.
- C. Hernández-Chico, J. L. San Millán, R. Kolter and F. Moreno, J. Bacteriol., 1986, 167, 1058–1065 CAS.
- V. de Lorenzo, J. L. Martínez and C. Asensio, J. Gen. Microbiol., 1984, 130, 391–400 CAS.
- Q. Gao, A. Fang and A. L. Demain, J. Ind. Microbiol. Biotechnol., 2001, 26, 341–344 CrossRef CAS.
- V. de Lorenzo, J. Antibiot., 1985, 38, 340–345 CAS.
- Q. Gao, A. Fang, D. L. Pierson, S. K. Mishra and A. L. Demain, Appl. Microbiol. Biotechnol., 2001, 56, 384–387 Search PubMed.
- D. E. Bohannon, N. Connell, J. Keener, A. Tormo, M. Espinosa-Urgel, M. M. Zambrano and R. Kolter, J. Bacteriol., 1991, 173, 4482–4492 CAS.
- L. Díaz-Guerra, F. Moreno and J. L. San Millán, J. Bacteriol., 1989, 171, 2906–2908 CAS.
- O. Lomovskaya, K. Lewis and A. Matin, J. Bacteriol., 1995, 177, 2328–2334 CAS.
- I. del Castillo, J. M. Gómez and F. Moreno, J. Bacteriol., 1990, 172, 437–445 CAS.
- K. Hantke, Curr. Opin. Microbiol., 2001, 4, 172–177 CrossRef CAS.
- R. A. Salomón and R. N. Farías, FEMS Microbiol. Lett., 1994, 121, 275–279 CrossRef CAS.
- C. Orellana and R. Lagos, FEMS Microbiol. Lett., 1996, 136, 297–303 CAS.
- M. R. Baquero, M. Bouzon, J. Varea and F. Moreno, Mol. Microbiol., 1995, 18, 301–311 CrossRef CAS.
- A. P. Pugsley, F. Moreno and V. de Lorenzo, J. Gen. Microbiol., 1986, 132, 3253–3259 CAS.
- M. Laviña, A. P. Pugsley and F. Moreno, J. Gen. Microbiol., 1986, 132, 1685–1693 CAS.
- R. A. Salomón and R. N. Farías, J. Bacteriol., 1993, 175, 7741–7742 CAS.
- R. A. Salomón and R. N. Farías, J. Bacteriol., 1995, 177, 3323–3325 CAS.
- M. Trujillo, E. Rodríguez and M. Laviña, Antimicrob. Agents Chemother., 2001, 45, 3128–3131 CrossRef CAS.
- A. D. Ferguson and J. Deisenhofer, Biochim. Biophys. Acta, 2002, 1565, 318–332 CAS.
- L. Letellier and M. Santamaria, Mini-Rev. Med. Chem., 2002, 2, 343–351 Search PubMed.
- A. D. Ferguson, E. Hofmann, J. W. Coulton, K. Diederichs and W. Welte, Science, 1998, 282, 2215–2220 CrossRef CAS.
- K. P. Locher, B. Rees, R. Koebnik, A. Mitschler, L. Moulinier, J. P. Rosenbusch and D. Moras, Cell, 1998, 95, 771–778 CrossRef CAS.
- S. K. Buchanan, B. S. Smith, L. Venkatramani, D. Xia, L. Esser, M. Palnitkar, R. Chakraborty, D. Van Der Helm and J. Deisenhofer, Nat. Struct. Biol., 1999, 6, 56–63 CrossRef CAS.
- R. Annamalai, B. Jin, Z. Cao, S. M. Newton and P. E. Klebba, J. Bacteriol., 2004, 186, 3578–3589 CrossRef CAS.
- F. Endriss and V. Braun, J. Bacteriol., 2004, 186, 4818–4823 CrossRef CAS.
- K. Postle and R. J. Kadner, Mol. Microbiol., 2003, 49, 869–882 CrossRef CAS.
- H. Killmann, C. Herrmann, H. Wolff and V. Braun, J. Bacteriol., 1998, 180, 3845–3852 CAS.
- P. A. Vincent, M. A. Delgado, R. N. Farías and R. A. Salomón, FEMS Microbiol. Lett., 2004, 236, 103–107 CrossRef CAS.
- S. Bieler, F. Silva, C. Soto and D. Belin, J. Bacteriol., 2006, 188, 7049–7061 CrossRef CAS.
- Z. Cao and P. E. Klebba, Biochimie, 2002, 84, 399–412 CrossRef CAS.
- M. Ferrario, B. R. Ernsting, D. W. Borst, D. E. Wiese, 2nd, R. M. Blumenthal and R. G. Matthews, J. Bacteriol., 1995, 177, 103–113 CrossRef CAS.
- K. Hantke, Mol. Gen. Genet., 1984, 197, 337–341 CrossRef CAS.
- L. Escolar, J. Perez-Martin and V. de Lorenzo, J. Bacteriol., 1998, 180, 2579–2582 CAS.
- F. Gerard, N. Pradel and L. F. Wu, J. Bacteriol., 2005, 187, 1945–1950 CrossRef CAS.
- N. A. Likhacheva, V. V. Samsonov and S. P. Sineoky, J. Bacteriol., 1996, 178, 5309–5315 CAS.
- V. V. Samsonov and S. P. Sineoky, Res. Microbiol., 2002, 153, 639–646 CrossRef CAS.
- P. Yorgey, J. Lee, J. Kordel, E. Vivas, P. Warner, D. Jebaratnam and R. Kolter, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4519–4523 CAS.
- R. E. de Cristóbal, J. O. Solbiati, A. M. Zenoff, P. A. Vincent, R. A. Salomón, J. Yuzenkova, K. Severinov and R. N. Farías, J. Bacteriol., 2006, 188, 3324–3328 CrossRef CAS.
- K. Matsuzaki, K. Sugishita, M. Harada, N. Fujii and K. Miyajima, Biochim. Biophys. Acta, 1997, 119–130, 1327.
- D. Gidalevitz, Y. Ishitsuka, A. S. Muresan, O. Konovalov, A. J. Waring, R. I. Lehrer and K. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6302–6307 CrossRef CAS.
- A. Bohling, S. O. Hagge, S. Roes, R. Podschun, H. Sahly, J. Harder, J. M. Schroder, J. Grotzinger, U. Seydel and T. Gutsmann, Biochemistry, 2006, 45, 5663–5670 CrossRef.
- M. R. Rintoul, B. F. de Arcuri, R. A. Salomón, R. N. Farías and R. D. Morero, FEMS Microbiol. Lett., 2001, 204, 265–270 CrossRef CAS.
- M. R. Rintoul, B. F. de Arcuri and R. D. Morero, Biochim. Biophys. Acta, 2000, 1509, 65–72 CAS.
- A. Bellomio, R. G. Oliveira, B. Maggio and R. D. Morero, J. Colloid Interface Sci., 2005, 285, 118–124 CrossRef CAS.
- R. Lagos, M. Wilkens, C. Vergara, X. Cecchi and O. Monasterio, FEBS Lett., 1993, 321, 145–148 CrossRef CAS.
- V. de Lorenzo and A. P. Pugsley, Antimicrob. Agents Chemother., 1985, 27, 666–669 CAS.
- N. Williams, D. K. Fox, C. Shea and S. Roseman, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 8934–8938 CrossRef CAS.
- B. Erni, B. Zanolari and H. P. Kocher, J. Biol. Chem., 1987, 262, 5238–5247 CAS.
- E. Rodríguez and M. Laviña, Antimicrob. Agents Chemother., 2003, 47, 181–187 CrossRef CAS.
- M. Herrero and F. Moreno, J. Gen. Microbiol., 1986, 132, 393–402 CAS.
- J. L. Vizán, C. Hernández-Chico, I. del Castillo and F. Moreno, EMBO J., 1991, 10, 467–476 CAS.
- F. J. del Castillo, I. del Castillo and F. Moreno, J. Bacteriol., 2001, 183, 2137–2140 CrossRef CAS.
- R. S. Roy, N. L. Kelleher, J. C. Milne and C. T. Walsh, Chem. Biol., 1999, 6, 305–318 CrossRef CAS.
- D. B. Zamble, D. A. Miller, J. G. Heddle, A. Maxwell, C. T. Walsh and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7712–7717 CrossRef CAS.
- O. A. Pierrat and A. Maxwell, J. Biol. Chem., 2003, 278, 35016–35023 CrossRef CAS.
- O. A. Pierrat and A. Maxwell, Biochemistry, 2005, 44, 4204–4215 CrossRef CAS.
- M. A. Delgado, M. R. Rintoul, R. N. Farías and R. A. Salomón, J. Bacteriol., 2001, 183, 4543–4550 CrossRef CAS.
- J. Yuzenkova, M. Delgado, S. Nechaev, D. Savalia, V. Epshtein, I. Artsimovitch, R. Mooney, R. Landick, R. N. Farías, R. Salomón and K. Severinov, J. Biol. Chem., 2002, 277, 50867–50875 CrossRef CAS.
- K. Adelman, J. Yuzenkova, A. La Porta, N. Zenkin, J. Lee, J. T. Lis, S. Borukhov, M. D. Wang and K. Severinov, Mol. Cell, 2004, 14, 753–762 CrossRef CAS.
- J. Mukhopadhyay, E. Sineva, J. Knight, R. M. Levy and R. H. Ebright, Mol. Cell, 2004, 14, 739–751 CrossRef CAS.
- E. Semenova, Y. Yuzenkova, J. Peduzzi, S. Rebuffat and K. Severinov, J. Bacteriol., 2005, 187, 3859–3863 CrossRef CAS.
- F. J. Van Der Wal, J. Luirink and B. Oudega, FEMS Microbiol. Rev., 1995, 17, 381–399 CrossRef CAS.
- C. Janion, Acta Biochim. Pol., 2001, 48, 599–610 CAS.
- B. Salles, J. M. Weisemann and G. M. Weinstock, J. Bacteriol., 1987, 169, 5028–5034 CAS.
- I. Kuhar and D. Zgur-Bertok, J. Bacteriol., 1999, 181, 7373–7380 CAS.
- H. Benedetti, M. Frenette, D. Baty, M. Knibiehler, F. Pattus and C. Lazdunski, J. Mol. Biol., 1991, 217, 429–439 CrossRef CAS.
- A. P. Pugsley and M. Schwartz, J. Bacteriol., 1983, 156, 109–114 CAS.
- D. Baty, R. Lloubes, V. Geli, C. Lazdunski and S. P. Howard, EMBO J., 1987, 6, 2463–2468 CAS.
- F. J. Van Der Wal, G. Koningstein, C. M. ten Hagen, B. Oudega and J. Luirink, Appl. Environ. Microbiol., 1998, 64, 392–398 CAS.
- R. James, C. N. Penfold, G. R. Moore and C. Kleanthous, Biochimie, 2002, 84, 381–389 CrossRef CAS.
- W. A. Cramer, J. B. Heymann, S. L. Schendel, B. N. Deriy, F. S. Cohen, P. A. Elkins and C. V. Stauffacher, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 611–641 CrossRef CAS.
- D. Duche, Biochimie, 2002, 84, 455–464 CrossRef CAS.
- W. Vollmer, H. Pilsl, K. Hantke, J. V. Holtje and V. Braun, J. Bacteriol., 1997, 179, 1580–1583 CAS.
- M. El Ghachi, A. Bouhss, H. Barreteau, T. Touze, G. Auger, D. Blanot and D. Mengin-Lecreulx, J. Biol. Chem., 2006, 281, 22761–22772 CrossRef CAS.
- S. Garneau, N. I. Martin and J. C. Vederas, Biochimie, 2002, 84, 577–592 CrossRef CAS.
- R. W. Jack and H. G. Sahl, Trends Biotechnol., 1995, 13, 269–278 CrossRef CAS.
- G. C. Patton and W. A. Van Der Donk, Curr. Opin. Microbiol., 2005, 8, 543–551 CrossRef CAS.
- E. Breukink, H. E. van Heusden, P. J. Vollmerhaus, E. Swiezewska, L. Brunner, S. Walker, A. J. Heck and B. de Kruijff, J. Biol. Chem., 2003, 278, 19898–19903 CrossRef CAS.
- H. E. Hasper, B. de Kruijff and E. Breukink, Biochemistry, 2004, 43, 11567–11575 CrossRef CAS.
- G. Fimland, L. Johnsen, B. Dalhus and J. Nissen-Meyer, J. Pept. Sci., 2005, 11, 688–696 CrossRef CAS.
- K. Dalet, Y. Cenatiempo, P. Cossart and Y. Hechard, Microbiology, 2001, 147, 3263–3269 CAS.
- M. Ramnath, S. Arous, A. Gravesen, J. W. Hastings and Y. Hechard, Microbiology, 2004, 150, 2663–2668 CrossRef CAS.
- A. H. Keeble and C. Kleanthous, J. Mol. Biol., 2005, 352, 656–671 CrossRef CAS.
- D. Smajs, P. Matejkova and G. M. Weinstock, FEMS Microbiol. Lett., 2006, 258, 108–113 CrossRef CAS.
- S. Schauder and B. L. Bassler, Genes Dev., 2001, 15, 1468–14680 CrossRef CAS.
- M. B. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55, 165–199 CrossRef CAS.
- N. C. Reading and V. Sperandio, FEMS Microbiol. Lett., 2006, 254, 1–11 CrossRef CAS.
- M. Kleerebezem and L. E. Quadri, Peptides, 2001, 22, 1579–1596 CrossRef CAS.
- P. E. Kristiansen, G. Fimland, D. Mantzilas and J. Nissen-Meyer, J. Biol. Chem., 2005, 280, 22945–22950 CrossRef CAS.
- M. A. Riley, C. M. Goldstone, J. E. Wertz and D. Gordon, J. Evol. Biol., 2003, 16, 690–697 CrossRef CAS.
- F. Baquero and C. Asensio, in New criteria for antimicrobial therapy: maintenance of digestive tract colonization resistance, ed. D. van der Waaij and J. Verhoef, Excerpta Medica, Amsterdam, 1979, pp. 90–94 Search PubMed.
- R. E. Wooley, P. S. Gibbs and E. B. Shotts, Jr., Avian Dis., 1999, 43, 245–250 Search PubMed.
- T. S. Frana, S. A. Carlson, D. C. Rauser, B. D. Jones, B. J. Fergen and R. W. Griffith, Am. J. Vet. Res., 2004, 65, 1616–1620 Search PubMed.
- M. A. Riley and J. E. Wertz, Annu. Rev. Microbiol., 2002, 56, 117–137 CrossRef CAS.
- M. A. Riley and J. E. Wertz, Biochimie, 2002, 84, 357–364 CrossRef CAS.
- O. Gillor, B. C. Kirkup and M. A. Riley, Adv. Appl. Microbiol., 2004, 54, 129–146 Search PubMed.
- B. C. Kirkup and M. A. Riley, Nature, 2004, 428, 412–414 CrossRef CAS.
- B. Kerr, M. A. Riley, M. W. Feldman and B. J. Bohannan, Nature, 2002, 418, 171–174 CrossRef CAS.
- A. Altenhoefer, S. Oswald, U. Sonnenborn, C. Enders, J. Schulze, J. Hacker and T. A. Oelschlaeger, FEMS Immunol. Med. Microbiol., 2004, 40, 223–229 CrossRef CAS.
- S. Kleta, H. Steinruck, G. Breves, S. Duncker, C. Laturnus, L. H. Wieler and P. Schierack, J. Appl. Microbiol., 2006, 101, 1357–1366 CrossRef CAS.
- L. Cursino, D. Smajs, J. Smarda, R. M. Nardi, J. R. Nicoli, E. Chartone-Souza and A. M. Nascimento, J. Appl. Microbiol., 2006, 100, 821–829 CrossRef CAS.
- M. Duarte, G. Cottenceau, V. Portrait and A. M. Pons, Can. J. Microbiol., 2001, 47, 877–882 CrossRef CAS.
- C. Hetz, M. R. Bono, L. F. Barros and R. Lagos, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2696–2701 CrossRef CAS.
- Y. Weinrauch and A. Zychlinsky, Annu. Rev. Microbiol., 1999, 53, 155–187 CrossRef CAS.
- X. Q. Qiu, H. Wang, X. F. Lu, J. Zhang, S. F. Li, G. Cheng, L. Wan, L. Yang, J. Y. Zuo, Y. Q. Zhou, H. Y. Wang, X. Cheng, S. H. Zhang, Z. R. Ou, Z. C. Zhong, J. Q. Cheng, Y. P. Li and G. Y. Wu, Nat. Biotechnol., 2003, 21, 1480–1485 CrossRef CAS.
- X. Q. Qiu, J. Zhang, H. Wang and G. Y. Wu, Antimicrob. Agents Chemother., 2005, 49, 1184–1189 CrossRef CAS.
- J. K. McCormick, T. R. Klaenhammer and M. E. Stiles, Lett. Appl. Microbiol., 1999, 29, 37–41 CrossRef CAS.
- M. Ghosh and M. J. Miller, Bioorg. Med. Chem., 1996, 4, 43–48 CrossRef CAS.
- B. A. Weissberger, G. K. Abruzzo, R. A. Fromtling, C. Gill, S. Ponticas, M. E. Valiant, D. L. Shungu and H. H. Gadebusch, J. Antibiot., 1989, 42, 795–806 CAS.
| This journal is © The Royal Society of Chemistry 2007 |