DOI:
10.1039/B617425F
(Paper)
New J. Chem., 2007,
31, 549-555
Chemoselectivity in σ bond activation by lanthanocene complexes from a DFT perspective: reactions of Cp2LnR (R = CH3, H, SiH3) with SiH4 and CH3–SiH3†‡
Received
(in Montpellier, France)
29th November 2006
, Accepted 18th January 2007
First published on 21st February 2007
Abstract
The pathways for the σ bond metathesis reactions between Cp2LnCH3 and SiH4 to give either Cp2LnSiH3 and CH4 (CH3/SiH3 exchange) or Cp2LnH and H3C–SiH3 (Si–C coupling) have been studied using DFT(B3PW91) calculations. It is shown that the nature of the lanthanide atom has essentially no influence on the free enthalpy profile. All reactions that could occur between H3C–SiH3, formed from the reaction between the initial reagents (Cp2LnCH3 and SiH4), and the lanthanocene complexes (Cp2LnH or Cp2LnSiH3), have been then studied for La only. The activation of the Si–H bond is preferred over that of Si–C or C–H bonds. In addition, in the reaction of Cp2LaH with SiH3CH3, the silyl group favours the formation of the C-bonded alkylsilyl complex. The activation of the Si–H bond is not selective, i.e. Si can be at either the α or β sites, with respect to the metal center, in the 4-center transition state. The reaction between Cp2LaSiH3 and SiH4 is preferentially the exchange of SiH3 groups over the Si–Si coupling, mostly for thermodynamic reasons. The Si–Si coupling is however not strongly disfavoured by thermodynamics and has an accessible activation energy.
Introduction
Since the discovery in the 1980’s by Watson et al.1,2 that Cp*2LuCH3 (Cp* = η5-C5Me5) activates inert σ bonds such as the methane C–H bond, organolanthanide complexes have gained attention in the chemical community.3 Experimental work has shown that various lanthanocene derivatives can activate not only C–H1,4–7 in alkanes and arenes but also the C–F bond8,9 in fluoroalkanes and fluoroarenes, as well as the Si–H bonds in alkyl- and arylsilanes.10–17 However, these complexes rarely lead to homocoupling products via C–C, Si–Si or cross coupling products via Si–C bond formation.16,17 Formation of C–C bonds is observed because the lanthanide complexes are also Ziegler–Natta type polymerization catalysts.18 However, examples of Si–C and Si–Si coupling/decoupling reactions are known. Silane dehydropolymerization has been proposed to occur via
σ bond metathesis.13,14,16 This reaction, which has been found for early transition metal and lanthanide complexes, is supposed to occur by successive Si–H bond activations. In the reaction of C6H5–SiH3 with Cp*2SmH, the trimer of Cp*2Sm–SiH3 is the main product formed together with redistribution products, such as C6H6, Ph2SiH2, Ph3SiH or SiH4, as well as some Si–Si coupling products. In this case, the redistribution products originate from a Si–C coupling reaction.16
Considerable amounts of information on the chemistry of silanes with various metals, including lanthanides, can be found in the review of Corey and Braddock-Wilking.19 However, the factors which control the reactivity preferences (selectivity) with lanthanide complexes are still unclear and a theoretical study is then the method of choice to investigate the elementary steps that are needed to account for the variety of products. Several reactions implying C–H bond activations have been studied computationally.20–22 In the case of silane, computational works have shown that the reaction of Cp2LnH with SiH4 to give Cp2LnSiH3 and H2 has a low energy barrier and is thermodynamically accessible. The thermodynamically neutral reaction that exchanges the hydrogen atoms of the lanthanide complex and the silane has also a low energy barrier.23 Calculations by Ziegler and Folga using the ADF program have shown that the reaction of Cl2ScSiH3 with SiH4 could lead to the formation of SiH3–SiH3 with an accessible energy barrier.24 In no case was the cross coupling reaction involving the formation and cleavage of an Si–C bond studied.
In this paper, we have used DFT calculations to determine the energy profiles for the reactions between Cp2LnCH3 and SiH4 to form Cp2LnSiH3 and CH4 (termed the CH3/SiH3 exchange) and to form Cp2LnH and CH3–SiH3 (termed the C–Si coupling). The influence of the nature of the lanthanide metal on the energy profiles has been also studied. Since the influence of the metal has been found to be small, reactions that could occur between Cp2LnH and the products obtained from the first set of reactions have been studied for La only. These reactions include the activations of the C–H, Si–H and C–Si bonds from CH3–SiH3. In addition, the calculation of the energy profile for the reaction of Cp2Ln–SiH3 with SiH4 allows discussion of some aspects of the dehydropolymerization of silanes. All reactions considered are shown in eqns (1)–(10). The reaction of Cp2LnH with SiH4, which has been previously published, will be also mentioned.23
| | | Cp2Ln–CH3 + H–SiH3
→ Cp2Ln–SiH3 + H–CH3 | (1) |
| | | Cp2Ln–CH3 + H–SiH3
→ Cp2Ln–H + CH3–SiH3 | (2) |
| | | Cp2Ln–H + H–SiH2CH3
→ Cp2Ln–H + H–SiH2CH3 | (3) |
| | | Cp2Ln–H + H–SiH2CH3
→ Cp2Ln–SiH2CH3 + H–H | (4) |
| | | Cp2Ln–H + H–CH2SiH3
→ Cp2Ln–H + H–CH2SiH3 | (5) |
| | | Cp2Ln–H + H–CH2SiH3
→ Cp2Ln–CH2SiH3 + H–H | (6) |
| | | Cp2Ln–H + H3Si–CH3
→ Cp2Ln–SiH3 + H–CH3 | (7) |
| | | Cp2Ln–H + H3Si–CH3
→ Cp2Ln–CH3 + H–SiH3 | (8) |
| | | Cp2Ln–SiH3 + H–SiH3
→ Cp2Ln–H + H3Si–SiH3 | (9) |
| | | Cp2Ln–SiH3 + H–SiH3
→ Cp2Ln–SiH3 + H–SiH3 | (10) |
Computational details
The lanthanide metal centers have been represented with the large core pseudo-relativistic effective core potential (RECP) from the Stuttgart group25–28 and the basis set associated with these RECPs has been augmented by an f polarization function. The carbon and hydrogen atoms are treated with an all-electron double-ζ, 6-31G(d,p), basis set.29 Silicon atom has been treated with an RECP with the adapted basis set,30 augmented by a d polarization function. All the calculations have been carried out with the Gaussian 98 suite of programs31 at the DFT level using the B3PW91 hybrid functional.32,33 The geometry optimizations have been performed without any symmetry constraints. The nature of all extrema has been verified by analytic determination of frequencies and the connections between transition states and minima have been established by an optimization of the structures distorted along the intrinsic reaction coordinate (IRC). The Gibbs energies (free enthalpies) have been calculated at 298.15 K within the harmonic approximation for frequencies. The coordinates, energy and free enthalpies of all calculated species in the case of La are given in the ESI.†
Results and discussion
Reactions of Cp2LaCH3 with SiH4
For convenience, [M] represents Cp2M. The variation of Gibbs energies, ΔG, relative to the separate reagents, is used for discussing the reaction profiles. It has been verified that using the variation of potential energies ΔE in place of ΔG does not modify the relative positions of transition states and minima for the reactions studied and thus leads to the same interpretation of the results. The entropic term has its main effect in reactions involving change of molecularity (formation of complexes from separate reactants and associated reverse reactions).
The calculated Gibbs energy profiles for reactions shown in eqn (1) and eqn (2) are presented in Fig. 1 and the structures of the extrema are shown in Fig. 2. The CH3/SiH3 exchange reaction (right-hand side of Fig. 1) yields [La]–SiH3 and CH4, 2, after formation of the first adduct ([La]–CH3)(SiH4), 7, passage through transition state 8 and decomposition of the adduct ([La]–SiH3)(CH4) 9. It should be noted that the relative potential energies (ΔE) of the adducts are lower than that of the separate reactants but that their relative Gibbs energies are higher due to the entropic factor associated with the change of molecularity resulting from the formation of one complex from two molecules. From the literature, it appears that the translational part of the entropic factor, which contributes most to the variation in entropy, may be exaggerated so that the free enthalpy of the adducts and transition state may be closer in energy to the separate reactants than indicated by the calculated ΔG.34 This leads to no modification of the results in this work. The SiH3–CH3 coupling reaction (left-hand side of Fig. 1) yields [La]–H and H3C–SiH3 (3) via the adduct 6, the transition state 5 and the adduct 4. The two reactions are exoergic, the larger free enthalpy of reaction being for the formation of [La]–SiH3 and CH4. The thermodynamic preference for formation of the silyl complex and methane over the hydride complex and methylsilane is in line with previous studies: the reaction of [La]–H with CH4 to form [La]–CH3 and H2 is endoergic while that of [La]–H with SiH4 to form [La]–SiH3 and H2 is exoergic.20,23,35 These thermodynamic features are associated with the variation in bond strength in the metallic complex and the non-metallic species. Due to the difficulty in calculating a homolytic Ln–X bond dissociation energy when using large core ECP, we only calculated the bond free energies (Si–H, C–H and Si–C) in SiH4, CH4 and SiH3–CH3. Previous studies on bond dissociation energies have shown that DFT performs correctly on relative bond dissociation energies even though some detailed substituent effects are more difficult to represent.36 The calculated bond dissociation free enthalpies BDG, and associated bond dissociation energies, BDE, are: BDG 78.8, BDE 91.8 for Si–H in SiH4, BDG 96.0, BDE 112.3 for C–H in CH4, and BDG = 69.6, BDE 86.4 kcal mol−1 for Si–C in CH3–SiH3. The energy of reaction of eqn (1) is therefore in large part determined by the difference of −17.2 kcal mol−1 in energy between the weaker Si–H and the stronger C–H bonds suggesting that the La–Si and La–C bond strengths are of similar magnitude since the energy of the bond metathesis reaction is −15.2 kcal mol−1. In contrast, for eqn (2), the variation in free energy of +9.2 kcal mol−1 associated with the loss of the Si–H bond and the formation of the Si–C bond would give an endoergic reaction. This is thus compensated by the variation in free energies in the lanthanide complexes and confirms that the La–H bond energy is stronger than the La–C(alkyl) bond energy. This analysis thus suggests that the La–H is stronger than the La–C and La–Si bonds, which are of similar strength.
 |
| | Fig. 1 Free enthalpy profiles (kcal mol−1) for the CH3/SiH3 exchange reaction (eqn (1)) and formation of CH3–SiH3 (eqn (2)). | |
 |
| | Fig. 2 Optimized structures of the extrema for the reactions of eqns (1) and (2). The distances are given in Å. | |
We describe qualitatively the geometries of the intermediates and transition state of the CH3/SiH3 exchange reaction (eqn (1)) because none of the values is remarkable. Quantitative geometrical features are given in the ESI.† The reaction starts with the formation of a σ-adduct of SiH47. In 7, two H of SiH4 are close to La, which indicates that SiH4 is η3-coordinated to La. SiH4 coordination is endoergic by 7 kcal mol−1, which is due, as mentioned earlier, to the loss of the translational entropy. The adduct 7 leads to a σ-bond metathesis transition state, 8, in which SiH3 and H are at the α and β positions, respectively of the 4-center transition state (Fig. 2). At the transition state 8, the carbon atom, the transferring H and the silicon atom are almost aligned. Similar situations have been obtained for the reactions of [Ln]–H or [Ln]–CH3 with H2, CH4 and SiH4.20–23,37 The alignment of C, H and Si clearly suggests that this elementary step is a proton transfer between the negatively charged groups, SiH3 and CH3, in the stabilizing field of the positively charged lanthanide atom. The calculated activation energy of 18.3 kcal mol−1 is relatively low, which suggests that this elementary step is energetically accessible. The activation energy is calculated to be significantly lower than that for CH4 activation by [La]–CH3, for which a value of 28.5 kcal mol−1 was found at the same level of methodology.22 The transition state 8 leads to a complex between [La]–SiH3 and CH4, 9, where the methane is η3-bonded to La. Even if CH4 is bonded to La with two C–H bonds, the SiH3 group distorts to establish one α-agostic Si–H bond. This contrasts with the structure of the SiH4 adduct 7 where the methyl group is not α-agostic. The Si–H bond has a greater propensity to become agostic than C–H bond due to the more hydridic character of the silicon hydride.38
The CH3–SiH3 coupling reaction (Fig. 1 left) is initiated by the formation of a silane adduct 6, which differs from 7 in the orientation of the Si–H bond relative to the La–C bond but has almost the same free enthalpy (6.3 kcal mol−1). In 6, SiH4 is η1-coordinated to La. This coordination mode, which is less rotationally restrained, has thus a lower free enthalpy than 7. Species 6 leads to the transition state 5, in which SiH3 is at the β position and H at the α position of the 4-center transition state 5 (Fig. 2). In 5, the silicon has a square pyramidal geometry as previously obtained in the reaction of [La]–H with SiH4.23 The transition state 5, which is 20.6 kcal mol−1 above the separated reactants, leads to a CH3–SiH3 adduct 4, in which CH3 is close to the metal. This adduct is almost at the same free enthalpy (ΔG = 2.6 kcal mol−1) as the separate reactants so that decoordination of the methylsilane further lowers the free enthalpy by 6.7 kcal mol−1.
The reactions of eqns (1) and (2) have comparable activation energies and therefore the two reactions should both be observed. This is due to the ability of the SiH3 group to occupy the α or the β positions of the 4-center transition state of a σ bond metathesis transition state with almost equal energies as noted previously for the reactions of Cp2LnH with SiH4.23 Formation of the metal hydride as well as of the metal silyl is thus expected. These two complexes can themselves react with the reactant or with the products obtained from these two reactions. A priori, one should study the reactions of the hydride and the silyl complexes with the C–H, Si–H and Si–C bonds of the various possible reactants. However, it now appears that the energy profiles of the reactions are qualitatively similar for the hydride and for the silyl complex; consequently, we have focused on the reactions of the various reactants with the hydride complex. Nevertheless, the reaction of the silyl complex with SiH4 will also be discussed because it can give information on the silane dehydropolymerization mechanism.
Influence of the metal centre
As shown in previous work, the nature of the lanthanide has little influence on the energy profile of the reaction20–23,37 We verified that this was also the case for these two reactions because they have similar activation barriers. The free enthalpy profiles were calculated for all lanthanide metals and the results are given in the ESI.† The activation energies vary from 18.3 to 23.0 kcal mol−1 for CH3/SiH3 exchange reaction (eqn (1)) and from 20.6 to 23.4 kcal mol−1 for the SiH3–CH3 coupling reaction (eqn (2)). Free enthalpies of reaction have a narrower energy range (≤2 kcal mol−1). The main results found for La apply to all the lanthanides: the activation energy for the CH3/SiH3 exchange reaction is marginally lower than for the CH3–SiH3 coupling reaction, the first reaction being more exoergic. There is a slight tendency for the activation energies to increase from La to Lu and for the reactions to become slightly less exoergic. However the variations are too small to be significant and it can be concluded that the results found for La apply to all Ln(III) metals. For this reason, all the following calculations have been carried out for La only.
Reactions of Cp2LaH with SiH3–CH3
The free enthalpy profiles for the σ bond metathesis reactions described by eqns (3) to (8) are shown in Fig. 3, 4 and 5.
The results for the reactions of eqn (3) (H/H exchange) and eqn (4) (formation of the SiH2CH3 complex and H2, termed H/Si exchange or silylation) are shown in Fig. 3 on the left and right hand sides, respectively. The free enthalpy profiles resemble those for eqns (1) and (2) and that obtained for the reaction of [La]–H with SiH4.23 The geometrical features are almost identical to what described before and thus attention will be focused on the energetic data.
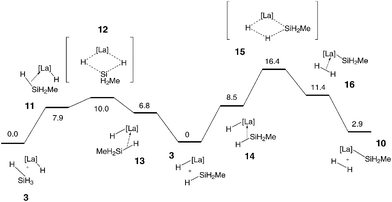 |
| | Fig. 3 Free enthalpy profiles (kcal mol−1) for the H/H exchange reaction (eqn (3)) and the silylation reaction (eqn (4)). | |
The H/H exchange is by definition an isoergic process whereas the H/Si exchange is calculated to be slightly endoergic (2.9 kcal mol−1). Considering the accuracy of the modelling these two reactions should be both considered as essentially isoergic. The activation energies are however somewhat different; the H/H exchange has a low activation energy of 10 kcal mol−1 while the silylation of the metal has a higher activation energy of 16.4 kcal mol−1. Similar trends were found in the reaction of [La]–H with SiH4, suggesting that the substituent on the silyl group does not have significant influence on the electronic aspects of the Si–H activation. To be sure, steric effects, which are not incorporated in these calculations (use of C5H5 ligand and of a small silylalkane) may play a role. At the present level of modelling, reactions (3) and (4), which both have low activation energies and comparable energy of reaction, should both occur. Thus, the Si–H bond can be activated either for non-productive bond metathesis or to form a silyl complex.
The relevant reactions are described in eqn (5) (H/H exchange) and eqn (6) (alkylation of the metal with associated formation of H2). The free enthalpy profiles are shown in Fig. 4.
 |
| | Fig. 4 Free enthalpy profiles (kcal mol−1) for the H/H exchange reaction (eqn (5)) and the alkylation reaction (eqn (6)). | |
The H/H exchange is isoergic whereas the methylation of the metal is endoergic by only 1.8 kcal mol−1. This value is small showing that alkylation of the metal by an alkylsilane is not disfavoured by thermodynamics. This is a significant difference from the results obtained for the reaction of [La]–H with CH4 to yield [La]–CH3 and H2, where the methylation of the lanthanide was endoergic by about 13 kcal mol−1. Considering that all groups coordinated to a lanthanide are strongly negatively charged, a silyl group, which stabilizes negative charge through delocalization into its σ*SiH bonds, significantly stabilizes the alkyl group next to the lanthanide. Previous studies have shown that Si–C bonds can also stabilize a negative charge so that this stabilizing effect should not be limited to the SiH3 group but should be present for all SiR3 groups.39,40
The activation energy (over 70 kcal mol−1) shows that the H/H exchange reaction is not accessible while that for alkylation of the metal is considerably lower (21.3 kcal mol−1). Only the latter reaction is thus kinetically accessible. The activation energy for reaction 4 is lower by 4 kcal mol−1 compared with the methylation reaction with CH4, which indicates that the stabilizing influence of the silyl group, already noted for the free enthalpy of reaction, also occurs at the transition state. This is due to the strong charges developed in the transition state on the groups in the vicinity of the metal, which induces the stabilizing effect of the silyl substituent to the alkyl group.
Comparison of Si–H and C–H activation
All reactions are permitted by thermodynamics and thus only the activation energies need be considered to access their feasibility. The main difference between Si–H and C–H activation is that the H/H exchange is not energetically accessible when the two H are bonded to a carbon while it is very accessible when they are bonded to Si. The activation energy of the Si–H bond to give a silyl complex is lower than that of the C–H bond to give a C-bonded alkylsilyl complex. Thus, over all, the activation of the Si–H bond should occur preferentially to that of a C–H bond in an alkylsilane. This is not surprising since it is well recognized that the Si–H bond is more reactive than a C–H bond because it is weaker and more polarizable. The interesting point is that the presence of a silyl group in the alkylsilane makes the C–H bond more reactive by stabilizing the C-bonded alkylsilyl complex and lowering the activation energy for C–H activation. In contrast, the methyl group has no significant influence on the energy barrier and energy of reaction for the Si–H activation, although, to be sure, steric factors could disfavour reactions at hindered bonds.
The free enthalpy profiles of the reactions corresponding to the activation of the Si–C bond are shown in Fig. 5. In these two reactions, the Si–C bond is cleaved to give either a silyl complex and an alkane (eqn (7)) or an alkyl complex and a silane (eqn 8). The formation of a silyl complex and an alkane (Fig. 5 left) is thermodynamically preferred to that of an alkyl complex and a silane (Fig. 5 right), because of the formation of the stronger alkane C–H bond in the former. Similar factors have explained the free energy of reaction for the reaction of eqn (1). However, the very high activation energy associated with the presence of a carbon at the β position of the 4-center transition state prevents this reaction from occurring. The same features have been found for eqn (5) (Fig. 4 left). Thus despite the thermodynamic driving force for the reaction, the transition state is energetically inaccessible. In contrast, the reaction which forms the methyl complex and the silane is slightly endoergic but has an energetically accessible transition state. It should be noted that the reaction is significantly more favoured thermodynamically than the reaction between [La]–H and CH4 to give [La]–CH3 and H2 because the cleaved Si–C bond is significantly weaker than a C–H bond (see BDEs reported earlier).
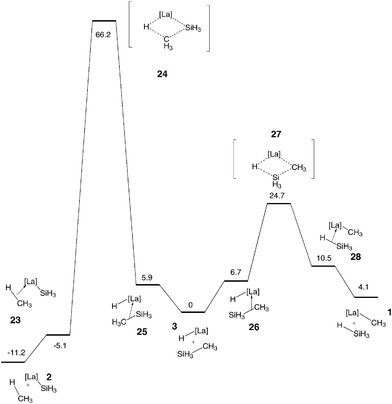 |
| | Fig. 5 Free enthalpy profiles (kcal mol−1) for the activation of CH3–SiH3 to give the silylation complex (eqn (7)) and the alkyl complex (eqn (8)). | |
The most favourable reactions of [La]–H with CH3–SiH3 are thus the two reactions involving the activation of the Si–H bond (eqns (3) and (4)). The transition states for these two reactions are lower than those associated with the activations of the Si–C and C–H bonds and the thermodynamics are essentially neutral for the two reactions. The next most preferred reaction, i.e. that of the C–H bond to form the alkylsilyl complex, has a moderately higher activation energy and is essentially thermoneutral. The direct σ bond metathesis of the Si–C bond to give the alkyl complex and silane requires a higher activation energy. However, the calculated value of 24 kcal mol−1 and the moderate endoergicity suggest that the reaction is accessible. These reactions have been mentioned to account for the formation of the products seen in the reaction of Ph–SiH3 with Cp*2Sm–H.16 A computational study of the reaction between these two specific species accounts for the large number of redistribution products and will be published separately.
Si–Si coupling: reactions of Cp2LaSiH3 with SiH4
The free enthalpy profiles, calculated for eqn (9) (SiH3/SiH3 exchange via Si–H activation) and eqn (10) (SiH3–SiH3 coupling) are shown in Fig. 6. The first reaction is isoergic with a low activation energy (17.7 kcal mol−1) (34), even slightly lower than the CH3/SiH3 exchange (eqn (1)); the transition state is reached from an SiH4 adduct 33, which is 6.9 kcal mol−1 above the separate reactants. As was the case for the reaction of eqn (1), the pathway of this reaction is best viewed as a proton transfer of the two silyl groups in the field of the positively charged lanthanide atom. The coupling of the two silyl groups corresponds to a reaction that has not been studied previously and the nature of the intermediates and transition states is discussed in more detail. The reaction starts with the formation of an SiH4 adduct 29 (Fig. 6), lying 7.3 kcal mol−1 above the separate reactants, to reach a transition state 30 with an activation energy of 17.7 kcal mol−1. This transition state leads to an SiH3–SiH3 adduct 31, 15.7 kcal mol−1 above the separated reactants which itself leads to another transition state 32 with a free enthalpy of 16.7 kcal mol−1. This latter transition state yields the final products [La]–H and SiH3–SiH3, 3. The reaction is endoergic by 6.1 kcal mol−1 in the direction of the formation of the Si–Si bond. The reactions described in eqns (9) and (10) thus have similar activation energies and differ only slightly in their energy of reaction.
 |
| | Fig. 6 Free enthalpy profiles (kcal mol−1) for the formation of the Si–Si bond (eqn (9)) and the Si/Si exchange (eqn (10)). | |
The geometry corresponding to the stationary points is presented in Fig. 7. The structure of 29, which is a SiH4 adduct to [La]–SiH3, shows that the SiH3 group is α-agostic while SiH4 is η2-bonded. This illustrates how the group with the highest negative charge (SiH3) maximizes its interaction with the electropositive metal. The α-agostic structure of the SiH3 group modifies the direction of the Si sp3 lone pair, which does not point toward the metal but towards the other Si atom. Thus, despite the long distance between the two Si in this adduct, 3.9 Å, the adduct is well prepared for establishing a new Si–Si bond. The geometry of the transition state 30 resembles that previously obtained for the reaction of [La]–H with SiH4. The β Si atom has square pyramidal geometry, with one apical H, typical for a hypervalent Si atom. The high-lying intermediate 31, which leads to 30, has a geometry close to that of 30 but with a shorter Si–Si distance and Siβ in a geometry close to a trigonal bipyramid. It leads to the other transition state 32, where Siβ also has a trigonal pyramidal geometry and which differs from 31 mostly by having a significantly lengthened Siβ–Hα bond. From 30 to 32, the main change has thus been a pseudorotation of the pentacoordinated Siβ associated with variations in the lengths of the bonds to be made and cleaved (Si–Si and Si–H). It is not surprising that these structures are close in energy. This reaction occurs with the hydrogens having interactions with both the metal and Si, during Si–H activation. This situation has been established experimentally and has been termed SiSHA (silicon secondary hydrogen attraction) by the group of Chaudret and Sabo-Etienne41 and IHI (interligand hypervalent interaction) by Nikonov.42
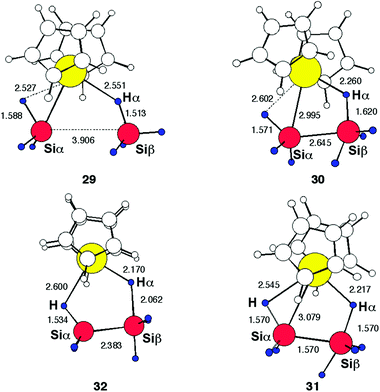 |
| | Fig. 7 Optimized structures of the extrema for the reaction in eqn (9). The distances are in Å. | |
The coupling reaction of the two silyl groups has an accessible activation energy but is slightly endoergic. The dehydropolymerization reaction is thus slightly disfavoured by thermodynamics. However, substituent effects may easily modify the energy of reaction which could then become feasible. Computational studies on Ph–SiH3 in a forthcoming paper will illustrate this point.43
Conclusions
DFT studies of the free enthalpy profiles of the σ bond metathesis reaction of [La]–R (R = CH3, H and SiH3) with SiH4 and SiH3–CH3 allow a better understanding the factors that control the reactivity in σ-bond metathesis processes. The Si–H bond can be activated by alkyl and hydride lanthanocene complexes to form either silyl or hydride derivatives and the associated products (alkane, silylalkane or H2). There is no significant preference for the reactions to go via a transition state with the silyl group at either the α or β positions. Consequently, a silyl complex can be formed from the hydride complexes. Substitution of the silane by an alkyl group does not modify the reactivity of the Si–H bond, though to be sure large steric effects have not been included. The activation of the Si–H bond is also kinetically accessible with a silyl complex but in this case there is a thermodynamic preference for the silyl exchange reaction over the formation of a new Si–Si bond. Dehydropolymerization does not require a high activation energy but is slightly unfavourable thermodynamically. However the cleavage of an Si–Si bond by a lanthanocene hydride is thermodynamically accessible.
The cleavage of an Si–C bond by a hydride complex requires a higher activation energy than the cleavage of the Si–H bond. Furthermore, in the elementary step directly cleaving the Si–C bond, the only reaction which has an accessible barrier and is not too disfavoured thermodynamically is the one which forms the alkyl complex and the free silane. The reaction which forms a silyl complex and the free alkane is strongly disfavoured because of the presence of an alkyl group at the highly unfavourable β position in the transition state. Overall, alkylsilanes prefer to react via the Si–H bond than via the Si–C bond. The activation of the C–H bond of alkylsilane obeys the rules already established for alkane activation. The reactions that would put the CH3 group at the β position in the transition state have very high activation energies so that the only possible products have the C-bonded alkylsilyl at the α position. It is remarkable that the silyl group lowers the activation energy and thermodynamically favours the formation of the alkyl complex from the reaction of the hydride lanthanocene and silylalkane. This reaction, which is significantly disfavoured in the case of non-substituted alkanes, benefits from the ability of the silyl group to stabilize the negatively charged alkyl group in a C-bonded alkylsilyl complex. The nature of the metal has been shown to have little influence on the energy profiles but one should remember that steric effects must also play a role.44 In addition, results obtained for alkylsilanes should not be extended to arylsilanes without caution because the small differences in the activation energies and energies of reaction which differentiate some of the reactions from this work can be influenced by the phenyl group. Therefore the study of the reaction between PhSiH3 and Cp2Sm–H will be published separately.43
Acknowledgements
The authors are grateful to CINES and CALMIP for a generous donation of computer time.
References
- P. L. Watson and G. W. Parshall, Acc. Chem. Res., 1985, 18, 51 CrossRef CAS.
- P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999 CrossRef.
- H. B. Kagan, Chem. Rev., 2002, 102, 1805 CrossRef . Special Issue on Frontiers in Lanthanide Chemistry.
- M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer and J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203 CrossRef CAS.
- W. J. Evans, Adv. Organomet. Chem., 1985, 24, 131 CAS.
- M. Booij, B. J. Deelman, R. Duchateau, D. S. Postma, A. Meetsma and J. H. Teuben, Organometallics, 1993, 12, 3531 CrossRef CAS.
- C. J. Schaverien, Adv. Organomet. Chem., 1994, 36, 283 CAS.
- L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279 CrossRef CAS.
- E. L. Werkema, E. Messines, L. Perrin, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 7781 CrossRef CAS.
- A. Z. Voskoboynikov, I. N. Parshina, A. K. Shestakova, K. P. Butin, I. P. Belestaya, L. G. Kuz’mina and J. A. K. Howard, Organometallics, 1997, 16, 4041 CrossRef CAS.
- P.-F. Fu, L. Brard, Y. Li and T. J. Marks, J. Am. Chem. Soc., 1995, 117, 7157 CrossRef CAS.
- G. Jeske, H. Lauke, H. Mauermann, P. N. Swepston, H. Schumann and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8091 CrossRef CAS.
- N. S. Radu and T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 5863 CrossRef CAS.
- N. S. Radu, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1992, 114, 8293 CrossRef CAS.
- N. S. Radu, T. D. Tilley and A. L. Rheingold, J. Organomet. Chem., 1996, 516, 41 CrossRef CAS.
- I. Castillo and T. D. Tilley, Organometallics, 2000, 19, 4733 CrossRef CAS.
- T. D. Tilley, Acc. Chem. Res., 1993, 26, 22 CrossRef CAS.
- J. A Gladysz, Chem. Rev., 2000, 100, 1167 CrossRef . Special issue on Metal Catalyzed Polymerization.
- J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1999, 99, 175 CrossRef CAS.
- L. Maron, L. Perrin and O. Eisenstein, J. Chem. Soc., Dalton Trans., 2002, 534 RSC.
- E. C. Sherer and C. J. Cramer, Organometallics, 2003, 22, 1682 CrossRef CAS.
- N. Barros, O. Eisenstein and L. Maron, Dalton Trans., 2006, 3052 RSC.
- L. Perrin, L. Maron and O. Eisenstein, Inorg. Chem., 2002, 41, 4355 CrossRef CAS.
- T. Ziegler and E. Folga, J. Organomet. Chem., 1994, 478, 57 CrossRef CAS.
-
M. Dolg and H. Stoll, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gscheinder, Jr and L. Eyring, Elsevier Science, Amsterdam, vol. 22, ch. 152, 1996 Search PubMed.
- M. Dolg, H. Stoll A. Savin and H. Preuss, Theor. Chim. Acta, 1989, 75, 173 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1993, 85, 441 CAS.
- M. Dolg, P. Fulde, W. Küchle, C.-S. Neumann and H. Stoll, J. Chem. Phys., 1991, 94, 3011 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431 CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.11), Gaussian, Inc., Pittsburgh PA, 2001 Search PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B, 1992, 45, 13244 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
-
(a) J. Cooper and T. Ziegler, Inorg. Chem., 2002, 41, 6614 CrossRef CAS;
(b) S. Sakaki, T. Takayama, M. Sumimoto and M. Sugimoto, J. Am. Chem. Soc., 2004, 126, 3332 CrossRef CAS;
(c) F. P. Rotzinger, Chem. Rev., 2005, 105, 2003 CrossRef CAS;
(d) B. O. Leung, D. L. Reidl, D. A. Armstrong and A. Rauk, J. Phys. Chem. A, 2004, 108, 2720 CrossRef CAS;
(e) D. Ardura, R. Lopez and T. L. Sordo, J. Phys. Chem. B, 2005, 109, 23618 CrossRef CAS.
-
L. Perrin, L. Maron and O. Eisenstein, Organometallic C–H Bond Activation, ed. A. Goldman and K. Goldberg, ACS Book Series 885, 2004, 116 Search PubMed.
-
(a) E. I. Izgorodina, M. L. Coote and L. Radom, J. Phys. Chem. A, 2005, 109, 7558 CrossRef CAS;
(b) E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2006, 128, 8350 CrossRef CAS.
- L. Maron and O. Eisenstein, J. Am. Chem. Soc., 2001, 123, 1036 CrossRef CAS.
- E. Clot and O. Eisenstein, Struct. Bonding, 2004, 113, 1 CAS , and references therein.
- L. Perrin, L. Maron, O. Eisenstein and M. F. Lappert, New J. Chem., 2003, 27, 121 RSC.
- E. D. Brady, D. L. Clark, J. C. Gordon, P. J. Hay, D. W. Keogh, R. Poli and J. G. Watkin, Inorg. Chem., 2003, 42, 6682 CrossRef CAS.
- I. Atheaux, F. Delpech, B. Donnadieu, S. Sabo-Etienne, B. Chaudret, K. Hussein, J.-C. Barthelat, T. Braun, S. B. Duckett and R. N. Perutz, Organometallics, 2002, 21, 5347 CrossRef CAS.
- G. I. Nikonov, Adv. Organomet. Chem., 2005, 53, 217 CAS.
-
L. Perrin, L. Maron and O. Eisenstein, to be submitted.
- N. Barros, O. Eisenstein, L. Maron and T. D. Tilley, Organometallics, 2006, 25, 5699 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1 to S6 of structural information for the reactions of eqns (1) and (2) for Ln = La to Lu. List of coordinates, energies and free enthalpies for all structures with La. See DOI: 10.1039/b617425f |
| ‡ The HTML version of this article has been enhanced with colour images. |
| § Present address: Protéines Membranaires Transductrices d’Energie, (URA 2096-CNRS), Bat. 528, DSV/DBJC/SBFM, CEA-Saclay, 91191 Gif-sur-Yvette Cedex, France. |
| ¶ Present address: Laboratoire de Nanophysique, Magnétisme et Optoélectronique, INSA, 135 avenue de Rangueil, 31077, Toulouse Cedex, France. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2007 |
Click here to see how this site uses Cookies. View our privacy policy here.