The tallysomycin biosynthetic gene cluster from Streptoalloteichus hindustanus E465-94 ATCC 31158 unveiling new insights into the biosynthesis of the bleomycin family of antitumor antibiotics
Meifeng
Tao
a,
Liyan
Wang
a,
Evelyn
Wendt-Pienkowski
a,
Nicholas P.
George
b,
Ute
Galm
a,
Guodong
Zhang
a,
Jane M.
Coughlin
c and
Ben
Shen
abcde
aDivision of Pharmaceutical Sciences, University of Wisconsin-Madison, 777 Highland Ave, Madison, Wisconsin 53705, USA
bMicrobiology Doctoral Training Program, University of Wisconsin-Madison, 777 Highland Ave, Madison, Wisconsin 53705, USA
cDepartment of Chemistry, University of Wisconsin-Madison, 777 Highland Ave, Madison, Wisconsin 53705, USA
dUniversity of Wisconsin National Drug Discovery Group, University of Wisconsin-Madison, 777 Highland Ave, Madison, Wisconsin 53705, USA
eDivision of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin-Madison, 777 Highland Ave, Madison, Wisconsin 53705, USA. E-mail: bshen@pharmacy.wisc.edu; Fax: +1 (608) 262-5345; Tel: +1 (608) 263-2673
First published on 28th November 2006
Abstract
The tallysomycins (TLMs) belong to the bleomycin (BLM) family of antitumor antibiotics . The BLM biosynthetic gene cluster has been cloned and characterized previously from Streptomyces verticillus ATCC 15003, but engineering BLM biosynthesis for novel analogs has been hampered by the lack of a genetic system for S. verticillus. We now report the cloning and sequencing of the TLM biosynthetic gene cluster from Streptoalloteichus hindustanus E465-94 ATCC 31158 and the development of a genetic system for S. hindustanus, demonstrating the feasibility to manipulate TLM biosynthesis in S. hindustanus by gene inactivation and mutant complementation. Sequence analysis of the cloned 80.2 kb region revealed 40 open reading frames (ORFs), 30 of which were assigned to the TLM biosynthetic gene cluster. The TLM gene cluster consists of nonribosomal peptide synthetase (NRPS) genes encoding nine NRPS modules, a polyketide synthase (PKS) gene encoding one PKS module, genes encoding seven enzymes for deoxysugar biosynthesis and attachment, as well as genes encoding other biosynthesis, resistance, and regulatory proteins. The involvement of the cloned gene cluster in TLM biosynthesis was confirmed by inactivating the tlmEglycosyltransferase gene to generate a TLM non-producing mutant and by restoring TLM production to the ΔtlmE::ermE mutant strain upon expressing a functional copy of tlmE. The TLM gene cluster is highly homologous to the BLM cluster, with 25 of the 30 ORFs identified within the two clusters exhibiting striking similarities. The structural similarities and differences between TLM and BLM were reflected remarkably well by the genes and their organization in their respective biosynthetic gene clusters.
Introduction
Nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) use a very similar biosynthetic strategy to catalyze the assembly of peptides and polyketides, two distinct classes of natural products, from amino acid and short carboxylic acid precursors, respectively. Hybrid peptide-polyketide metabolites refer to natural products that are biosynthetically derived from amino acids and short carboxylic acids, catalyzed by hybrid NRPS-PKS systems. The tallysomycins (TLMs)1–3 belong to the bleomycin (BLM)4–6 family of glycopeptide antitumor antibiotics , which are prominent members of the hybrid peptide-polyketide natural products, and other members of this family include the phleomycins4,5,7 and the zorbamycins.8BLMs and related compounds are thought to exert their biological effects through a sequence selective, oxidative cleavage of DNA and RNA in the presence of oxygen and a reduced transition metal ion.9–12 However, the BLMs are the only members of this family that are currently used clinically in combined chemotherapy for the treatment of several types of tumors. Under the trade name of Blenoxane®, the clinical drug contains BLM A2 and B2 as the principle constituents13 (Fig. 1). Almost uniquely among anticancer drugs, BLMs exhibit low myelo- and immunosuppression , prompting their wide application in combination chemotherapy.14,15 Early development of drug resistance and cumulative pulmonary toxicity are the major limitations of BLMs in chemotherapy.13
 | ||
| Fig. 1 Structures of bleomycin (BLM) A2 and B2 and tallysomycin (TLM) A and B. Structural differences between the BLMs and TLMs are boxed. | ||
The TLMs have been shown to exhibit activity similar to that of BLMs in preclinical studies,16 but failed to yield the desired response in phase II clinical trials due to poor cell penetration.17,18 Recently, the uptake problems associated with TLM delivery have been addressed by conjugation of TLM S10b to an internalizing antibody, thus increasing the potency of TLM S10b by up to 875-fold. This clearly shows the promise of further developing TLMs into clinically useful anticancer agents.19
The structures of TLM A and B were initially determined in 19772 and later revised according to the structural revision of the BLMs.4 The TLMs consist of four different functional domains: (i) the pyrimidoblamic acid subunit along with the adjacent β-hydroxyl histidine constitutes the metal-binding domain responsible for DNA cleavage; (ii) the bithiazole and C-terminal amine are thought to help facilitate DNA binding and may also contribute to polynucleotide recognition and the DNA cleavage selectivity; (iii) the (3S,4R)-4-amino-3-hydroxypentanoic acid subunit not only provides the connectivity between the metal-binding and DNA-binding sites but also plays an important role in the efficiency of DNA cleavage by TLMs; (iv) the sugar moieties are likely to participate in cell recognition by TLMs and possibly in cellular uptake and metal-ion coordination (Fig. 1).
Structurally, the TLMs are closely related to the BLMs. They share the same chromophore with the BLMs but differ from the BLMs in (i) the (3S,4R)-4-amino-3-hydroxypentanoic acid moiety, (ii) the C-terminal amine moiety, and (iii) the presence of an additional sugar moiety, 4-amino-4,6-dideoxy-L-talose, attached to the hybrid peptide–polyketide aglycone via a unique glycosylcarbinolamide linkage (Fig. 1).
We have previously cloned and characterized the BLM gene cluster from Streptomyces verticillus ATCC15003, unveiling hybrid NRPS-PKS machinery for BLM biosynthesis.20–22 However, all efforts to develop a genetic system for S. verticillus have been met with little success. We therefore set out to clone the TLM gene cluster to gain additional insights into the molecular mechanism for the biosynthesis of this family of important antitumor antibiotics and search for an alternative system to engineer novel TLM and BLM analogs by combinatorial biosynthetic methods. Here we report (i) the cloning and DNA sequence analysis of the TLM biosynthetic gene cluster from Streptoalloteichus hindustanus E465-94 ATCC 31158, (ii) functional assignments of the TLM gene products and comparison to the BLM proteins, (iii) the development of a genetic system for S. hindustanus by spore electroporation or intergeneric conjugation between Esherichia coli and S. hindustanus, and (iv) the demonstration of the ability to manipulate TLM biosynthesis by gene inactivation and mutant complementation in S. hindustanus. The TLM gene cluster is highly homologous to the BLM gene cluster with 25 of the 30 ORFs identified within the two clusters exhibiting 56–79% similarities.
Results
Cloning of the TLM biosynthetic gene cluster
The TLM resistance gene tlmA, also known as Sh ble, from S. hindustanus has been previously cloned and sequenced.23 The gene product Sh Ble was characterized as a BLM binding protein that has been extensively used as a BLM resistance marker in eukaryotic and prokaryotic cells.23–31 Since genes encoding antibiotic production usually are clustered with their respective resistance determinants,32 as was indeed the case for the blmA gene in the BLM biosynthetic gene cluster,20 the tlmA gene was considered to be a suitable probe to identify the TLM biosynthetic gene cluster from S. hindustanus.The TLM biosynthetic gene cluster was isolated as a set of overlapping cosmids by screening a cosmid library of S. hindustanus first using the tlmA probe (probe 1) and subsequently chromosomally walking from the tlmA locus using probes 2 and 3 (Fig. 2A). An 80.2 kb contiguous region of S. hindustanus DNA, as represented by pBS8001, pBS8002, and pBS8003, was finally sequenced (Fig. 2A).
 | ||
| Fig. 2 (A) Restriction map of the 80.2 kb DNA region from S. hindustanus ATCC 31158 as represented by the three overlapping cosmids pBS8001, pBS8002, and pBS8003 and (B) genetic organization of the TLM biosynthetic gene cluster. Proposed functions for individual ORFs are summarized in Table 1. B, BamHI. | ||
Sequence analysis of the TLM gene cluster
The overall GC content of the sequenced region is 72.4%. Sequence analysis of the 80.2 kb locus revealed 40 open reading frames (Fig. 2B and Table 1), 30 of which were assigned to the TLM biosynthetic gene cluster. The proposed functions for all of the ORFs identified are summarized in Table 1. Among them, we identified one PKS gene (tlmVIII), nine NRPS genes (tlmX, tlmIX, tlmVII, tlmVI, tlmV, tlmIV, tlmIII, tlmII, and tlmI), seven sugar biosynthesis genes (tlmC, tlmD, tlmE, tlmF, tlmG, tlmJ, and tlmK), seven genes encoding other biosynthesis enzymes (orf7, 8, 10, 11, 16, 21, and tlmH), as well as three resistance genes (tlmA, tlmB, and tlmT) and three regulatory genes (tlmR1, tlmR2, and tlmR3) (Fig. 2B and Table 1).| Gene | Length (amino acids) | Proposed functiona | Sequence homologsb | TLM–BLM Comparisonc |
|---|---|---|---|---|
| a Abbrevations for NRPS and PKS domains are: A, adenylation; ACP, acyl carrier protein; AL, acyl CoA ligase; AT, acyltransferase; C and C′, condensation; Cy, cyclization; KR, ketoreductase; KS, ketosynthase; Ox, oxidation; PCP, peptidyl carrier protein. b Protein accession numbers are given in parentheses. c Amino acid comparison of homologs identified from the TLM and BLM clusters as expressed in % identity/%similarity. | ||||
| orf38 | 1234 | Nitrate reductase (alpha chain) | NarG (NP_630616) | — |
| orf37 | 523 | Nitrate reductase (beta chain) | NarH (NP_630615) | — |
| orf36 | 221 | Nitrate reductase (delta chain) | NarJ (NP_630614) | — |
| orf35 | 247 | Nitrate reductase (gamma chain) | NarI (NP_630613) | — |
| orf34 | 188 | CBS-domain membrane protein | Rxyl_2733 (YP_645460) | — |
| orf33 | 351 | Zinc-binding dehydrogenase | AdhC (YP_119726) | — |
| orf32 | 453 | Oxidase | Fas5 (AAW49304) | — |
| orf31 | 280 | Hypothetical protein | SAV7303 (NP_828479) | — |
| — | Predicted upstream boundary of the TLM gene cluster | — | — | |
| tlmT | 599 | ABC transporter | AurT (AAK73550) | — |
| tlmX | 2132 | NRPS (C/A/PCP/C/A/PCP) | BlmX | 64/73 |
| tlmIX | 1088 | NRPS (C/A/PCP) | BlmIX | 55/64 |
| tlmVIII | 1533 | PKS (KS/AT/KR/ACP) | BlmVIII | 57/65 |
| tlmVII | 1141 | NRPS (C/A/PCP) | BlmVII | 55/65 |
| tlmVI | 2742 | NRPS (AL/PCP/C/A/PCP/C/A) | BlmVI | 56/65 |
| tlmV | 606 | NRPS (PCP/C') | BlmV | 56/65 |
| tlmG | 330 | NAD-dependent sugar epimerase | BlmG | 74/79 |
| tlmF | 633 | Glycosyl transferase and hydroxylase | BlmF | 51/59 |
| orf21 | 628 | Gln-dependent amino transferase | Blm-Orf18 | 63/74 |
| tlmIV | 2620 | NRPS (C/A/PCP/Cy/A/PCP/Cy) | BlmIV | 59/69 |
| tlmIII | 843 | NRPS (A/PCP/Ox) | BlmIII | 51/61 |
| tlmR3 | 348 | SyrP-like protein for regulation | Blm-Orf15 | 61/74 |
| tlmII | 490 | NRPS, type II C | BlmII | 48/56 |
| orf16 | 189 | Unknown, MbtH-like protein | Blm-Orf13 | 63/74 |
| tlmE | 388 | Glycosyl transferase | BlmE | 61/72 |
| tlmD | 538 | Carbamoyltransferase | BlmD | 73/83 |
| tlmI | 89 | Type II PCP | BlmI | 56/71 |
| tlmC | 498 | NTP-sugar synthase | BlmC | 59/68 |
| orf11 | 424 | SAM-dependent oxidase or methyl transferase | Blm-Orf8 | 70/81 |
| orf10 | 308 | α-Ketoglutarate-dependent hydroxylase | Blm-Orf1 | 69/82 |
| tlmA | 124 | Sh Ble binding protein | BlmA | 59/68 |
| orf8 | 197 | Ankyrin-like protein | Blm-Orf3 | 58/65 |
| orf7 | 129 | Peptide deformylase | Blm-Orf2 | 53/62 |
| tlmH | 324 | α-Ketoglutarate-dependent hydroxylase | Blm-Orf1 (AAB00457) | — |
| tlmJ | 399 | Aminotransferase | SpnR (AAG23279) | — |
| tlmK | 520 | Glycosyl transferase | StrH (CAA68520) | — |
| tlmB | 292 | N-acetyl transferase | BlmB | 57/64 |
| tlmR2 | 399 | Putative transcription regulator | Nfa9450 (YP_117154) | — |
| tlmR1 | 120 | Putative transcription regulator | ArsR (NP_924383) | — |
| — | Predicted downstream boundary of the TLM gene cluster | — | — | |
| orf-1 | 160 | Hypothetical protein | MM0500 (NP_632524) | — |
| orf-2 | 433 | Asp-tRNA synthase | AspRS (Q9Y9U7) | — |
The gene products of 25 of the 30 ORFs identified within the TLM gene cluster showed striking similarities to the proteins involved in BLM biosynthesis (ranging from 48–74% identity and 56–83% similarity, Table 1). Moreover, all of these ORFs were arranged in the same order and direction as in the BLM biosynthetic gene cluster. Based on comparison of the TLM cluster to the BLM biosynthetic gene cluster, tlmT, encoding an ABC multi-drug transporter, and tlmR1, encoding a putative transcription regulator, were predicted to be the upstream and downstream boundaries of the TLM cluster. As in the BLM cluster, all the ORFs within the TLM cluster, with the exception of tlmR2 that encodes another putative transcription regulator, are transcribed in the same direction, and most of the ORFs are thought to be translationally coupled, forming six operons (Fig. 2B). Possible promoter regions are located upstream of tlmT, tlmX, orf8, tlmH, tlmR1, and tlmR2.
The tlm NRPS genes
The tlmX, tlmIX, tlmVII, tlmVI, tlmV, tlmIV, and tlmIII genes encode modular NRPSs characterized by the typical domains known to be responsible for selection and activation of the amino acid precursors and for elongation of the growing peptide intermediates. Their deduced products show 51–64% identity and 61–73% similarity to their respective BLM homologs. These genes are organized in the same order and direction as in the BLM cluster and show the same characteristics and predicted amino acid specificities as the BLM enzymes (Table 2 and Fig. 3A). Both the N-terminal module of TlmVI (NRPS-5) and the so-called starter module of BlmVI (NRPS-5) are comprised of the same unusual acyl CoA ligase (AL) and acyl carrier protein (ACP)-like domains, which are thought to be involved in the biosynthesis of the β-aminoalaninamide moiety. Another characteristic feature of the BLM cluster, the absence of both His residues of the conserved HHxxxDG active site of the C domain in NRPS-3, was also found in TlmV (NRPS-3) in the TLM cluster. Since this critical mutation is in the well conserved active site, this C domain is expected to be non-functional for normal transpeptidation. The A domain in TlmIII (NRPS-0) appears to be inactive as well, lacking the crucial A3-A6 motifs, and this is consistent with its predicted nonfunctional role in bithiazole biosynthesis at the C-terminus of TLMs (Fig. 3A), as has been demonstrated for BlmIII (NRPS-0).33![(A) A linear model for the TLM hybrid NRPS-PKS templated assembly of the TLM aglycone from nine amino acids and one acetate in comparison with that for the BLM aglycone. Abbrevations for NRPS and PKS domains are: A, adenylation; ACP, acyl carrier protein; AL, acyl CoA ligase; AT, acyltransferase; C and C′, condensation, Cy, cyclization; KR, ketoreductase; KS, ketosynthase; MT, methyltransferase, Ox, oxidation; PCP, peptidyl carrier protein. (B) Proposed pathway for TLM biosynthesis in comparison with that for BLM. [?] indicates a step whose enzyme activity could not be identified within the sequenced TLM and BLM clusters. While all intermediates for TLM biosynthesis are hypothetical, those for BLM, except the ones in brackets, have been identified from S. verticillusfermentation as the corresponding free acids.](/image/article/2007/MB/b615284h/b615284h-f3.gif) | ||
| Fig. 3 (A) A linear model for the TLM hybrid NRPS-PKS templated assembly of the TLM aglycone from nine amino acids and one acetate in comparison with that for the BLM aglycone. Abbrevations for NRPS and PKS domains are: A, adenylation; ACP, acyl carrier protein; AL, acyl CoA ligase; AT, acyltransferase; C and C′, condensation, Cy, cyclization ; KR, ketoreductase; KS, ketosynthase; MT, methyltransferase, Ox, oxidation; PCP, peptidyl carrier protein. (B) Proposed pathway for TLM biosynthesis in comparison with that for BLM. [?] indicates a step whose enzyme activity could not be identified within the sequenced TLM and BLM clusters. While all intermediates for TLM biosynthesis are hypothetical, those for BLM, except the ones in brackets, have been identified from S. verticillusfermentation as the corresponding free acids. | ||
| Domain | 235 | 236 | 239 | 278 | 299 | 301 | 322 | 330 | 331 | 517 | Similarity (%)a |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Similarity is calculated by AlignX in the Vector NTI Advance™ 10 program from Invitrogen. b X indicates a variable amino acid within the determined code. | |||||||||||
| L-Cys(2) | D | L | Y | N | L | S | L | I | W | K | — |
| TlmIII (NRPS0) | G | F | Y | H | L | G | L | L | W | R | 60 |
| BlmIII (NRPS0) | P | L | Y | H | L | G | L | P | W | R | 60 |
| TlmIV (NRPS1) | D | L | Y | N | M | S | L | I | W | K | 100 |
| BlmIV (NRPS1) | D | L | Y | N | L | S | L | I | W | K | 100 |
| β-Ala b | V | D | X | V | I | S | X | G | D | K | — |
| TlmIV (NRPS2) | V | D | W | V | V | S | L | A | D | K | 80 |
| BlmIV (NRPS2) | V | D | W | V | I | S | L | A | D | K | 80 |
| L-Asn | D | L | T | K | L | G | E | V | G | K | — |
| TlmVI (NRPS3) | D | L | T | K | V | G | E | V | G | K | 100 |
| BlmVI (NRPS3) | D | L | T | K | V | G | E | V | G | K | 100 |
| TlmX (NRPS9) | D | L | T | K | V | G | E | V | G | K | 100 |
| BlmX (NRPS9) | D | L | T | K | V | G | E | V | G | K | 100 |
| L-Ser | D | V | W | H | L | S | L | I | D | K | — |
| TlmVI (NRPS4) | D | V | W | H | V | S | L | V | D | K | 100 |
| BlmVI (NRPS4) | D | V | W | H | V | S | L | V | D | K | 100 |
| L-Thr | D | F | W | N | I | G | M | V | H | K | — |
| TlmVII (NRPS6) | D | F | W | G | V | G | M | V | H | K | 90 |
| BlmVII (NRPS6) | D | F | W | S | V | G | M | I | H | K | 90 |
| L-Ala | D | L | F | N | N | A | L | T | Y | K | — |
| TlmIX (NRPS7) | D | L | F | N | N | A | L | T | Y | K | 100 |
| BlmIX (NRPS7) | D | L | F | N | N | A | L | T | Y | K | 100 |
| L-His b | D | S | X | L | X | A | E | V | X | K | — |
| TlmX (NRPS8) | D | S | A | L | V | A | E | V | W | K | 70 |
| BlmX (NRPS8) | D | S | A | L | I | A | E | V | W | K | 70 |
The tlmI and tlmII genes, like blmI (56% identity and 71% similarity) and blmII (48% identity and 56% similarity), encode discrete proteins homologous to individual PCP and C domains of type I NRPSs. BlmI has been characterized as a type II PCP,34 but its role in BLM biosynthesis remains unknown. BlmII, together with another homolog, BlmXI, were assigned the function of catalyzing the coupling reaction between the NRPS-bound (i.e., at the PCP domain of NRPS-0) full length BLM peptide intermediate and the terminal amines.20 While a similar role could be proposed for TlmII (Fig. 3A), the TLM cluster does not contain a gene homologous to blmXI, a finding that suggests blmXI might not be involved in BLM biosynthesis, and hence may not belong to the BLM cluster.
The tlmPKS gene
The tlmVIII gene encodes a PKS module consisting of a ketoacyl synthase (KS), an acyltransferase (AT), a ketoreductase (KR), and an ACP domain. TlmVIII shows 57% identity and 65% similarity to BlmVIII, but is 308 amino acids shorter than the latter and does not contain any of the three core motifs of AdoMet-dependent methyltransferases (MTs).35 Since MT domains are typically 260 amino acids in length, TlmVIII apparently lacks the MT domain predicted to be involved in AdoMet-dependent methylation of the malonyl CoA derived PKS extender unit in BLM biosynthesis (Fig. 3A).20 These findings are in perfect agreement with the structural difference between TLMs and BLMs, since TLMs lack the methyl group in the PKS-derived 4-amino-3-hydroxypentanoic acid moiety (Fig. 1).The tlm sugar biosynthesis genes
Seven sugar biosynthesis genes, tlmC, tlmD, tlmE, tlmF, tlmG, tlmJ, and tlmK were identified within the tlm cluster. Five of them exhibit significant similarity to their respective BLM homologs: blmC (59% identity and 68% similarity), blmD (73% identity and 83% similarity), blmE (61% identity and 72% similarity), blmF (51% identity and 59% similarity), and blmG (74% identity and 79% similarity). These five genes are located at the same relative positions in the TLM cluster as in the BLM cluster, and their gene products are expected to catalyze the same reactions in the formation of the L-gulose-3-O-carbamoyl-D-mannose disaccharide and its attachment to the aglycone as proposed for the BLM biosynthetic pathway20 (Figs. 4A and 4C).![Proposed biosynthetic pathways for (A) the disaccharide moiety of TLM from d-mannose in comparison with that for BLM and (B) the talose moiety of TLM from d-glucose and (C) a unified model for BLM and TLM biosynthesis by sequential glycosylation of the BLM and TLM aglycones. [?] indicates steps whose enzyme activities could not be identified within the sequenced TLM cluster.](/image/article/2007/MB/b615284h/b615284h-f4.gif) | ||
| Fig. 4 Proposed biosynthetic pathways for (A) the disaccharide moiety of TLM from D-mannose in comparison with that for BLM and (B) the talose moiety of TLM from D-glucose and (C) a unified model for BLM and TLM biosynthesis by sequential glycosylation of the BLM and TLM aglycones. [?] indicates steps whose enzyme activities could not be identified within the sequenced TLM cluster. | ||
Two additional genes, tlmJ and tlmK, that have no homologs in the BLM cluster, were found close to the downstream end of the TLM cluster. The gene product of tlmJ is closely related to SpnR (54% identity and 67% similarity), a pyridoxal 5′-phosphate (PLP) dependent aminotransferase from Saccharopolyspora spinosa,36 and that of tlmK exhibits low similarity to StrH (27% identity and 42% similarity), a putative glycosyltransferase from Streptomyces griseus.37 They might therefore be involved in the biosynthesis of the 4-amino-4,6-dideoxy-L-talose and its attachment to the TLM aglycone (Figs. 4B and 4C).
Resistance, regulatory, and other genes
The tlmA, tlmB, and tlmT genes encode proteins that presumably confer TLM resistance via three different mechanisms. Prior to cloning the TLM cluster, a BLM binding protein encoded by tlmA, known as Sh ble, had already been identified and characterized to confer resistance to the BLM family of antibiotics by drug sequestering.38 As expected, tlmA was indeed found within the TLM biosynthetic gene cluster and located at the same relative position as blmA, the homolog identified from the BLM cluster that encodes the BLM binding protein (59% identity and 68% similarity).39 The deduced gene product of tlmB shows similarity to N-acetyltransferases, and the closest homolog, BlmB (57% identity and 64% similarity) from the BLM biosynthetic gene cluster, has been shown to confer BLM resistance to the BLM producer S. verticillus by drug modification.40,41 TlmB presumably provides TLM resistance by the same mechanism as BlmB. The gene product of tlmT is closely related to the ATP dependent transporter AurT (25% identity and 48% similarity) of the aureocin 70 biosynthetic gene cluster from Staphylococcus aureus A70.42Drug transport is a common resistance mechanism found in antibiotic producing microorganisms,43 hence TlmT could contribute to TLM resistance by transporting the drug out of the cells.In addition to resistance genes, antibiotic biosynthetic gene clusters in Streptomyces usually contain pathway specific regulatory genes. Three regulatory genes, tlmR1, tlmR2, and tlmR3, were identified within the TLM cluster. The deduced product of tlmR3 shows a high degree of similarity to Blm-Orf15 (61% identity and 74% similarity) and to SyrP from Pseudomonas syringae pv syringae that controls syringomycin production.44 The tlmR1 gene encodes a protein that is closely related to the ArsR family of regulators such as ArsR (58% identity and 76% similarity) from Gloeobacter violaceus.45 The gene product of tlmR2 resembles Nfa9450 (32% identity and 45% similarity), a putative transcription regulator from Nocardia farcinica.46 No homologs to these genes, however, could be found within the BLM biosynthetic gene cluster, indicative of different regulatory mechanisms for BLM and TLM production in the two organisms.
The remaining genes of the TLM cluster, orf7, 8, 10, 11, 1621, and tlmH, are proposed to be involved in precursor biosynthesis or tailoring reactions, or the function of their gene products could not be predicted by sequence comparison alone. The orf7 product and its homolog from the BLM cluster, Blm-Orf2 (53% identity and 62% similarity), are weakly related to peptide deformylases, and their role in TLM and BLM biosynthesis has not been proposed yet. The gene product of orf8, like the product of blm-orf3 (58% identity and 65% similarity), shows similarity to ankyrin-like proteins.47 It is not clear what the function of this type of protein in TLM and BLM biosynthesis might be.39 The deduced product of orf10 is highly similar to Blm-Orf1 (69% identity and 82% similarity) and to α-ketoglutarate-dependent Asp/Asn hydroxylases.48–50 Blm-ORF1 has been proposed to be the putative His β-hydroxylase.39 A similar function could be proposed for Tlm-Orf10, hydroxylating the His residue of the TLM peptide-polyketide backbone, thereby providing the required hydroxyl group for the attachment of the disaccharide moiety (Fig. 3B). The orf11 product and its homolog from the BLM cluster, Blm-orf8 (70% identity and 81% similarity), belong to the radical SAM superfamily and show significant similarity with many coproporphyrinogen III oxidases such as the Bacillus subtilis HemN (31% identity and 45% similarity).51 They might be involved in methylation or oxidation reactions in the TLM and BLM biosynthetic pathways (Fig. 3B). The gene product of orf16, like Blm-Orf13 (63% identity and 74% similarity), resembles MbtH, which is a protein of unknown function found in the mycobactin and many other antibiotic biosynthetic gene clusters.52–54 While the role of MbtH and its homologs in antibiotic biosynthesis remains unknown, bal-orf1, an mbtH homolog in the balhimycin gene cluster, was recently demonstrated not to be involved in glycopeptide biosynthesis.55 The product of orf21 is a putative glutamine-dependent transaminase highly similar to Blm-Orf18 (63% identity and 74% similarity), which could be involved in the biosynthesis of the pyrimidoblamic acid moiety as proposed previously20 (Fig. 3B). Finally, although tlmH has no counterpart in the BLM cluster, its deduced gene product shows significant sequence homology to Blm-Orf1 (44% identity and 61% similarity) and other α-ketoglutarate-dependent hydroxylases.48–50 TlmH therefore serves as a candidate to hydroxylate the β-Ala residue of the TLM backbone, thereby providing the hydroxy group for the attachment of the 4-amino-4,6-dideoxy-L-talose moiety (Fig. 3B).
A genetic system for S. hindustanus by E. coli–S. hindustanus conjugation or S. hindustanusspore electroporation
We used the pSET152 derived plasmid pBS8004, which carried the ΦC31 integration function and the thiostrepton resistance gene tsr for selection, to establish and optimize the genetic system for S. hindustanus. By intergeneric conjugation between E. coli and S. hindustanus using E. coli ET12567 (pUZ8002/pBS8004) as a donor, pBS8004 was introduced into S. hindustanus at a frequency of 1 × 10–8 per colony-forming-unit (cfu). The frequency could be increased 10-fold by supplementing ISP4 medium with MgCl2 to a final concentration of 28 mM. An additional 2-fold increase was achieved by extending the time of 50 °C heat shock treatment of the S. hindustanusspores prior to conjugation to 2 h. The maximum conjugation frequency under the best conditions was 2 × 10–7 per cfu. Other standard procedures such as protoplast transformation and protoplast electroporation failed to yield any transformants.56 However a respectable transformation frequency (1 × 10–6 per cfu) was achieved by electroporating S. hindustanusspores with unmethylated DNA. When methylated DNA was used, the electroporation frequency dropped by 10-fold, suggesting that restriction on methylated DNA operates in S. hindustanus.Inactivation of tlmE and complementation of the ΔtlmE::ermE mutant in S. hindustanus
The tlmE gene encodes a glycosyltransferase, as does its homolog, blmE, which has been proposed to play a role in the formation of the disaccharide or its attachment to the BLM aglycone20 (Figs. 4A and 4C). To inactivate tlmE, the mutated cosmid pBS8005, in which the tlmE gene was replaced by an ermE-neo resistance cassette, was first constructed by gene replacement using the REDIRECT method.57 A ΔtlmE::ermE mutant was then isolated in S. hindustanus by allelic exchange using pBS8005 via homologous recombination. Introduction of pBS8005 into S. hindustanus by electroporation, followed by antibiotic selection resulted in the isolation of several erythromycin resistant and thiostrepton sensitive colonies, named S. hindustanus SB8001 (Fig. 5A). The genotype of S. hindustanus SB8001 was confirmed first by PCR experiments (data not shown) and subsequently by Southern analysis (Fig. 5B). While the wild-type strain showed a distinct band at 3 kb, this fragment was shifted to 5 kb in the SB8001 mutant strains, as would be expected when replacing the tlmE gene with the ΔtlmE::ermEallele by a double-crossover homologous recombination event (Fig. 5A). To complement the ΔtlmE::ermE mutation in the SB8001 mutant strain, the tlmE expression plasmid pBS8006 was contructed in the integrative vector pBS8004 by placing the ORFp1 promoter58 in front of tlmE. Introduction of pBS8006 into SB8001 by conjugation followed by selection for thiostrepton resistance afforded strain SB8002 in which pBS8006 is integrated into the S. hindustanus chromosome, and the expression of tlmE is under the control of the constitutive ORFp1 promoter.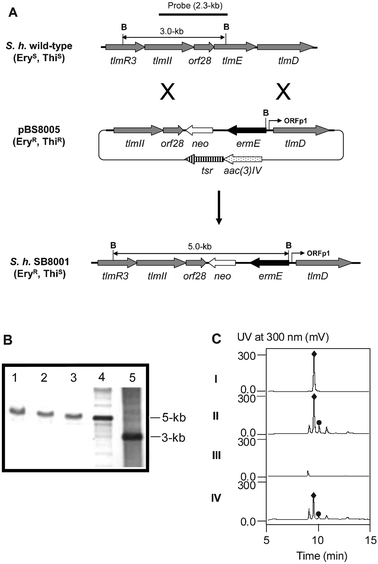 | ||
| Fig. 5 Inactivation of tlmE by gene replacement and ΔtlmE::ermE mutant complementation. (A) Construction of the tlmE gene replacement mutant and restriction map of S. hindustanus wild-type and SB8001 mutant strains showing fragment sizes upon BamHI digestion. B, BamHI; EryR, erythromycin resistant; EryS, erythromycin sensitive; ThiR, thiostrepton resistant; ThiS, thiostrepton sensitive. (B) Southern analysis of SB8001 genomic DNAs (lanes 1–3 are three individual isolates) and cosmids harbouring the wild-type (lane 5) and mutated tlmE (lane 4) loci as controls digested with BamHI using the 2.3 kb PCR-amplified fragment as a probe. (C) HPLC analysis of TLM A (♦) and TLM B (●) production in wild-type (II) and recombinant strains SB8001 (III) and SB8002 (IV) with authentic TLM A as a standard (I). | ||
TLM production in S. hindustanus wild-type, SB8001 mutant, and SB8002 complementation strains
Conditions for TLM isolation, purification, and detection were established using the S. hindustanus wild-type strain. Fermentation of S. hindustanus under the standard conditions produced TLMs as a mixture of congeners, differing structurally at the C-terminal amines of the glycopeptides (Fig. 1). Fig. 5C shows a typical profile of the partially purified TLMs after Amberlite® IRC50 and Diaion HP20 chromatography upon analysis by high performance liquid chromatography (HPLC). The isolated yield for TLM A ranged from 18 to 20 mg/l. The identity of TLMs was confirmed by HPLC comparison to an authentic TLM A standard, electrospray ionization (ESI) and MALDI-FTMS mass spectrometry (MS) analyses, and 1H NMR spectroscopy. Upon ESI-MS analysis, TLM A-Cu and TLM B-Cu complexes showed molecular ion peaks (m/z) at 896.8 and 832.8, consistent with the M2+ ions for the molecular formula of C68H110N22O27S2Cu (calculated 1793.5) and C62H98N20O26S2Cu (calculated 1665.5), respectively. Upon MALDI-FTMS MS analysis, Cu-free TLM A showed a molecular ion peak (m/z for [M + H]+) at 1731.7970, consistent with the molecular formula of C68H110N22O27S2 (calculated 1730.7425). The 1H NMR data of Cu-free TLM A agreed well with those reported in the literature.59The S. hindustanus SB8001 mutant and SB8002 complementation strains were fermented under the same conditions along with the wild-type strain as a control. HPLC analysis of the fermentations showed that inactivation of tlmE completely abolished TLM production in SB8001 and TLM production was partially restored (∼50% of that of the wild-type strain) by expressing a functional copy of tlmE in SB8002 (Fig. 5C). TLMs produced by the SB8002 strain were similarly isolated and confirmed by ESI-MS, yielding the identical masses as those produced by the wild-type strain.
Discussion
Cloning of the TLM cluster from S. hindustanus
The TLMs and BLMs are closely related antitumor antibiotics that exert their biological effect through a similar mechanism of oxidative cleavage of DNA and RNA.9–12 While the BLMs are currently used clinically for the treatment of several types of tumors, early development of drug resistance and cumulative pulmonary toxicity have limited their widespread application. The TLMs have been shown to have stronger activity than BLMs, but they also exhibited high toxicity and poor bioavailability.1,60 Consequently, there have been continued efforts to develop new BLM and TLM analogs to define the structure and activity relationship (SAR) of individual subunits of the BLM and TLM scaffolds, to search for drugs with better clinical efficacy and lower toxicity, and to develop new drug delivery technologies to improve bioavailability.19Complementary to drug delivery technologies and SAR studies by conventional medicinal chemistry, we set out to explore the feasibility of engineering novel analogs of the BLM family of antitumor antibiotics by manipulation of genes governing their biosynthesis. Previously we cloned and characterized the BLM biosynthetic gene cluster from S. verticillus ATCC 15003 but failed to engineer BLM biosynthesis due to the lack of a genetic system for this strain. To circumvent this technical difficulty, we explored the feasibility of carrying out combinatorial biosynthesis experiments in other BLM or related metabolite-producing organisms. We systematically tested several other strains, including S. verticillus ATCC 31307 (BLM-producing), S. verticillus ATCC 21890 (phleomycin-producing), Streptomyces flavoviridis ATCC 21892 (zorbamycin-producing), and S. hindustanus ATCC 31158 (TLM-producing), for antibiotic production and genetic amenability and successfully developed genetic systems for both the S. flavoviridis (data not shown) and S. hindustanus strains.
In this study, we report the cloning and sequencing of an 80.2 kb contiguous DNA region adjacent to the previously described tlmA (also called Sh ble) gene from S. hindustanus. Among the 40 ORFs unveiled within the sequenced region, 30 of them were assigned to constitute the TLM cluster and govern TLM production in S. hindustanus (Fig. 2 and Table 1). This conclusion was based on the facts that: (i) S. hindustanus has been proven to be a TLM-producing strain (Fig. 5C); (ii) the sequenced DNA region was found to be clustered with the previously described resistance gene, tlmA, encoding a BLM binding protein (Fig. 2B); (iii) 25 of the 30 ORFs assigned to the TLM cluster showed strikingly high similarity to their homologs in the BLM cluster (Table 1), a finding that correlates well with the structural similarity of the produced compounds; (iv) the differences between the TLM and the BLM structures, such as the lack of a methyl group in the polyketide portion and the presence of an additional sugar (Fig. 1) were consistent with the lack of an MT domain in the PKS (Fig. 3) and the extra sugar biosynthesis genes (Fig. 4); and (v) the ΔtlmE::ermE mutant srain SB8001 completely lost TLM production, and its ability to produce TLM was restored by expressing tlmE (Fig. 5).
Boundaries of the TLM gene cluster
The TLM and BLM biosynthetic gene clusters were found to be very similar regarding their organization and genes encoding their biosynthetic machinery (Fig. 1 and Table 1). Nearly all of the ORFs were oriented in the same direction and were in the same relative order in both gene clusters. The genes encoding biosynthetic enzymes were flanked by genes encoding proteins predicted to be responsible for regulatory processes (tlmR1 and tlmR2 for the TLM cluster and orf30 for the BLM cluster) and drug transport (tlmT for the TLM cluster and orf7 and orf29 for the BLM cluster). The proteins encoded by ORFs located upstream and downstream of the aforementioned genes seemed to be strain specific as they did not have direct counterparts in the corresponding clusters. Since these genes were not considered to be related to the TLM and BLM biosynthetic pathways, tlmT and blm-orf29 were likely to define the upstream and tlmR1 and blm-orf7 the downstream boundaries of the TLM and BLM clusters, respectively. It cannot be excluded, however, that additional biosynthetic enzymes encoded at different loci of the genome may also contribute to TLM production, such as the three missing genes implicated in 4-amino-4,6-dideoxy-L-talose biosynthesis (Fig. 4B).Assembly of the TLM peptide-polyketide aglycone
The TLM cluster contains the genes tlmX, tlmIX, tlmVII, tlmVI, tlmV, tlmIV, and tlmIII, for which homologs exist in the BLM cluster (blmX, blmIX, blmVII, blmVI, blmV, blmIV, and blmIII) (Table 1). For BLM biosynthesis, the different NRPS modules encoded by these genes have been proposed to activate their respective amino acids (Table 2) and catalyze the formation of the peptide backbone of BLM.20 Some of the NRPS modules involved in this pathway have been characterized in vitro and were shown to activate the predicted amino acids,20,33,61 yet other biosynthetic steps like formation of the pyrimidoblamic acid moiety are still poorly understood. Taking our results for the TLM cluster into consideration (Table 2), we suggest that biosynthesis of the TLM peptide backbone is performed in the same manner as described earlier for BLM with subtle deviations (Fig. 3).20Two further NRPS related genes were also found within the TLM cluster, tlmI and tlmII. The TlmI homolog, BlmI, has been shown to be a type II PCP.34 The role of BlmI and its counterpart TlmI in BLM and TLM biosynthesis is not clear, although one can speculate that a discrete PCP protein could be necessary to tether the respective biosynthetic intermediates while a different enzyme performs a biosynthetic reaction on the protein-bound compound. A possible candidate enzyme for this scenario might be TlmII and BlmII, a discrete C domain found in both clusters. These proteins could be involved in attaching the various terminal amines sequestered from the production media to the peptide–polyketide backbone (Fig. 3). Another discrete C domain encoded by blmXI, residing at the upstream end of the BLM cluster, was also speculated to play a role in the terminal amine coupling steps,20 but no blmXI counterpart could be identified within the TLM cluster, hence arguing against its involvement in BLM biosynthesis.
The PKS protein in the TLM pathway, TlmVIII, is composed of KS, AT, KR and ACP domains and shows a high degree of similarity to BlmVIII consisting of KS, AT, KR, MT and ACP domains (Table 1). The obvious difference is the absence of an MT domain in TlmVIII, and therefore, the PKS derived portion of the TLM backbone is suggested to be incorporated into the molecule as described earlier for BLM using malonyl-CoA as the precursor, but without the subsequent methylation step (Fig. 3A).20 This difference between TlmVIII and BlmVIII agrees perfectly with the TLM structure in that, in comparison to BLM, it lacks the methyl group in the polyketide-derived moiety (Fig. 1 and 3). Previously we speculated that the MT domain of BlmVIII might also function in methylating the pyrimidine ring of pyrimidoblamic acid, since no other definitive AdoMet-dependent MT could be found within the BLM cluster.20 The pyrimidoblamic acid moiety of TLM contains the methyl group but the TLM cluster does not harbor an extra AdoMet-dependent MT either, a fact that would argue against the earlier proposal that the MT domain of BlmVIII is responsible for methylating the pyrimidine ring. We now favor assigning Tlm-Orf11 and its homolog Blm-Orf8 as the MTs and propose that they catalyze the C-methylation of the pyrimidine ring in TLM and BLM biosynthesis, respectively (Fig. 3B).
Tailoring by sugar biosynthesis and attachments
The sugar attachments are suggested to be the final steps in TLM biosynthesis as has been described for the BLM pathway (Fig. 4).20 For the L-gulose and 3-O-carbamoyl-D-mannose moieties, the gene products of tlmCDEFG, like the products of blmCDEFG, are assumed to be responsible for their biosynthesis and attachment to the TLM aglycone (Figs. 4A and 4C). Two additional sugar biosynthesis genes, tlmJ and tlmK, along with tlmH were found in the TLM cluster as a putative operon that was absent in the BLM cluster. While TlmH has been proposed to catalyze the penultimate step for TLM aglycone biosynthesis (Fig. 3B), TlmJ and TlmK were presumably responsible for the biosynthesis of the 4-amino-4,6-dideoxy-L-talose moiety and its attachment to the TLM aglycone (Figs. 4B and 4C). Although the genes encoding proteins responsible for the first three steps of 4-amino-4,6-dideoxy-L-talose biosynthesis, an NDP-D-glucose synthetase, a 4,6-dehydratase, and a 3,5-epimerase, were not found within the TLM cluster (Fig. 4B), one can imagine these fairly common enzymes to be shared between different biosynthetic pathways for secondary metabolites . The exact timing for these steps in TLM biosynthesis has to be determined by future experiments.Feasibility to engineer TLM biosynthesis for structural diversity in S. hindustanus
Until we can reliably produce natural products in model heterologous hosts by expression of gene clusters encoding their biosyntheses, the development of genetic systems for the manipulation of natural product biosynthetic machineries in their native producers is the best alternative. Most natural product-producing strains were discovered for their ability to produce a specific compound, and therefore, little is known about their physiology, microbiology, and genetic amenability. The development of a genetic system for a new natural product-producing organism remains to be an empirical rather than scientific endeavor.In spite of the unparalleled structural diversity observed for natural products, their biosynthetic machinery appears to be remarkably conserved. Recent progress in genetic and biochemical characterization of natural product biosynthetic machinery clearly suggests that permutations to a given biosynthetic pathway can often account for the formation of the entire family of metabolites. This opened up the possibility of screening a pool of organisms to select one with the most desirable genetic, biochemical, and chemical traits as a biosynthetic platform to access structural diversity for a given natural product scaffold by combinatorial biosynthesis methods.
Since the cloning and sequencing of the blmAB resistance locus in 199439,40 and the entire BLM gene cluster in 200020 from S. verticillus, little progress has been made in developing a genetic system for this organism in spite of considerable efforts from several laboratories over the past decade. Recognizing that members of the BLM family of antitumor antibiotics most likely will share a common biosynthetic machinery, we screened several strains known to produce BLM-like compounds and succeeded in developing a genetic system for the TLM producing S. hindustanus as well as the zorbamycin producing S. flavoviridis (data not shown). The feasibility of manipulating TLM biosynthesis in S. hindustanus was demonstrated by inactivating the tlmE gene to generate the mutant strain SB8001 and by complementing the SB8001 strain to restore TLM production. While it was not clear why the SB8001 strain failed to accumulate any TLM intermediates, the fact that expression of a functional copy of tlmE restored TLM production excluded the possibility of potential polar effects on downstream genes from tlmE inactivation. The development of a genetic system for S. hindustanus provides an opportunity, for the first time, to apply combinatorial biosynthetic methods to the biosynthetic machinery of this important family of antitumor antibiotics to engineer novel TLM and BLM analogs with better clinical efficacy and lower toxicity.
Materials and methods
Bacterial strains, plasmids, and culture conditions
S. hindustanus E465-94 (ATCC 31158, American Type Culture Collection, Rockville, MD, USA) was cultivated at 30 °C on ISP4 agar or in YEME liquid medium.56 For production of TLMs, S. hindustanus was cultured in 250 ml baffled flasks containing 50 ml of the seed medium composed of 1.5% glucose, 0.2% yeast extract, 0.5% peptone, 0.05% K2HPO4, 0.05% MgSO4·7H2O, 0.5% CaCO3, adjusted to pH 7.2 with 1.0 M HCl. After growth at 30 °C and 250 rpm for 2 days, 5 ml of the seed cultures were inoculated into 250 ml baffled flasks with 50 ml of production medium containing 2.5% sucrose, 0.5% glucose, 3% cotton-seed meal, 3% distiller's grains and solubles (Sigma-Aldrich, Milwaukee WI.), 0.3% (NH4)2SO4, 0.01% CuSO4·5H2O. The resultant cultures were fermented at 30 °C and 250 rpm for 4 to 6 days.E. coli XL1 Blue MR (Stratagene, La Jolla, CA) and ET1256762 were grown at 37 °C in Luria-Bertani (LB) medium.63E. coli ET12567 was used to propagate unmethylated plasmid DNA for transformation of S. hindustanus. E. coli ET12567 (pUZ8002)64 was used as a donor in E. coli–S. hindustanus conjugation. E. coli BW25113 (pIJ790) was the host for gene replacement by the REDIRECT Technology.57
SuperKos, a modified version of SuperCos 1 (Stratagene) that lacks the kanamycin resistance marker, was used to construct the S. hindustanus genomic library. pSET15265 carrying the ΦC31 integration function was modified by inserting the thiostrepton resistance gene tsr between the aac(3)IV and oriT elements to afford pBS8004, which was used to establish and optimize the genetic system for S. hindustanus. pIJ4026,58 SuperCos 1, and pPAC-S166 were the sources of resistance genes for erythromycin (ermE), kanamycin (neo), and thiostrepton (tsr), respectively.
ISP4 medium supplemented with MgCl2 to a final concentration of 28 mM was used for S. hindustanussporulation, S. hindustanusspore electroporation, and E. coli–S. hindustanus conjugation. R2YE56 was used for S. hindustanusprotoplast transformation and protoplast electroporation. S. hindustanus wild-type and recombinant strains were all cultivated at 30 °C.
Ampicillin (150 µg ml–1), apramycin (100 µg ml–1), chloramphenicol (25 µg ml–1), and neomycin (50 µg ml–1) were used for plasmid selection in E. coli. Thiostrepton (10 µg ml–1) and erythromycin (100 µg ml–1) were used for selection of S. hindustanus recombinant strains. Trimethoprim (50 µg ml–1) was used to select against E. coli conjugal donors after E. coli–S. hindustanus conjugation.
Genetic manipulation of S. hindustanus
E. coli–S. hindustanus conjugation conditions were optimized according to Choi and co-workers.67 Briefly, E. coli ET12567 (pUZ8002) donor cells carrying the desired plasmid were prepared by growing at 37 °C in 50 ml of LB supplemented with kanamycin, chloramphenicol, and apramycin for 6 hrs to OD600 = 0.4–0.6, washing twice with 50 ml of LB, and resuspending in 0.5 ml of LB. S. hindustanusspores (1 × 109–1010) as conjugation recipients were suspended in 0.5 ml of TSBY (103 g sucrose, 30 g Bacto tryptic soy broth, and 5 g yeast extract per liter) and heat shocked at 50 °C for 10 min, cooled to room temperature, mixed with the donor cells, and centrifuged briefly to remove most of the supernatant. The resultant pellet was resuspended, spread on ISP4 plates supplemented with 28 mM MgCl2, and grown 16–20 h at 30 °C. The plate was covered with 1 ml of water containing the appropriate antibiotics to select for S. hindustanus exconjugants and trimethoprim to select against E. coli donor cells.S. hindustanus protoplast transformation and protoplast electroporation were carried out according to Kieser and co-workers.56S. hindustanusspore electroporation was conducted as follows: spores (1 × 108–109) were resuspended in 1 ml of TSBY, heat shocked for 10 min at 50 °C in a water bath, cooled on ice, washed 4 times with ice cold H2O, resuspended in ice cold H2O to 100 µl, and mixed with 1–5 µl of plasmid or cosmid DNA. The resulting solution was subjected to electroporation in a 0.2 cm ice-cold gene pulser cuvette using a BioRad GenePulser II set to 200 Ω, 25 µF, and 2.5 kV.
DNA isolation and manipulation
Plasmid preparations were carried out using commercial kits (Qiagen). Total S. hindustanus DNA was isolated according to literature protocols.56Restriction enzymes and other molecular biology reagents were from commercial sources, and digestion and ligation followed standard methods.63 For Southern analysis, digoxigenin labeling of DNA probes, hybridization, and detection were performed according to the protocols provided by the manufacturer (Boehringer Mannheim Biochemicals, Indianapolis, IN).Construction and screening of the S. hindustanuscosmid library
Chromosomal DNA of S. hindustanus was partially digested with Sau3AI, dephosphorylated, and ligated into the BamHI site of SuperKos. The ligation products were packaged with Gigapack III XL (Stratagene) and transduced into E. coli XL1 Blue MR.The tlmA probe (probe 1) containing the TLM resistance gene Sh ble of S. hindustanus was prepared by PCR, using primers R1 (5′-CCAAGTTGACCAGTGCCGTTCC-3′) and R2 (5′-CGTCGGTCAGTCCTGCTCCTCG-3′). The cosmid library was screened by colony hybridization on Hybond N+ (Amersham, Little Chalfont, Bucks, UK) as recommended by the manufacturer, and cosmid pBS8001 was isolated. Two more probes, the 3.6 kb EcoRV fragment excised from pBS8001 (probe 2), and the 4.2 kb SanDI fragment from pBS8002 (probe 3), were used to isolate the cosmid (ie, pBS8001, pBS8002 and pBS8003) covering the entire cluster.
DNA sequencing and analysis
Double-stranded DNA sequencing of the two cosmids, pBS8001 and pBS8003, was performed by the dideoxynucleotide chain termination method using a shotgun library (Lucigen, Middleton, WI) with DNA fragments of approximately 1.5 to 2.0 kb in length. A third cosmid, pBS8002 was chosen to fill-in the 1034 bp gap between pBS8001 and pBS8003. Sequencing reactions were run using Big Dye Terminator mix (Applied Biosystems, Foster City, CA), cleaned using CleanSeq magnetic beads (Agencourt Biosciences, Beverly, MA), and sequenced by the University of Wisconsin Biotechnology Center (Madison, WI). Samples were electrophoresed on an Applied Biosystems 3700 automated DNA sequencing instrument, and data were analyzed using PE-Biosystems version 3.7 for Sequence Analyses. Sequence assembly and contig alignments were done using the Seqman program in the Lasergene software package (DNASTAR Inc. Madison, WI). ORF assignments were accomplished with the Codon Preference program from the GCG software package (Biocomp, University of Wisconsin, Madison, WI) using the STM7074 codon table, the GeneMark.hmm, version 2.4 online program using S. avermitillis and S. coelicolor matrices, and Frameplot 2.2 analyses.Construction of aac(3)IV-tsr and ermE-neo tandem resistance gene cassettes by the REDIRECT method
S. hindustanus showed a high level of resistance to many antibiotics including apramycin, kanamycin, viomycin, and streptomycin, all of which are routinely used in the REDIRECT Technology gene replacement kit.57S. hindustanus was sensitive to thiostrepton and erythromycin, which therefore can be used to select for S. hindustanus recombinant strains. However, the resistance genes for these two antibiotics are not selectable in E. coli, hence they can not be used directly for construction of gene replacement vectors by the REDIRECT method.To combine the thiostrepton resistance gene tsr with the E. coli selectable apramycin resistance gene aac(3)IV, the tsr gene was amplified by PCR from pPAC-S165 as an SgrAI–ApaI fragment using primers tsr-r3-SgrAI (5′-GAGGTAGCGCCGGCGTCCGAGGAACAGAGGCG-3′) and tsr-f-ApaI (5′-CGTGGCGGGCCCGATCAAGGCGAATACTTCATATG-3′). The resultant PCR product was sequenced to verify PCR fidelity and ligated into the SgrAI–ApaI sites of pSET15264 to afford pBS8004. pBS8004 carries the aac(3)IV-tsr-oriTallele with aac(3)IV for selection in E. coli, tsr for selection in S. hindustanus, and oriT for E. coli–S. hindustanus conjugation.
To construct the ermE-neo resistance cassette, oligonucleotides ermE-neo-1 (5′-GTTCTGTCGAGAGGAATCAGAGGTTGATGTCGGCCCGGAGCTTCACGCTGCCGCAAGCAC-3′) and ermE-neo-2 (5′-GCGAGCGGTGGAGATCGGCTCGTCGTCGCGCGACATCGACTCGAAATCTCGTGATGGCAG-3′) were used to first amplify the kanamycin resistance gene neo from SuperCos 1 and subsequently place it in tandem with the erythromycin resistance gene ermE in pIJ402658 by the REDIRECT method to afford pBS8007. The fidelity of the ermE-neo cassette in pBS8007 was confirmed by DNA sequencing.
Constuction of pBS8005 by the REDIRECT method for tlmE inactivation by gene replacement
To modify the vector backbone of cosmid pBS8001, the β-lactamase gene (bla) of pBS8001 was replaced with the aac(3)IV-tsr-oriT cassette from pBS8004 by the REDIRECT method using oligonucleotides f-pMT3 (5′-CTCACGTTAAGGGATTTTGGTCATG-3′) and r-pMT3 (5′-GGAAGAGTATGAGTATTCAACATTTCCGTGTCGCCCTTATGCATGCCTGCAGGTCGACGG-3′), affording pBS8008. The tsr gene in pBS8008 serves as a selection marker in S. hindustanus, while the oriT function in pBS8008 mediates E. coli-S. hindustanus conjugation.pBS8008 was then modified by the REDIRECT method using oligonucleotides tlmE-neoem2 (5′-AGTTGACGGACAGGATGGTGCGGACGCGTTCCACGCTCATGGTATATCTCCTTCTTGCGCCCGATGCTAGTCGCG-3′) and tlmE-neoem1 (5′-GCCGCCTGGAGCGGGTGGCCTGAGGCGACCGGGGCGGACGGTTGGGCGTCGCTTGGTCGG-3′) to yield pBS8005. This replaced the entire tlmE gene with the ermE-neo resistance cassette pBS8007, hence inactivating the tlmE gene in pBS8005. A conserved ribosomal binding site (RBS) sequence was designed into the tlmE-neoem2 primer so that an RBS was introduced between the ORFp158 promoter of ermE and the ATG translational start codon of tlmD in pBS8005 to prevent potential polar effects on genes downstream of tlmE.
Isolation of the S. hindustanus SB8001 mutant strain by tlmE replacement in S. hindustanus
Plasmid pBS8005, carrying the ΔtlmE::ermEallele , was introduced into S. hindustanus by spore electroporation followed by antibiotic selection. Colonies that were thiostrepton sensitive and erythromycin resistant were isolated as S. hindustanus SB8001, in which the tlmE gene has been replaced with the the ΔtlmE::ermEallele . The genotype of the S. hindustanus SB8001 mutant strain was first analyzed by PCR using oligonucleotides f-tlmE (5′-CTGGAGCGGGTGGCCTGAGG-3′) and r-tlmE (5′-GACGGACAGGATGGTGCGG-3′) and subsequently confirmed by Southern analysis. A 2287-bp fragment harboring a portion of tlmII, tlm-orf28, and a portion of tlmE was amplified from pBS8001 by PCR using oligonucleotides Mbth-up (5′-GGTCGTGCACTACCTGCCC-3′) and r-tlmE (5′-GACGGACAGGATGGTGCGG-3′) and used as a probe in Southern analysis.Construction of pBS8006 and complementation of the ΔtlmE::ermE mutation in S. hindustanus SB8001
To construct the tlmE expression plasmid pBS8006, the ORFp1 promoter and RBS from pBS8005 was first inserted into pBS8008 in front of tlmE by the REDIRECT method using oligonucleotides Mbthneoem1 (5′-GTGGACAGGAACACCGCGCACGAGAACGAGGAGTGACCCGTCGCTTGGTCGGTCATTTCG-3′) and Mbthneoem2 (5′-CGGATCGCCGGGTTGTCGGTCATGCCACCTCCCCTCGCCATGGTATATCTCCTTCTTG-3′) to yield pBS8009. The modified tlmE locus in pBS8009 was cloned into the NotI site of pBS8004 by ligation-independent cloning68 using oligonucleotides NotI-tlmE1 (5′-TCGCGCGCGGCCAACTGCTCACCGCTGGATCC-3′) and NotI-tlmE2 (5′-TCCGCGGCCAACCGTTCGGTGTTGACGTAG-3′) to afford pBS8006, in which the expression of tlmE was under the control of the ORFp1 promoter.Introduction of pBS8006 into S. hindustanus SB8001 was carried out by spore electroporation. Transformants that were resistant to thiostrepton were selected as S. hindustanus SB8002. The ΦC31 site-specific integration function, int and attP, of pBS8006 was expected to mediate its integration into the chromosome of S. hindustanus SB8001, and maintenance of pBS8006 in the resultant S. hindustanus SB8002 strain was verified by PCR using oligonucleotides f-pMT3 and r-pMT3.
Isolation and purification of TLMs from S. hindustanusfermentation
To isolate TLMs from S. hindustanus wild-type or recombinant strains, a typical fermentation broth (50 ml) was adjusted to pH 7.0 with 1.0 N HCl and loaded onto an Amberlite® IRC50 column (24 ml). After washing the column with ten bed volumes of H2O, TLMs were eluted with 50 ml of 20% NaCl. The resultant Amberlite® IRC50 eluate was mixed with 1/8 volume of Diaion® HP-20 resin and incubated at room temperature with gentle agitation for 45 min. The Diaion® HP-20 resin was then packed into a column, washed with ten bed volumes of H2O, and drained of excess water. The column was then eluted with eight bed volumes of 80% methanol, and fractions containing TLMs were combined and concentrated in vacuo to 1 ml. TLMs isolated at this step were about 80% pure, and the estimated yield of TLMs was ∼18–20 mg l–1. Analytical HPLC was carried out on an Apollo C-18 column (5 µl, 250 × 4.6 mm, Alltech Associates, Inc., Deerfield, IL). The column was equilibrated with 100% solvent A (99.8% H2O, 0.2% acetic acid) and 0% solvent B (99.8% methanol, 0.2% acetic acid) and developed with a linear gradient (0–5 min, linear gradient from 100% A/0% B to 90% A/10% B; 5–30 min, linear gradient from 90%A/10% B to 0% A/100% B; 30–35 min, 0% A/100% B) at a flow rate of 0.7 ml min–1 and UV detection at 300 nm using a Varian Prostar 330 PDA detector (Varian, Palo Alto, CA). Under these conditions, TLM A and TLM B were eluted with retention times of 9.1 and 9.8 min, respectively. Final purification of TLMs was achieved by semipreparative HPLC on an Altima C18 column (5µ, 250 × 10 mm, Alltech Associates, Inc., Deerfield, IN). HPLC was carried out on the same instrument and with the same detector under the following conditions: the column was equilibrated with 100% solvent A (99.9% H2O, 0.1% TFA) and 0% solvent B (99.9% methanol, 0.1% TFA) and developed with a linear gradient from 100% A/0% B to 50% A/50% B in 15 min at a flow rate of 3 ml min–1 and UV detection at 300 nm. Upon removal of the solvent, TLM A or TLM B was purified as a blue powder of a TLM A-Cu or TLM B-Cu complex, respectively. Cu-free TLM A was obtained as a pale yellow powder by treating the TLM A-Cu complex with 0.5 M EDTA-Na (pH 7.3) solution followed by HPLC purification.Analysis of TLMs as TLM A-Cu and TLM B-Cu complexes and Cu-free TLM A
LC-ESI-MS analysis of the TLM A-Cu and TLM B-Cu complexes was performed on an Agilent 1100 HPLC-MSD SL quadrupole mass spectrometer or an Agilent 1100 HPLC-MSD SL ion trap mass spectrometer with an authentic TLM A-Cu complex as a standard. MALDI-FTMS analysis of Cu-free TLM A was performed on an IonSpec HiResMALDI FT-Mass spectrometer. NMR data were acquired on a VARIAN Inova-500 (500 MHz) spectrometer. The sample was dissolved in D2O (pH unadjusted) with sodium 3-trimethyl silyl [2,2,3,3,–2H4] propionate [TSP] as an internal standard. 1H NMR (D2O), δ (ppm) 1.15 (d, J = 6.8 Hz, 3H, Me-39), 1.17 (d J = 6.9 Hz, 3H, Me-32), 1.28 (d, J = 7.0 Hz, Me-56), 1.64–1.80 (m, 10H, H-58,59,67,68), 1.88 (t, 2H, H-64), 1.96 (s, 3H, Me-11), 2.43 (dd J = 15.3, 3.0, 1H, H-34a), 2.50 (dd, J = 15.0, 9.5 Hz, 1H, H-34b), 2.62 (dd, J = 16.0, 5.0 Hz, 1H, H-61a), 2.71 (dd, J = 16.0, 10.0 Hz, 1H, H-61b), 2.91 (dd, J = 16.0, 7.6 Hz, 1H, H-5a), 2.95–3.10 (m, 7H, H-5b, 65, 66, 69), 3.28 (m, 2H, H-63), 3.41–3.50 (m, 5H, H-3, H-54, 57), 3.54 (m, 2H, H-50), 3.69 (m, 1H, H-60), 3.75–3.90 (m, 6H, H-17, 23, 24, 25, 52), 3.91–4.00 (m, 2H, H-31, 33), 4.05 (m, 3H, H-16,19, 21), 4.11–4.19 (2H, m, H-18, 53), 4.19–4.25 (2H, m, H-38, 55), 4.35 (1H, d, J = 4.5 Hz, H-37), 4.40 (1H, t, J = 5.5 Hz, H-2), 4.48 (1H, t, J = 5.0 Hz, H-6), 4.74 (1H, overlapped by H2O, H-22), 5.04 (1H, s, H-51), 5.05 (1H, s, H-20), 5.13 (1H, d, J = 8.0 Hz, H-13), 5.27 (1H, d, J = 3.5 Hz, H-42), 5.41 (1H, d, J = 8.0 Hz, H-14), 5.74 (1H, d, J = 3.5 Hz, H-41), 7.63 (1H, s, H-28), 8.20 (1H, s, H-44), 8.22 (1H, s, H-47), 8.82 (1H, s, H-29).Nucleotide sequence accession number
The nucleotide sequence reported in this study is available in the GenBank database under accession no. EF032505.Acknowledgements
We thank the Analytical Instrumentation Center of the School of Pharmacy, UW-Madison, for support in obtaining MS and NMR data and the John Innes Center, Norwich, United Kingdom, for providing the REDIRECT Technology kit.This work was supported in part by the NIH grant CA94426. N.P.G is supported in part by NIH grant T32 GM08505, U.G is a Postdoctoral Fellow of the Deutsche Forschungsgemeinschaft (DFG), and B.S. is the recipient of an NIH Independent Scientist Award (AI51687).
References
- H. Kawaguchi, H. Tsukiura, K. Tomita, M. Konishi, K. Saito, S. Kobaru, K. Numata, K. Fujisawa, T. Miyaki, M. Hatori and H. Koshiyama, J. Antibiot., 1977, 30, 779–788 CAS.
- M. Konishi, K. Saito, K. Numata, T. Tsuno, K. Asama, H. Tsukiura, T. Naito and H. Kawaguchi, J. Antibiot., 1977, 30, 789–805 CAS.
- K. Tomita, Y. Uenoyama, E. I. Numata, T. Sasahira, Y. Hoshino, K. I. Fujisawa, H. Tsukiura and H. Kawaguchi, J. Antibiot., 1978, 31, 497–510 CAS.
- T. Takita, Y. Muraoka, T. Nakatani, A. Fujii, Y. Umezawa and H. Naganawa, J. Antibiot., 1978, 31, 801–804 CAS.
- T. Takita, Y. Muraoka, T. Yoshioka, A. Fujii and K. Maeda, J. Antibiot., 1972, 25, 755–757 CAS.
- H. Umezawa, K. Maeda, T. Takeuchi and Y. Okami, J. Antibiot., 1966, 19, 200–209 CAS.
- K. Maeda, H. Kosaka, K. Yagishita and H. Umezawa, J. Antibiot., 1956, 9, 82–85 CAS.
- A. D. Argoudelis, M. E. Bergy and T. R. Pyke, J. Antibiot., 1971, 24, 543–557 CAS.
- D. L. Boger and H. Cai, Angew. Chem., Int. Ed., 1999, 38, 448–476 CrossRef CAS.
- R. M. Burger, Chem. Rev., 1998, 98, 1153–1170 CrossRef CAS.
- S. M. Hecht, J. Nat. Prod., 2000, 63, 158–168 CrossRef.
- J. Stubbe and J. W. Kozarich, Chem. Rev., 1987, 87, 1107–1136 CrossRef CAS.
- B. I. Sikic, M. Rosenzweig and S. K. Chater, Bleomycin chemotherapy, Academic Press, New York, 1985 Search PubMed.
- S. S. Boggs, G. P. Sartiano and A. DeMezza, Cancer Res., 1974, 34, 1938–1942 CAS.
- D. E. Lehane, E. Hurd and M. Lane, Cancer Res., 1975, 35, 2724–2728 CAS.
- J. E. Schurig, W. C. Rose, R. S. Hirth, A. Schlein, J. B. Huftalen, A. P. Florczyk and W. T. Bradner, Cancer Chemother. Pharmacol., 1984, 13, 164–170 CrossRef CAS.
- C. Nicaise, J. Ajani, P. Goudeau, M. Rozencweig, B. Levin and I. Krakoff, Cancer Chemother. Pharmacol., 1990, 26, 221–222 CrossRef CAS.
- C. Nicaise, W. K. Hong, W. Dimery, J. Usakewicz, M. Rozencweig and I. Krakoff, Invest. New Drugs, 1990, 8, 325–328 CAS.
- M. A. Walker, H. D. King, R. A. Dalterio, P. Trail, R. Firestone and G. M. Dubowchik, Bioorg. Med. Chem. Lett., 2004, 14, 4323–4327 CrossRef CAS.
- L. Du, C. Sanchez, M. Chen, D. J. Edwards and B. Shen, Chem. Biol., 2000, 7, 623–642 CrossRef CAS.
- B. Shen, L. Du, C. Sanchez, D. J. Edwards, M. Chen and J. M. Murrell, J. Ind. Microbiol. Biotechnol., 2001, 27, 378–385 CrossRef CAS.
- B. Shen, L. Du, C. Sanchez, D. J. Edwards, M. Chen and J. M. Murrell, J. Nat. Prod., 2002, 65, 422–431 CrossRef CAS.
- D. Drocourt, T. Calmels, J. P. Reynes, M. Baron and G. Tiraby, Nucleic Acids Res., 1990, 18, 4009 CAS.
- M. Baron, J. P. Reynes, D. Stassi and G. Tiraby, Gene, 1992, 114, 239–243 CrossRef CAS.
- R. P. Bennett, C. A. Cox and J. P. Hoeffler, Biotechniques, 1998, 24, 478–482 CAS.
- R. Gautier, D. Drocourt and T. Jaffredo, Exp. Cell Res., 1996, 224, 291–301 CrossRef CAS.
- S. E. Gold, G. Bakkeren, J. E. Davies and J. W. Kronstad, Gene, 1994, 142, 225–230 CrossRef CAS.
- D. Jefferies, P. Tebabi, D. Le Ray and E. Pays, Nucleic. Acids Res., 1993, 21, 191–195 CAS.
- P. Mulsant, A. Gatignol, M. Dalens and G. Tiraby, Somat. Cell Mol. Genet., 1988, 14, 243–252 Search PubMed.
- P. Perez, G. Tiraby, J. Kallerhoff and J. Perret, Plant Mol. Biol., 1989, 13, 365–373 CrossRef CAS.
- M. Santos, I. Vallejo, L. Rebordinos, S. Gutierrez, I. G. Collado and J. M. Cantoral, FEMS Microbiol. Lett., 1996, 137, 153–158 CrossRef CAS.
- D. A. Hopwood, Chem. Rev., 1997, 97, 2465–2497 CrossRef CAS.
- L. Du, M. Chen, Y. Zhang and B. Shen, Biochemistry, 2003, 42, 9731–9740 CrossRef CAS.
- L. Du and B. Shen, Chem. Biol., 1999, 6, 507–517 CrossRef CAS.
- R. M. Kagan and S. Clarke, Arch. Biochem. Biophys., 1994, 310, 417–427 CrossRef CAS.
- C. Waldron, P. Matsushima, P. R. Rosteck, Jr., M. C. Broughton, J. Turner, K. Madduri, K. P. Crawford, D. J. Merlo and R. H. Baltz, Chem. Biol., 2001, 8, 487–499 CrossRef CAS.
- K. Mansouri and W. Piepersberg, Mol. Gen. Genet., 1991, 228, 459–469 CAS.
- A. Gatignol, H. Durand and G. Tiraby, FEBS Lett., 1988, 230, 171–175 CrossRef CAS.
- M. J. Calcutt and F. J. Schmidt, Gene, 1994, 151, 17–21 CrossRef CAS.
- M. Sugiyama, C. J. Thompson, T. Kumagai, K. Suzuki, R. Deblaere, R. Villarroel and J. Davies, Gene, 1994, 151, 11–16 CrossRef CAS.
- M. Sugiyama, T. Kumagai, M. Shionoya, E. Kimura and J. E. Davies, FEMS Microbiol. Lett., 1994, 121, 81–85 CrossRef CAS.
- D. J. Netz, H. G. Sahl, R. Marcolino, J. S. Nascimento, S. S. Oliveira, M. B. Soares and M. C. F. Bastos, J. Mol. Biol., 2001, 311, 939–949 CrossRef CAS.
- C. Mendez and J. A. Salas, FEMS Microbiol. Lett., 1998, 158, 1–8 CrossRef CAS.
- J. H. Zhang, N. B. Quigley and D. C. Gross, Appl. Environ. Microbiol., 1997, 63, 2771–2778 CAS.
- Y. Nakamura, T. Kaneko, S. Sato, M. Mimuro, H. Miyashita, T. Tsuchiya, S. Sasamoto, A. Watanabe, K. Kawashima, Y. Kishida, C. Kiyokawa, M. Kohara, M. Matsumoto, A. Matsuno, N. Nakazaki, S. Shimpo, C. Takeuchi, M. Yamada and S. Tabata, DNA Res., 2003, 10, 137–145 Search PubMed.
- J. Ishikawa, A. Yamashita, Y. Mikami, Y. Hoshino, H. Kurita, K. Hotta, T. Shiba and M. Hattori, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14925–14930 CrossRef CAS.
- V. Bennett, J. Biol. Chem., 1992, 267, 8703–8706 CAS.
- S. Jia, W. J. VanDusen, R. E. Diehl, N. E. Kohl, R. A. Dixon, K. O. Elliston, A. M. Stern and P. A. Friedman, J. Biol. Chem., 1992, 267, 14322–14327 CAS.
- L. Lavaissiere, S. Jia, M. Nishiyama, S. de la Monte, A. M. Stern, J. R. Wands and P. A. Friedman, J. Clin. Invest., 1996, 98, 1313–1323 CrossRef CAS.
- J. E. Dinchuk, N. L. Henderson, T. C. Burn, R. Huber, S. P. Ho, J. Link, K. T. O'Neil, R. J. Focht, M. S. Scully, J. M. Hollis, G. F. Hollis and P. A. Friedman, J. Biol. Chem., 2000, 275, 39543–39554 CrossRef CAS.
- G. Homuth, M. Heinemann, U. Zuber and W. Schumann, Microbiology, 1996, 142, 1641–1649 CrossRef CAS.
- Y. Q. Cheng, G. L. Tang and B. Shen, J. Bacteriol., 2002, 184, 7013–7024 CrossRef CAS.
- M. Sosio, H. Kloosterman, A. Bianchi, P. de Vreugd, L. Dijkhuizen and S. Donadio, Microbiology, 2004, 50, 95–102 CrossRef.
- S. Pelzer, R. Sussmuth, D. Heckmann, J. Recktenwald, P. Huber, G. Jung and W. Wohlleben, Antimicrob. Agents Chemother., 1999, 43, 1565–1573 CAS.
- E. Stegmann, C. Rausch, S. Stocker, D. Burkert and W. Wohlleben, FEMS Microbiol. Lett., 2006, 262, 85–92 CrossRef CAS.
- T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater and D. A. Hopwood, Practical Streptomyces Genetics, John Innes Foundation, Norwich, UK, 2000 Search PubMed.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541–1546 CrossRef CAS.
- M. J. Bibb, G. R. Janssen and J. M. Ward, Gene, 1985, 38, 215–226 CrossRef CAS.
- A. M. Calafat, H. Won and L. G. Marzilli, J. Am. Chem. Soc., 1997, 119, 3656–3664 CrossRef CAS.
- H. Imanishi, M. Ohbayashi, Y. Nishiyama and H. Kawaguchi, J. Antibiot., 1978, 31, 667–674 CAS.
- L. Du, M. Chen, C. Sanchez and B. Shen, FEMS Microbiol. Lett., 2000, 189, 171–175 CrossRef CAS.
- D. J. MacNeil, J. L. Occi, K. M. Gewain, T. MacNeil, P. H. Gibbons, C. L. Ruby and S. J. Danid, Gene, 1992, 155, 119–125 CrossRef CAS.
- J. Sambrook and D. W. Russell, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, New York, 2001 Search PubMed.
- F. Flett, V. Mersinias and C. P. Smith, FEMS Microbiol. Lett., 1997, 155, 223–229 CrossRef CAS.
- M. Bierman, R. Logan, K. O'Brien, E. T. Seno, R. N. Rao and B. E. Schoner, Gene, 1992, 116, 43–49 CrossRef CAS.
- M. Sosio, F. Giusino, C. Cappellano, E. Bossi, A. M. Puglia and S. Donadio, Nat. Biotechnol., 2000, 18, 343–345 CrossRef CAS.
- S. U. Choi, C. K. Lee, Y. I. Hwang, H. Kinoshita and T. Nihira, Arch. Microbiol., 2004, 181, 294–298 CrossRef CAS.
- C. Aslanidis, P. J. de Jong and G. Schmitz, PCR Methods Appl., 1994, 4, 172–177 Search PubMed.
| This journal is © The Royal Society of Chemistry 2007 |