Encapsulation and functionalization of nanoparticles in crosslinked resorcinarene shells†
Ramjee
Balasubramanian
,
Young-Gil
Kwon
and
Alexander
Wei
*
Department of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA
First published on 3rd November 2006
Abstract
Two resorcinarene-derived tetrathiols with terminal alkene sidechains (tetraarylthiol cavitand 3 and tetrabenzylthiol cavitand 4) were determined to be efficient at extracting colloidal gold nanoparticles from aqueous solutions and stabilizing their dispersion in organic solvents. Treatment of these nanoparticle dispersions with the Grubbs olefin metathesis catalyst resulted in crosslinked resorcinarene shells that were highly resistant to alkanethiol-induced desorption at high temperatures. Nanoparticles in crosslinked shells of tetrabenzylthiol cavitand 4 were particularly robust, and could be precipitated and redispersed many times with minimal attrition. These shells could also withstand oxidative conditions and were amenable to synthetic modifications involving epoxidation and dihydroxylation.
Introduction
Recent years have witnessed an exponential growth in fundamental and applied studies of metal nanoparticles with size- and shape-dependent properties.1 Much of this has been propelled by readily available methods of surface functionalization, in particular by the chemisorption of organic thiols on gold to produce monolayer-protected nanoparticles.2 Ironically, the benefits (and limitations) of organic surface functionalization may have also contributed toward a dichotomy in nanoparticle research as a function of particle size. Alkanethiol-passivated gold nanoparticles are typically prepared in the 2–10 nm size range by the reduction of gold chloride in the presence of the surfactant, such as the popular two-phase protocol first introduced by Brust et al.3 Nanoparticles of this size can be readily dispersed in organic or aqueous solvents, depending largely on the terminal groups of the chemisorptive monolayer. On the other hand, colloidal gold nanoparticles (>10 nm), which are stabilized electrostatically in aqueous dispersions, are susceptible to kinetic aggregation upon treatment with simple alkanethiols regardless of their interfacial surface properties.4 Dispersion control of these larger metal nanoparticles is important for their subsequent application as functional nanostructures; for example, gold and silver nanostructures are well known for their size-dependent optical resonances generated by localized surface plasmons, which have been applied with great effect in numerous chemical and bioanalytical sensing modalities5 as well as in nanophotonics.6 The factors which determine dispersion stability or surfactant robustness are often overlooked or dismissed as simple colloidal processing issues, but these are in fact of fundamental importance to materials synthesis and fabrication methodologies, and remain critical bottlenecks in the development of functionalized nanomaterials.We and others have addressed such issues by developing surfactant systems based on resorcinarenes and the closely related cavitands, whose anisotropic structures are well suited for the encapsulation and dispersion of metal nanoparticles in organic solvents.7–10 For example, thiolated cavitands such as 1 and 2 (see Fig. 1) are effective at extracting gold nanoparticles in the 20–100 nm size range from aqueous solution,9 and enabling their self-organization into 2D arrays at the air/water interface.11 Tetrabenzylthiol cavitand 2 is especially noteworthy for its capacity to stabilize dispersions of gold nanoparticles in organic solvents.9 However, these dispersions are slowly degraded when exposed to alkanethiols, which compete with the resorcinarene-based surfactants for adsorption onto the nanoparticle surface.
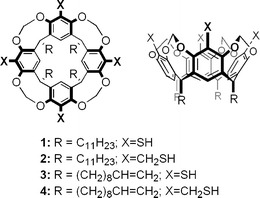 | ||
| Fig. 1 Resorcinarene-derived cavitands 1–4, top and side views. | ||
To further improve the stability of the surfactant coating, one can either increase the chemisorptive strength of the passivating ligand, or develop a covalent crosslinking methodology to produce a nondesorptive coating. With respect to the former, we have recently established that metal surfaces passivated by dithiocarbamate ligands are strongly resistant to displacement by competing thiols.12 Such ligands are readily formed by condensation of the corresponding amine with CS2, and permit a diversity of structures to be attached to metal surfaces.13 With respect to surface crosslinking, one can expect the resulting covalent shell to have a mechanical robustness similar to that of grafted polymers. Numerous crosslinking methods have been established in recent years; notable examples include the stabilization of self-assembled amphiphilic copolymers into polymeric nanostructures,14,15 the covalent encapsulation of carbon nanotubes16 and metal nanoparticles,17,18 and the formation of hollow polymer nanoshells by dissolution of the core material.19–22 Ruthenium-catalyzed olefin metathesis is an especially mild and compatible method for crosslinking surfactant termini or for cross-metathesis with functionalized monomers, and has been used successfully with dendrimers21 and inorganic nanoparticles.17,20,23
As part of our effort to develop robust platforms for the synthetic modification of nanoparticle surfaces, we have prepared two novel tetrathiolated cavitands 3 and 4 for the encapsulation of gold nanoparticles and their subsequent transformation into nondesorptive crosslinked shells. The tailgroups of these surfactants possess terminal alkenes for covalent crosslinking by olefin metathesis, but their thiolated headgroups can potentially interfere with the metal catalyst. We show that a robust crosslinked surfactant coating can still be achieved in spite of this susceptibility, with excellent resistance to thermal desorption and displacement by competing surfactants while remaining amenable to further chemical functionalization.
Results and discussion
Synthesis
The preparations of tetrathiolated tetraenes 3 and 4 are described in Scheme 1. Resorcinarene C10 tetraene 5 was synthesized using literature procedures24 and cleanly transformed into tetrabrominated tetraene 6, followed by methylenation of the phenolic hydroxyl groups to produce cavitand 7. Metal–halogen exchange with n-BuLi and treatment with elemental sulfur9b,25 produced tetraarylthiol cavitand 3 in good yield. The tetraanionic cavitand was also treated with methyl chloroformate26 to produce tetraester 8, which was reduced to tetraol 9 and transformed in two steps into tetrabenzylthiol cavitand 4. Although both compounds could be obtained in analytically pure form, it is worth noting that neat tetrabenzylthiol 4 was susceptible to polymerization by thiol addition to alkenes, presumably initiated by light or air oxidation.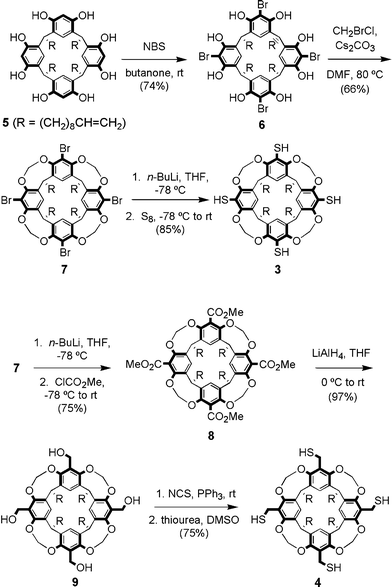 | ||
| Scheme 1 Synthesis of tetrathiolcavitand tetraenes 3 and 4. | ||
Crosslinking by olefin metathesis
We have previously used the “first generation” Grubbs metathesis catalyst (G-1; Cl2(PCy3)2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh) to crosslink weakly adsorbed monolayers of resorcinarene tetraene (octamethyl ether of 5) into nondesorptive surfactant shells around gold nanoparticles, produced by an aerosol method.17 This greatly increased the nanoparticles’ robustness to subsequent chemical processing (e.g., size-exclusion chromatography), but their dispersibility was compromised due to excessive crosslinking of the surfactant tailgroups. In this study, tetrathiols 3 and 4 enabled us to extract gold nanoparticles from aqueous solutions (see below), but the presence of free thiols gave rise to concerns regarding their compatibility with the crosslinking agent, as the sensitivity of the G-1 catalyst to thiols is well documented.27 This issue was examined by NMR studies on the ring-closing metatheses (RCM) of tetraenes 3, 4, and 7 to evaluate the performance of the G-1 catalyst in the presence of excess thiol (see Scheme 2 and Fig. 2). RCM of resorcinarene tetraenes into polycyclic dienes such as 10 and 11 also provides a useful model for 2D crosslinking on nanoparticle surfaces, which can be expected to proceed with a significant level of intermolecular crosslinking as suggested by our earlier studies with resorcinarene ether derivatives adsorbed on gold nanoparticles.17
CHPh) to crosslink weakly adsorbed monolayers of resorcinarene tetraene (octamethyl ether of 5) into nondesorptive surfactant shells around gold nanoparticles, produced by an aerosol method.17 This greatly increased the nanoparticles’ robustness to subsequent chemical processing (e.g., size-exclusion chromatography), but their dispersibility was compromised due to excessive crosslinking of the surfactant tailgroups. In this study, tetrathiols 3 and 4 enabled us to extract gold nanoparticles from aqueous solutions (see below), but the presence of free thiols gave rise to concerns regarding their compatibility with the crosslinking agent, as the sensitivity of the G-1 catalyst to thiols is well documented.27 This issue was examined by NMR studies on the ring-closing metatheses (RCM) of tetraenes 3, 4, and 7 to evaluate the performance of the G-1 catalyst in the presence of excess thiol (see Scheme 2 and Fig. 2). RCM of resorcinarene tetraenes into polycyclic dienes such as 10 and 11 also provides a useful model for 2D crosslinking on nanoparticle surfaces, which can be expected to proceed with a significant level of intermolecular crosslinking as suggested by our earlier studies with resorcinarene ether derivatives adsorbed on gold nanoparticles.17
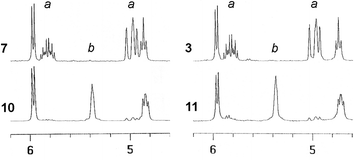 | ||
| Fig. 2 1H NMR spectra (300 MHz, CDCl3) showing intramolecular crosslinking of resorcinarene-derived tetraenes 7 and 3 (top) into dienes 10 and 11 (bottom) by treatment with G-1 catalyst. a, Terminal alkene protons; b, cis-alkene protons. | ||
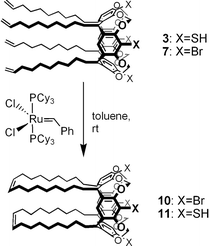 | ||
| Scheme 2 Ring-closing metathesis of resorcinarene-based tetraenes 3 and 7 into polycyclic dienes (11 and 10, respectively). | ||
Dilute solutions of tetrabromocavitand tetraene 7 (0.7 mM in CDCl3) were treated with 30 mol% of the G-1 catalyst, with the expectation of efficient RCM into polycyclic diene 10. This was indeed the case: in situ NMR analysis revealed that 95% of the terminal alkenes were crosslinked into disubstituted alkenes (see Fig. 2, left), with 75% conversion within just 5 min. Tetraarylthiol tetraene 3 appeared to have surprisingly little effect on G-1 activity, and could be transformed by RCM into polycyclic diene 11 with a crosslinking efficiency just slightly less than that observed for 7 (see Fig. 2, right). In contrast, G-1 catalyzed RCM of tetrabenzylthiol tetraene 4 was sluggish with only 10% crosslinking of the alkene groups after overnight treatment, strongly suggestive of catalyst deactivation. RCM studies with the second-generation Grubbs catalyst28 were also conducted, but neither 3 nor 4 could be converted into their respective RCM products in significant quantities. The efficient RCM of 3 can be attributed to the relatively low reactivity of the arenethiols; in addition to their reduced Lewis basicity, the sulfur atoms are recessed within the cavitand headgroup, whereas the benzylthiol groups in 4 are clearly exposed.
Nanoparticle encapsulation
Colloidal gold nanoparticles (17–20 nm) were preconditioned using a mixed-bed ion-exchange resin, treated with millimolar solutions of 3 or 4 in THF, then extracted into toluene. In the case of tetraarylthiol 3, micromolar quantities of tetraoctylammonium bromide (TOAB) were needed to enable full extraction of the nanoparticles into the organic phase, as observed previously with saturated tetraarylthiol 1.9 The chemisorptive properties of tetrabenzylthiol 4 were superior to those of 3, and 4 could extract gold nanoparticles without the addition of TOAB. In both cases, the resorcinarene-encapsulated nanoparticles could be separated from free tetrathiol by centrifugation and redispersion in fresh solvent, but multiple washes often resulted in a significant loss of material. Furthermore, attempts to purify resorcinarene-encapsulated nanoparticles by size-exclusion chromatography (BioBeads SX-1, in toluene) resulted in their irreversible adsorption onto the polystyrene resin. Therefore, most of the surface crosslinking studies were performed in the presence of excess 3 or 4.Dispersion stability and functionalization of nanoparticles in crosslinked resorcinarene shells
Gold nanoparticles encapsulated by tetraarylthiol 3 were treated with G-1 for 5 min at different catalyst loadings (3–30 mol%, or 10–100 µM), followed by addition of ethyl vinyl ether to quench the catalyst. To our great satisfaction, the nanoparticle dispersions were extremely stable after treatment with G-1, a major improvement to our earlier crosslinking studies.17 The crosslinked monolayer of 3 likely contains a mixture of intermolecular and intramolecular crosslinked chains, as well as some uncrosslinked tailgroups. In this context, it is worth mentioning that attempts to transfer Au nanoparticles into the organic phase with polycyclic diene 11 were unsuccessful, implying that such coatings were structurally distinct from the crosslinked resorcinarene monolayers.The efficacy of crosslinking was evaluated by exposing nanoparticle dispersions to excess dodecanethiol (C12SH) at different temperatures, and monitoring optical changes by absorption spectroscopy (see Fig. 3). These systems were also compared with nanoparticles encapsulated in 3 but not crosslinked with G-1 catalyst. We have previously shown that displacement of resorcinarene-based surfactants from the surfaces of colloidal gold particles by C12SH results in precipitation and loss of optical density.8,9 However, all dispersions in this study proved to be stable in the presence of millimolar concentrations of C12SH at ambient temperature over a 140 h period, and could also survive a 2 h exposure at 70 °C (t = 67–69 h). The latter treatment revealed minor differences in dispersion stability as a function of catalyst loading, but more pronounced differences were observed upon further heating of these dispersions over a 16 h period (t = 142–158 h). Nanoparticles coated with uncrosslinked surfactants were completely precipitated from solution, whereas those treated with 3 mol% catalyst retained approximately 25% of their original optical density, but with noticeable broadening and redshifting of the plasmon resonance peak indicative of flocculation. In comparison, nanoparticles treated with 30 mol% catalyst were essentially unaffected, demonstrating that a higher degree of crosslinking in the surfactant shell was essential to resist displacement by C12SH. Subsequent TEM analysis of these dispersions revealed varying degrees of particle aggregation, which correlated strongly with the loss of optical absorption and flocculation (see Fig. 4). It is worth noting that nanoparticles encapsulated in uncrosslinked 3 without competing surfactant were unaffected by prolonged heating, indicating that crosslinking was not necessary for stability at 70 °C.
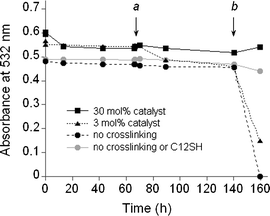 | ||
| Fig. 3 Dispersion stability of Au nanoparticles in toluene encapsulated by tetraarylthiol 3 and crosslinked with G-1 catalyst at variable loadings, followed by exposure to 40 mM C12SH. Dispersions were maintained at ambient temperatures except at point a (70 °C, t = 67–69 h) and point b (70 °C, t = 142–158 h). The thermal stability of nanoparticles encapsulated by uncrosslinked 3 in the absence of competing surfactant (grey circles) was also evaluated. | ||
 | ||
| Fig. 4 TEM images (Philips EM-400, 80 keV) of 20 nm Au particles encapsulated in tetraarylthiol 3 with varying degrees of crosslinking, after exposure to C12SH for 16 h at 70 °C. a, No crosslinking; b, treated with 3 mol% G-1 catalyst; c, treated with 30 mol% G-1 catalyst. TEM samples were prepared by depositing toluene dispersions onto Formvar-coated Cu grids (300 mesh), then were slowly dried in air; in the case of a, the precipitated particles were sonicated briefly prior to deposition. Scale bars = 150 nm. | ||
Gold nanoparticles encapsulated with tetrabenzylthiol 4 were also treated with 30 mol% G-1 catalyst for 5 min, followed by addition of ethyl vinyl ether. Only the highest loading was used because of the catalyst’s attenuated activity in the presence of 4. Remarkably, these crosslinked nanoparticles not only formed stable dispersions at elevated temperatures and in the presence of competing surfactants (see above), but could also be repeatedly precipitated and redispersed with minimal attrition. Nanoparticles encapsulated in crosslinked 4 were subjected to 10 cycles of high-speed centrifugation and redispersion by a brief exposure to a sonicating bath, with less than 10% fluctuation in optical density (see Fig. 5). Nanoparticles encapsulated in uncrosslinked 4 did not hold up as well to these conditions; the optical absorbance dropped to a third of its initial intensity after 4 cycles with an observable redshift in plasmon resonance, indicating the onset of aggregation. We note that nanoparticles encapsulated in crosslinked 3 could also survive several rounds of centrifugation and redispersion, but suffered a greater loss in optical absorbance compared with those stabilized in crosslinked shells of 4. This is likely related to the residual charge associated with incomplete surface passivation by 3, which can result in electrostatic aggregation. Ironically, despite the poor reactivity of the G-1 catalyst in the presence of free 4, the metathesis conditions yielded nanoparticles with superior dispersion characteristics. We surmise that a high catalyst loading increases the probability of initiating olefin metathesis on adsorbed molecules of 4, followed by rapid crosslinking around the nanoparticle surface.
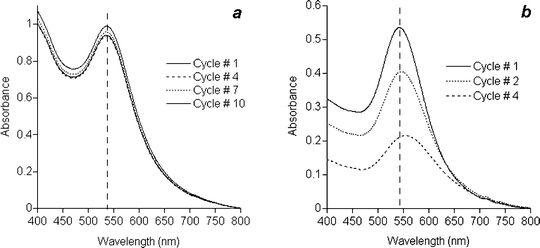 | ||
| Fig. 5 Effect of surfactant crosslinking on the dispersion stability of Au nanoparticles encapsulated by tetrabenzylthiol 4. a, Nanoparticles stabilized in crosslinked shells of 4 can withstand multiple centrifugation–redispersion cycles with minimal attrition. b, Nanoparticles without crosslinked shells are prone to aggregation after a few cycles. Vertical dashed line marks initial plasmon resonance of nanoparticle dispersion. | ||
Nanoparticles encapsulated in crosslinked 4 were subjected to additional chemical reactions to evaluate their capacity for multistep synthetic modifications. Given the high density of cis-alkenes on the crosslinked surfactant shells, we chose to examine two types of functionalization: (i) epoxidation followed by nucleophilic ring opening to produce polyhydroxylated species, and (ii) dihydroxylation followed by acetylation to produce nanoparticles with multiple ester groups (see Scheme 3). Epoxide generation entails the use of strongly oxidizing agents, and provides a stringent test of chemical stability. With respect to chemical characterization, the submicrogram quantities and large hydrodynamic radii of the functionalized nanoparticles preclude a straightforward analysis by NMR spectroscopy, but simple derivatization (e.g., acetylation) provides a convenient handle for structural analysis by infrared spectroscopy.
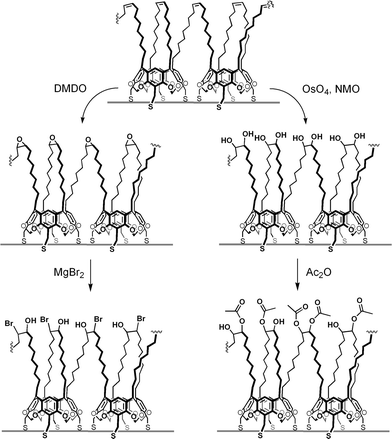 | ||
| Scheme 3 Synthetic modifications of crosslinked resorcinarene shells. | ||
Crosslinked and uncrosslinked species were treated at room temperature with excess dimethyldioxirane (DMDO), a reagent which can also oxidize thiols at ambient conditions and abolish their chemisorptive properties. The functionalized nanoparticles were gently precipitated by methanol and centrifuged, then redispersed in toluene after a brief exposure to a sonicating bath. Optical absorption spectra revealed that over 60% of the nanoparticles in crosslinked surfactant shells survived the oxidation step without loss of dispersibility (see Fig. 6), whereas nearly all of the nanoparticles without crosslinking were flocculated as a result of surfactant degradation. The epoxide-functionalized nanoparticles could then be dispersed in Et2O and treated with MgBr2 to produce a poly(halohydrin) coating, resulting in the formation of a pink–red precipitate. The reaction mixture was subjected to a typical aqueous workup, followed by centrifugation for recovery of the nanoparticles. These polyhydroxylated particles could not be resuspended in toluene, but could be successfully dispersed in methanol to form an indefinitely stable suspension.
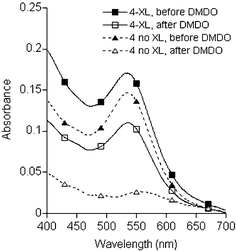 | ||
| Fig. 6 Effect of DMDO oxidation on Au nanoparticles encapsulated by tetrabenzylthiol 4. Nanoparticles stabilized in crosslinked shells (solid lines, squares) retained good dispersion characteristics after treatment with DMDO, whereas those without prior crosslinking (dashed lines, triangles) were almost completely flocculated. | ||
Nanoparticles encapsulated in crosslinked 4 were also treated with OsO4 in a toluene–acetone mixture, using N-methylmorpholine oxide (NMO) as the stoichiometric oxidant. These nanoparticles were centrifuged and formed stable suspensions in methanol or pyridine, and could be acetylated using Ac2O with catalytic 4-dimethylaminopyridine (DMAP). Transmission IR spectra of the isolated nanoparticles in KBr pellets revealed both the presence of a broad vibrational band for O–H stretching centered at 3450 cm−1 and the absence of C–H stretching modes above 3000 cm−1; in the case of the polyacetylated nanoparticles, a carbonyl stretching peak near 1740 cm−1 was also present (see ESI†). These studies respectively indicated the gross conversion of the alkenes into 1,2-diols followed by partial acetylation.
Experimental
Synthesis
THF, toluene, and DMF were dried and distilled according to standard procedures. All other chemicals were purchased from commercial sources and used as such unless otherwise specified. All organic reactions were carried out under argon atmosphere. Citrate-stabilized gold colloids were synthesized by a standard protocol.29Resorcinarene C10 tetraene 524
Resorcinol (14.3 g, 130 mmol) and 10-undecenal (27.5 mL, 132 mmol) were dissolved in ethanol (165 mL) at 0 °C and treated by the dropwise addition of 12 N HCl (20 mL). The reaction mixture was heated for 12 h at 65 °C, then cooled to rt and poured into 450 mL water to produce a yellow precipitate, which was filtered off and washed extensively with hot water (60–80 °C) for the complete removal of acid. The precipitate was dissolved in hot acetonitrile (350 mL, 65 °C), then reprecipitated by the dropwise addition of water (50–60 mL). The mixture was cooled first to rt then to 4 °C to induce further precipitation, followed by suction filtration and drying in air to yield compound 5 (21.4 g, 63%) as a light yellow powder.Tetrabromoresorcinarene tetraene 6
Compound 5 (9.37 g, 9 mmol) was dissolved in 2-butanone (90 mL) and treated with N-bromosuccinimide (9.61 g, 54 mmol), and allowed to stir for 12 h at rt under an inert atmosphere with protection from light. The reaction mixture was concentrated by rotary evaporation, then stirred for 1 h with saturated aqueous sodium thiosulfate (25 mL), followed by saturated sodium chloride (25 mL) and extraction with ethyl acetate (2 × 50 mL). The combined organic extracts were dried over MgSO4, concentrated and resuspended in MeOH (100 mL). This was collected by filtration and washed with additional MeOH, then dried in air to yield compound 6 (9.04 g, 74%) as a pale yellow solid. δH (300 MHz, DMSO-d6): 9.04 (8 H, s), 7.32 (4 H, s), 5.80 (4 H, m), 4.95 (8 H, m), 4.36 (4 H, t, J 7.5 Hz), 2.14 (8 H, m), 1.99 (8 H, q, J 6.9 Hz), 1.30 (48 H, m).Tetrabromocavitand tetraene 7
Tetrabromoresorcinarene 6 (3.81 g, 2.81 mmol), bromochloromethane (8.76 mL, 134.8 mmol), and Cs2CO3 (16.41 g, 50.64 mmol) were suspended in anhydrous DMF (90 mL) and heated for 12 h at 80 °C in a heavy-walled pressure reaction vessel. The reaction mixture was poured into 150 mL of 2 N HCl and extracted several times with ethyl ether, until the aqueous layer was clear and colorless. The combined organic phase was washed with water and brine, dried over MgSO4, then concentrated to a brown oil. Purification by silica gel chromatography (0–50% CH2Cl2 in hexanes) yielded compound 7 (2.60 g, 66%) as an amorphous white solid. δH (300 MHz, CDCl3): 7.04 (4 H, s), 5.97 (4 H, d, J 7.2 Hz), 5.8 (4 H, m), 4.97 (8 H, m), 4.86 (4 H, t, J 8.1 Hz), 4.40 (4 H, d, J 7.5 Hz), 2.2 (8 H, m), 2.05 (8 H, q, J 6.6 Hz), 1.3 (48 H, m).Tetraarylthiolcavitand tetraene 3
Tetrabromocavitand 7 (450 mg, 0.32 mmol) was azeotroped 3 times in toluene in a three-necked round-bottomed flask, and dried under vacuum overnight in the presence of P2O5, with a magnetic stirring bar and a solid addition tube containing sublimed sulfur (164 mg, 5.13 mmol). Compound 7 was then dissolved in anhydrous, degassed THF (15 mL) and cooled to −78 °C under an argon atmosphere, then treated with n-BuLi (1.34 mL of a 2.8 M solution in hexanes). The reaction mixture was stirred for 10 min, then treated with excess sulfur and allowed to warm over a 16 h period to rt. The pale yellow reaction mixture was quenched with aqueous saturated Na2SO3 (5 mL), then partially concentrated until a precipitate appeared. The reaction mixture was diluted with water (60 mL) and extracted with EtOAc (3 × 70 mL), then washed with water and brine and dried over MgSO4. Purification by silica gel chromatography (5–20% EtOAc in hexanes) yielded compound 3 (330 mg, 85%) as a pale yellow solid. δH (300 MHz, CDCl3): 6.85 (4 H, s), 5.97 (4 H, d, J 7.1 Hz), 5.84 (4 H, m), 4.97 (8 H, m), 4.73 (4 H, t, J 7.9 Hz), 4.37 (4 H, d, J 7.0 Hz), 3.78 (4 H, s), 2.15 (8 H, m), 2.03 (8 H, m), 1.3 (48 H, m); δC (75 MHz, CDCl3): 150.11, 139.09, 138.13, 121.65, 116.10, 114.14, 98.21, 37.13, 33.78, 29.78, 29.65, 29.60, 29.46, 29.10, 28.93, 27.72; m/z (MALDI) 1219 (M + H)+.Tetra(carbomethoxy)cavitand tetraene 8
Tetrabromocavitand 7 (1.093 g, 0.78 mmol) was dried as previously described without the solid addition tube, dissolved in anhydrous THF (35 mL) and cooled to −78 °C under an Ar atmosphere, then treated with n-BuLi (2.3 mL of a 2.8 M solution in hexanes). The reaction mixture was stirred for 10 min, then treated with freshly distilled methyl chloroformate (2.5 mL, 30.6 mmol) and allowed to warm over a 12 h period to rt. The reaction mixture was quenched with aqueous NH4Cl (10 mL), extracted with ethyl acetate, washed with water and brine, and dried over MgSO4. Purification by silica gel chromatography (0–60% EtOAc in hexanes) yielded compound 8 (769 mg, 75%) as a white solid. δH (300 MHz, CDCl3): 7.16 (4 H, s), 5.8 (4 H, m), 5.66 (4 H, d, J 7.5 Hz), 5.00 (8 H, m), 4.77 (4 H, t, J 8.1 Hz), 4.58 (4 H, d, J 7.5 Hz), 2.2 (8 H, m), 2.05 (8 H, q, J 6.6 Hz), 1.3 (48 H, m); δC (75 MHz, CDCl3): 165.48, 151.31, 139.00, 138.16, 123.41, 121.46, 114.08, 99.54, 52.55, 36.08, 33.71, 29.68, 29.55, 29.51, 29.37, 29.03, 28.85, 27.61.Tetra(hydroxymethyl)cavitand tetraene 9
LiAlH4 (440 mg, 11.6 mmol) was suspended in anhydrous THF (27 mL) in an oven-dried flask, then cooled to −5 °C under an Ar atmosphere. Tetraester 8 (1.02 g, 0.77 mmol) was dissolved in anhydrous THF (10 mL) and transferred by cannula to the reaction flask, and the reaction mixture was stirred for 5 h at 0 °C. The reaction mixture was quenched by adding water (30 mL) followed by 5 M NaOH (30 mL), and stirred for 15 min at 0 °C then 20 min at rt. The reaction mixture was brought to pH 6 with 5% HCl and stirred overnight at rt, then extracted with EtOAc (3 × 75 mL); the aqueous phase was then further treated with 5% Na,K-tartrate solution, then extracted again with EtOAc. The combined organic extracts were washed with brine and dried over MgSO4. Purification by silica gel chromatography (5–40% acetone in hexanes) yielded compound 9 (900 mg, 97%) as a colorless oil. δH (300 MHz, CDCl3): 7.12 (4 H, s), 5.92 (4 H, d, J 7.2 Hz), 5.8 (4 H, m), 4.97 (8 H, m), 4.80 (4 H, t, J 8.1 Hz), 4.55 (8 H, s), 4.43 (4 H, d, J 7.2 Hz), 2.2 (8 H, m), 2.05 (8 H, q, J 6.9 Hz), 1.3 (48 H, m).Tetrabenzylthiolcavitand tetraene 4
Triphenylphosphine (451 mg, 1.72 mmol) was dissolved in THF (10 mL), cooled to 0 °C, treated with N-chlorosuccinimide (239 mg, 1.79 mmol), and stirred for 10 min under an inert atmosphere. Tetrabenzylalcohol 9 (108 mg, 0.09 mmol) was added to this reaction mixture, which was stirred for 12 h at rt. The reaction mixture was quenched with ethanol (4 mL), then concentrated under reduced pressure. Purification by silica gel column chromatography (0–15% ethyl acetate–hexanes) afforded the corresponding tetrabenzylchloride in quantitative yield. δH (300 MHz, CDCl3): 7.17 (4 H, s), 6.00 (4 H, d, J 7.2 Hz), 5.8 (4 H, m), 4.96 (8 H, m), 4.80 (4 H, t, J 7.8 Hz), 4.54 (12 H, m), 2.2 (8 H, m), 2.05 (8 H, q, J 6.9 Hz), 1.3 (48 H, m).The purified tetrabenzylchloride was dissolved in degassed DMSO (8 mL), then treated with thiourea (50 mg, 0.66 mmol) and stirred for 12 h at rt. The reaction mixture was treated with 1 N NaOH (30 mL) and stirred for 1 h, then treated with HCl until the reaction mixture reached pH 4. The reaction mixture was extracted with CH2Cl2 and the organic phase was washed with water, then dried over MgSO4. Purification by silica gel chromatography (0–20% EtOAc in hexanes) yielded compound 4 (84.5 mg, 75%) as an off-white solid. Note: this compound is susceptible to polymerization. δH (300 MHz, CDCl3): 7.06 (4 H, s), 5.97 (4 H, d, J 7.2 Hz), 5.8 (4 H, m), 5.00 (8 H, m), 4.76 (4 H, t, J 7.8 Hz), 4.47 (4 H, d, J 6.9 Hz), 3.59 (8 H, d, J 7.2 Hz), 2.2 (8 H, m), 2.05 (8 H, q, J 6.9 Hz), 1.90 (4 H, t, J 7.2 Hz), 1.3 (48 H, m); δC (75 MHz, CDCl3): 152.81, 139.13, 138.09, 127.16, 119.24, 114.15, 99.82, 36.90, 33.80, 30.13, 29.76, 29.64, 29.45, 29.13, 28.95, 27.86, 18.12.
Nanoparticle encapsulation and crosslinking studies
Protocols for the conditioning and extraction of gold nanoparticles (d ∼ 20 nm, O.D. ∼ 2) are essentially identical to those reported previously.9 All glassware involved in the handling of resorcinarene-encapsulated nanoparticles was silanized (Siliclad, Gel-est) to minimize loss by nonspecific adsorption. For efficient nanoparticle extraction, the best results could be obtained by treatment of the colloidal gold suspensions with a mixed-bed ion-exchange resin (Amberlite MB-3, Mallinckrodt), which was washed and soaked with deionized water at least 3 times (10 minutes each) prior to its addition. Aqueous nanoparticle suspensions were mixed with an equal portion of 3 or 4 in THF (1–1.5 mM solution), then extracted with two or more aliquots of toluene. In the case of 3, the encapsulated gold colloid also contained 10 µM TOAB for effective phase transfer, followed by vigorous agitation using a vortex mixer. The gold nanoparticle extractions were dried for 1 h over prewashed 4Å molecular sieves. THF was removed by two cycles of azeotropic distillation with toluene using rotary evaporation, and the nanoparticle suspension was concentrated until a final optical density between 0.5 and 1.0 was achieved.G-1 catalyst was obtained from Strem and used without further purification. For the RCM studies, resorcinarene tetraenes 3, 4, and 7 were dissolved as 0.7 mM solutions in CDCl3 and treated with 30 mol% G-1 catalyst, then monitored by 1H NMR spectroscopy at rt. For the crosslinking of resorcinarene-encapsulated nanoparticles, a typical reaction involved treating the suspensions with 30 mol% of G-1 catalyst (approximately 0.1 mM) for 5 min at rt with occasional agitation, then quenching with 100 µL ethyl vinyl ether. The dispersion was concentrated to approximately 0.3 mL, then diluted with 8 mL of MeOH to induce precipitation. The nanoparticles were centrifuged at 11000 rpm (24000 g) for 20 min, then redispersed in fresh solvent by briefly immersing the sample in an ultrasonic cleaning bath. Dispersion stability was monitored by optical absorption spectroscopy, using a Cary 300 Bio spectrophotometer with a spectral resolution of 1 nm. TEM analysis was carried out using a Philips CM-10 or a Philips EM-400 operating at 80 keV. TEM samples were prepared on 200 mesh Cu grids overlaid with carbon-coated Formvar, which were dipped briefly (15 s) into gold nanoparticle dispersions, then immediately blotted with an absorptive tissue and allowed to dry overnight in air.
Epoxidation of crosslinked resorcinarene shells, followed by ring opening
Toluene dispersions (1 mL) of gold nanoparticles (d = 17 nm) encapsulated in crosslinked 4 were treated with DMDO (0.1 M in acetone, 2 mL) and stirred for 12 h at rt. MeOH was added to induce precipitation, and the nanoparticles were recovered by centrifugation (24000 g, 20 min) and redispersion in toluene (3 mL) after mild sonication. The epoxidized nanoparticles were diluted with an equal portion of dry Et2O, then treated with MgBr2·Et2O (20 mg) and agitated for 20 min with a vortex mixer under an Ar atmosphere (a pink precipitate formed within minutes). The reaction was quenched with 2 mL of aqueous NH4Cl solution; the organic phase was separated, and the nanoparticles were recovered by centrifugation of the aqueous layer (24000 g, 20 min). The precipitate could be easily dispersed in MeOH after mild sonication.Dihydroxylation of crosslinked resorcinarene shells, followed by acetylation
Toluene (1.5 mL) dispersions of gold nanoparticles (d = 20 nm) encapsulated in crosslinked 4 were treated with OsO4 (10 µL of a 5 mM solution in toluene) and NMO (14.5 mg) in acetone (1.5 mL). The reaction mixture was stirred for 12 h at rt; the functionalized nanoparticles were recovered by 2 cycles of centrifugation (24000 g, 20 min) and redispersion in MeOH. Alternatively, the polyhydroxylated nanoparticles were azeotroped twice with toluene, dispersed in pyridine (0.8 mL), then treated with Ac2O (0.5 mL) and catalytic DMAP (0.1 mg) and stirred for 12 h at rt. The reaction mixture was quenched by addition of ethanol and concentrated. The polyacetylated nanoparticles were recovered by redispersion in MeOH, followed by 2 cycles of centrifugation (24000 g, 20 min) and redispersion in toluene. Infrared spectra were collected from KBr pellets containing dried nanoparticles, using a Perkin-Elmer Spectrum 2000 FT-IR spectrometer. Selected IR frequencies of polyhydroxylated resorcinarene shells (cm−1, see ESI†): 3446, 2928, 2854, 1458; selected IR frequencies of partially acetylated resorcinarene shells: 3435, 2923, 2851, 1739, 1648; 1468.Conclusion
Tetrathiolcavitand tetraenes 3 and 4 can extract colloidal gold nanoparticles from aqueous solutions into organic solvents, then be crosslinked by olefin metathesis into nondesorptive monolayers around the nanoparticle surface. Nanoparticles in crosslinked resorcinarene shells form stable dispersions and are highly resistant to alkanethiol-induced precipitation and other forms of chemical degradation. Although tetraarylthiol 3 appears to be more compatible with the metathesis catalyst than tetrabenzylthiol 4, the latter can still be efficiently crosslinked on nanoparticle surfaces, and in fact provides superior resistance to chemically induced flocculation. Partial degradation upon oxidation indicates that the crosslinking conditions used in this study does not always result in a fully continuous shell, leaving room for further improvement. Lastly, the crosslinked resorcinarene monolayers are amenable to epoxidation or dihydroxylation reactions, providing new opportunities for synthetic modification of nanoparticle surfaces and their applications toward ligand-directed assembly, bioconjugate recognition, and as multifunctional platforms for chemical sensing and catalysis.Acknowledgements
We gratefully acknowledge financial support from the National Science Foundation (BES-0228143, CHE-0243496), the National Institutes of Health (GM-06982-01), and a postdoctoral fellowship from the Korea Science and Engineering Foundation (Y.-G. K.).References
- Recent reviews: (a) M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS; (b) A. C. Grimsdale and K. Muellen, Angew. Chem., Int. Ed., 2005, 44, 5592 CrossRef CAS; (c) C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS; (d) N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS; (e) A. Fu, W. Gu, C. Larabell and A. P. Alivisatos, Curr. Opin. Neurobiol., 2005, 15, 568 CrossRef CAS; (f) E. Roduner, Chem. Soc. Rev., 2006, 35, 583 RSC.
- (a) A. C. Templeton, M. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27 CrossRef CAS; (b) J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- C. S. Weisbecker, M. V. Merritt and G. M. Whitesides, Langmuir, 1996, 12, 3763 CrossRef CAS.
- (a) J. Yguerabide and E. E. Yguerabide, Anal. Biochem., 1998, 262, 157 CrossRef CAS; (b) A. Wei, in Plasmonic Nanomaterials: Enhanced Optical Properties From Metal Nanoparticles and their Ensembles, ed. V. M. Rotello, Kluwer Academic, New York, 2004 Search PubMed.
- (a) S. A. Maier, P. G. Kik and H. A. Atwater, Appl. Phys. Lett., 2002, 81, 1714 CrossRef CAS; (b) Y. Kang, K. J. Erickson and T. A. Taton, J. Am. Chem. Soc., 2005, 127, 13800 CrossRef CAS.
- A. Wei, Chem. Commun., 2006, 1581 RSC.
- (a) K. B. Stavens, S. V. Pusztay, S. Zou, R. P. Andres and A. Wei, Langmuir, 1999, 15, 8337 CrossRef CAS; (b) A. Wei, B. Kim, S. V. Pusztay, S. L. Tripp and R. Balasubramanian, J. Inclusion Phenom. Macrocycl. Chem., 2001, 41, 83 Search PubMed.
- (a) R. Balasubramanian, J. Xu, B. Kim, B. Sadtler and A. Wei, J. Dispersion Sci. Technol., 2001, 22, 485 CrossRef CAS; (b) R. Balasubramanian, B. Kim, S. L. Tripp, X. Wang, M. Lieberman and A. Wei, Langmuir, 2002, 18, 3676 CrossRef CAS.
- T. K. Misra, T. S. Chen and C. Y. Liu, J. Colloid Interface Sci., 2006, 297, 584 CrossRef CAS.
- (a) B. Kim, S. L. Tripp and A. Wei, J. Am. Chem. Soc., 2001, 123, 7955 CrossRef CAS; (b) A. Wei, B. Kim, B. Sadtler and S. L. Tripp, ChemPhysChem, 2001, 2, 743 CrossRef CAS; (c) B. Kim, M. A. Carignano, S. L. Tripp and A. Wei, Langmuir, 2004, 20, 9360 CrossRef CAS; (d) B. Kim, R. Balasubramanian, W. Pérez-Segarra, A. Wei, B. Decker and J. Mattay, Supramol. Chem., 2005, 17, 173 CrossRef CAS.
- Y. Zhao, W. Pérez-Segarra, Q. Shi and A. Wei, J. Am. Chem. Soc., 2005, 127, 7328 CrossRef CAS.
- (a) J. M. Wessels, H.-G. Nothofer, W. E. Ford, F. von Wrochem, F. Scholz, T. Vossmeyer, A. Schroedter, H. Weller and A. Yasuda, J. Am. Chem. Soc., 2004, 126, 3349 CrossRef CAS; (b) M. S. Vickers, J. Cookson, P. D. Beer, P. T. Bishop and B. Thiebaut, J. Mater. Chem., 2006, 16, 209 RSC; (c) P. Morf, F. Raimondi, H.-G. Nothofer, B. Schnyder, A. Yasuda, J. M. Wessels and T. A. Jung, Langmuir, 2006, 22, 658 CrossRef CAS; (d) T. B. Huff, M. H. Hansen, Y. Zhao, J.-X. Cheng and A. Wei, submitted.
- (a) V. Bütun, A. B. Lowe, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1999, 121, 4288 CrossRef; (b) K. Tajima and T. Aida, Chem. Commun., 2000, 2399 RSC; (c) A. Mueller and D. F. O’Brien, Chem. Rev., 2002, 102, 727 CrossRef CAS; (d) T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS; (e) W. Jin, T. Fukushima, A. Kosaka, M. Niki, N. Ishii and T. Aida, J. Am. Chem. Soc., 2005, 127, 8284 CrossRef CAS.
- (a) K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239 CrossRef; (b) J. Ding and G. Liu, Macromolecules, 1998, 31, 6554 CrossRef CAS; (c) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684 CrossRef CAS; (d) M. J. Joralemon, R. K. O’Reilly, C. J. Hawker and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 16892 CrossRef CAS; (e) K. L. Wooley and C. J. Hawker, Top. Curr. Chem., 2005, 245, 287 CAS.
- Y. Kang and T. A. Taton, J. Am. Chem. Soc., 2003, 125, 5650 CrossRef CAS.
- S. V. Pusztay, A. Wei, K. B. Stavens and R. P. Andres, Supramol. Chem., 2002, 14, 291 CrossRef CAS.
- (a) C. D. Jones, M. J. Serpe, L. Schroeder and L. A. Lyon, J. Am. Chem. Soc., 2003, 125, 5292 CrossRef CAS; (b) B.-S. Kim, J.-M. Qiu, J.-P. Wang and T. A. Taton, Nano Lett., 2005, 5, 1987 CrossRef CAS; (c) Y. Kang and T. A. Taton, Angew. Chem., Int. Ed., 2005, 44, 409 CrossRef CAS.
- (a) H. Huang, E. E. Remsen, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1999, 121, 3805 CrossRef; (b) S. M. Marinakos, J. P. Novak, L. C. Brousseau, A. B. House, E. M. Edeki, J. C. Feldhaus and D. L. Feldheim, J. Am. Chem. Soc., 1999, 121, 8518 CrossRef CAS.
- M. L. Wu, S. A. O'Neill, L. C. Brousseau, W. P. McConnell, D. A. Shultz, R. J. Linderman and D. L. Feldheim, Chem. Commun., 2000, 775 RSC.
- (a) M. S. Wendland and S. C. Zimmerman, J. Am. Chem. Soc., 1999, 121, 1389 CrossRef CAS; (b) N. G. Lemcoff, T. A. Spurlin, A. A. Gewirth, S. C. Zimmerman, J. B. Beil, S. L. Elmer and H. G. Vandeveer, J. Am. Chem. Soc., 2004, 126, 11420 CrossRef CAS; (c) J. B. Beil, N. G. Lemcoff and S. C. Zimmerman, J. Am. Chem. Soc., 2004, 126, 13576 CrossRef CAS.
- L. Sun, R. M. Crooks and V. Chechik, Chem. Commun., 2001, 259 RSC.
- (a) K. J. Watson, J. Zhu, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 1999, 121, 462 CrossRef CAS; (b) H. Skaff, M. F. Ilker, E. B. Coughlin and T. Emrick, J. Am. Chem. Soc., 2002, 124, 5729 CrossRef CAS.
- E. U. T. van Velzen, J. F. J. Engbersen and D. N. Reinhoudt, Synthesis, 1995, 989 CrossRef.
- B. C. Gibb, A. R. Mezo, A. S. Causton, J. R. Fraser, F. C. S. Tsai and J. C. Sherman, Tetrahedron, 1995, 51, 8719 CrossRef CAS.
- J. R. Moran, S. Karbach and D. J. Cram, J. Am. Chem. Soc., 1982, 104, 5826 CrossRef CAS.
- (a) J. A. Smulik, A. J. Giessert and S. T. Diver, Tetrahedron Lett., 2002, 43, 209 CrossRef CAS; (b) G. Spagnol, M.-P. Heck, S. P. Nolan and C. Mioskowski, Org. Lett., 2002, 4, 1767 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of resorcinarenes 3–9; IR spectra of polyhydroxylated and partially acetylated crosslinked resorcinarene shells. See DOI: 10.1039/b614295h |
| This journal is © The Royal Society of Chemistry 2007 |