DOI:
10.1039/B610111A
(Paper)
J. Mater. Chem., 2007,
17, 167-173
Highly efficient white-electrophosphorescent devices based on polyfluorene copolymers containing charge-transporting pendent units†
Received
14th July 2006
, Accepted 26th September 2006
First published on 9th October 2006
Abstract
We have developed an efficient green-emitting polymer (FTO-BT5) through the incorporation of low-bandgap 2,1,3-benzothiadiazole (BT) moieties into the backbone of a blue-light-emitting polyfluorene copolymer (PF-TPA-OXD), which contains hole-transporting triphenylamine (TPA) and electron-transporting oxadiazole (OXD) pendent groups. A polymer light-emitting device based on this BT-containing copolymer exhibited very bright green emission having Commission Internationale d'Énclairage (CIE) chromaticity coordinates (0.32, 0.61); the maximum brightness was 10232 cd m−2 and the maximum luminance efficiency was 8.9 cd A−1. In addition, a highly efficient three-band white-light-emitting diode was prepared using PF-TPA-OXD as a blue-light-emitting host doped with this green-light-emitting FTO-BT5 copolymer and a red-light-emitting Os phosphor Os(fppz). Without co-doping any other electron- and/or hole-transporting molecules, this white-light-emitting device reached a maximum external quantum efficiency of 4.1% (8.3 cd A−1) at a luminance of 402 cd m−2 and a current density of 4.8 mA cm−2. When the luminance increased from 26 cd m−2 (0.36, 0.32) to 5258 cd m−2 (0.33, 0.31), the corresponding CIE chromaticity coordinates remain almost unaltered and very close to the emission of pure white light; the value of ηext remained above 3% in this high-luminance region.
Introduction
Recently, organic light-emitting diodes (OLEDs) have attracted considerable attention because of their potential applications in solid-state lighting and backplane lighting for liquid crystal displays.1 Among these devices, white light OLEDs that are based on semiconductor polymers (PLEDs) are of particular interest because their solution processability is expected to be less expensive than fabrication techniques that require high-vacuum deposition of small molecules. Various approaches toward—and the challenges associated with—realizing white PLEDs have been reported.2 The light from a dye-doped polymer acting as a single emissive layer can be tailored to emit a white color through control of the doping level.1b,2b,d–f,i This single-layer polymer blend approach results in devices having simple structures and, therefore, it is utilized widely to generate white light.
Polymers having large bandgaps can be used either as blue light sources in full-color displays or as host materials for lower-energy fluorescent or phosphorescent dyes. Polyfluorenes (PFs) emit in the blue region and are very promising candidates for light-emitting materials because of their high photoluminescence (PL) and electroluminescence (EL) efficiencies, high thermal stabilities, and ready color-tuning through the introduction of low-bandgap co-monomers.3 In addition, PFs can be used as host materials to generate other colors through energy transfer to lower-energy emitters in blends with other conjugated polymers, fluorescent dyes, and organometallic triplet emitters.4 Consequently, PFs can function as both the host and the blue emitter in white-light-emitting PLEDs.
In this article, we prepared an efficient green-emitting polyfluorene copolymer FTO-BT5 through covalently binding 5 mol% of 2,1,3-benzothiadiazole (BT) units into the backbone of a bipolar charge-transporting polyfluorene derivative, PF-TPA-OXD,5 and the resultant electroluminescent device can reach a high luminance efficiency (LE) of 8.9 cd A−1 at a luminance of 622 cd m−2. Moreover, we realized an efficient and stable white PLED using PF-TPA-OXD as a blue-light-emitting host, doped with this green-light-emitting benzothiadiazole-containing FTO-BT5 copolymer and a red-light-emitting Os phosphor Os(fppz). The three-band white-light-emitting diode exhibited a maximum LE of 8.3 cd A−1 and its emission covered the entire visible range with a full-width at half-maximum (FWHM) of 221 nm.
Experimental
Materials
4,7-Dibromo-2,1,3-benzothiadiazole,6PF-TPA-OXD,5Os(fppz),7 and 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBI)8 were prepared according to reported procedures. The solvents were dried using standard procedures. All other reagents were used as received from commercial sources, unless otherwise stated.
Characterization
1H and 13C NMR spectra were recorded on Varian UNITY INOVA AS500 (500 MHz) and Bruker-DRX 300 (300 MHz) spectrometers. Mass spectra were obtained using a JEOL JMS-HX 110 mass spectrometer. Size exclusion chromatography (SEC) was performed using a Waters chromatography unit interfaced with a Waters 410 differential refractometer; three 5 µm Waters styragel columns (300 × 7.8 mm) were connected in series in order of decreasing pore size (104, 103, and 102
Å); THF was the eluent. Standard polystyrene samples were used for calibration. Differential scanning calorimetry (DSC) was performed using a SEIKO EXSTAR 6000DSC unit at a heating rate of 20 °C min−1 and a cooling rate of 40 °C min−1. Samples were scanned from 30 to 300 °C, cooled to 0 °C, and then scanned again from 30 to 300 °C. The glass transition temperatures (Tg) were determined from the second heating scan. Thermogravimetric analysis (TGA) was undertaken using a DuPont TGA 2950 instrument. The thermal stabilities of the samples were determined under nitrogen atmosphere by measuring their weight loss while heating at a rate of 20 °C min−1. UV-Vis spectra were measured using an HP 8453 diode-array spectrophotometer. PL spectra were obtained from a Hitachi F-4500 luminescence spectrometer.
Fabrication of light-emitting devices
LED devices were fabricated in the structure ITO/poly(styrene sulfonate)-doped poly(3,4-ethylenedioxythiophene) (PEDOT) (35 nm)/polymer emitting layer (80–100 nm)/TPBI (30 nm)/Mg : Ag (100 nm)/Ag (100 nm). The PEDOT was spin coated directly onto the ITO glass and dried at 80 °C for 12 h under vacuum to improve hole injection and the substrate's smoothness. The light-emitting layer was spin coated on top of the PEDOT layer using chlorobenzene as the solvent and then the sample was dried for 3 h at 60 °C under vacuum. Prior to film casting, the polymer solution was filtered through a Teflon filter (0.45 µm). The TPBI layer, which was used as an electron transporting layer that would also block holes and confine excitons, was grown by thermal sublimation in a vacuum of 3 × 10−6 Torr.9 Subsequently, the cathode Mg : Ag (10 : 1, 100 nm) alloy was deposited by co-evaporation onto the TPBI layer; this process was followed by placing an additional layer of Ag (100 nm) onto the alloy as a protection layer. The current–voltage–luminance relationships were measured under ambient conditions using a Keithley 2400 source meter and a Newport 1835C optical meter equipped with an 818ST silicon photodiode.
Preparation of FTO-BT5
Aqueous potassium carbonate (2.0 M, 1.0 mL) and Aliquat 336 (ca. 20 mg) were added to a mixture of monomers 1 (64 mg, 73 µmol), 2 (75 mg, 73 µmol), 3 (4.7 mg, 16 µmol), and 4 (104 mg, 162 µmol) in distilled toluene (2.0 mL). The mixture was degassed and Pd(PPh3)4 (ca. 5 mg) was added in one portion under N2. The solution was then heated at 110 °C for 72 h. The end groups were then capped by heating the mixture under reflux sequentially with benzeneboronic acid (39 mg, 320 µmol) and bromobenzene (50 mg, 320 µmol), each for 12 h. The reaction mixture was cooled to room temperature and precipitated into a mixture of MeOH and H2O [1 : 1 (v/v), 100 mL]. The crude polymer was collected and washed with excess MeOH. This product was dissolved in THF and reprecipitated into MeOH before being washed with acetone for 48 h using a Soxhlet apparatus. Drying under vacuum gave FTO-BT5 (153 mg, 84.8%). 1H NMR (300 MHz, CDCl3): δ 0.71–0.75 (m, 20 H), 0.89 (t, 12 H J = 7.0 Hz, 20 H), 1.06 (br, 40 H), 1.34 (br, 26 H), 1.54 (br, 8 H), 2.03 (br, 8 H), 2.51 (br, 8 H), 6.89–7.16 (m, 24H), 7.50–7.83 (m, 30H), 7.96–8.11 (m, 10H). 13C NMR (75 MHz, CDCl3): δ 14.0, 14.1, 22.4, 22.5, 23.9, 29.1, 30.0, 31.1, 31.7, 33.6, 35.0, 35.1, 40.3, 55.3, 64.7, 65.8, 120.3, 120.8, 121.0, 121.3, 121.5, 121.8, 122.9, 124.6, 126.1, 126.2, 126.7, 127.2, 127.3, 127.6, 128.8, 128.9, 129.1, 137.5, 137.6, 138.5, 139.0, 140.0, 140.2, 140.4, 141.0, 141.8, 145.3, 146.7, 149.2, 150.8, 151.7, 151.9, 152.9, 155.4, 164.0, 164.7. Anal. Calcd: C, 86.86; H, 7.92; N, 3.63. Found: C, 85.57; H, 7.93; N, 3.33%.
Results and discussion
Synthesis and characterization of FTO-BT5
Scheme 1 illustrates the synthetic route used to prepare polyfluorene derivatives possessing bipolar pendent groups. The OXD monomer 1, TPA monomer 2, 4,7-dibromo-2,1,3-benzothiadiazole (3), and diboronate 4 were prepared according to reported procedures.5,6,10 We synthesized the green-emitting copolymer FTO-BT5 by performing a Suzuki coupling reaction between the dibromides 1, 2, and 3 and the diboronate 4 in a mole ratio of 9 : 9 : 2 : 20. The copolymerization was undertaken using Pd(PPh3)4 as the catalyst in a mixture of toluene and aqueous K2CO3 (2.0 M) in the presence of Aliquat 336 as a phase-transfer reagent.11 When polymerization was complete, the end groups of the polymer chain were capped—by heating the mixture under reflux sequentially with phenylboronic acid and bromobenzene. We prepared the blue-light-emitting polymer PF-TPA-OXD through the copolymerization of monomers 1, 2, and 4, as reported previously.5
 |
| | Scheme 1 Synthesis of FTO-BT5 and PF-TPA-OXD copolymers. | |
The polyfluorene copolymer FTO-BT5 is readily soluble in common organic solvents, such as chloroform, THF, toluene, and chlorobenzene. The molecular weights of the polymer were determined by gel permeation chromatography (GPC) using THF as the eluent and calibrating against polystyrene standards. This polymer possesses a weight-average molecular weight (Mw) of ca. 5.8 × 104 g mol−1, with a polydispersity index of 2.3. We investigated the thermal properties of FTO-BT5 through differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). A distinct glass transition temperature (Tg) was observed at 172 °C. This relatively high value of Tg may help to suppress morphological changes from occurring upon exposure to heat—an essential characteristic of polymers intended for use as emissive materials in light emitting applications.12 As revealed by TGA, FTO-BT5 also exhibited good thermal stability: its 5% and 10% weight losses occurred at 373 °C and 409 °C, respectively.
Optical properties
Fig. 1a compares the absorption and PL spectra of FTO-BT5 with that of PF-TPA-OXD in diluted THF solution. FTO-BT5 exhibits absorption spectral features that are similar to those observed of PF-TPA-OXD—two major absorption bands at around 300 and 390 nm, which originate from the absorptions of the charge-transporting pendants (TPA and OXD groups)5 and of the conjugated polyfluorene backbone, respectively. Besides these, the incorporation of 5 mol% of BT units into the PF backbone leads to a weak absorption at ca. 445 nm,6b which overlaps well with the blue emission arising from the polyfluorene segments. This overlap should enable energy transfer to occur from the excited polyfluorene segments to lower-energy sites containing the BT units. Upon photoexcitation at an excitation wavelength of 390 nm, the PL spectrum of FTO-BT5 is quite different from that of PF-TPA-OXD: it shows a PF emission at 420 nm as well as a longer-wavelength emission at 540 nm. We attribute the additional emission band, observed in the green region, to the narrow-bandgap BT units. The effect that the incorporation of BT units into the main chain has on the luminescence properties is more dramatic, however, in thin films. As shown in Fig. 1b, the PF emission is barely observable; instead, the PL spectrum exhibits its emission arising predominantly from the BT units. The lack of PF emission from the FTO-BT5 film indicates that efficient Förster energy transfer is facilitated through both intra- and inter-chain interactions, which result from the shorter distances between the polymer chains in the solid state.
 |
| | Fig. 1 Absorption and PL spectra of FTO-BT5 (solid lines) and PF-TPA-OXD (dashed lines) in dilute THF solutions (a) and in the solid state (b). | |
Electroluminescence device based on FTO-BT5
To investigate the EL characteristics of FTO-BT5, we fabricated devices having the configuration ITO/PEDOT/FTO-BT5/TPBI/Mg : Ag/Ag. Fig. 2 presents the current density–voltage–luminance characteristics of the device employing FTO-BT5 as the emitting layer. After turn-on at ca. 7.5 V (corresponding to 1 cd m−2), the green electroluminescence with maximum emission intensity at 531 nm was observed from this device and a peak brightness of 10232 cd m−2 was obtained at an applied voltage of 19 V. As shown in the inset of Fig. 2, the resulting EL spectra are nearly independent of the driving voltages varied from 9 V to 19 V and quite similar to the PL spectrum of the FTO-BT5 film; these results indicate that both the PL and EL originate from the same radiative decay process of BT excited states. Moreover, the maximum external quantum efficiency (EQE) and luminance efficiency were found to be 2.4% and 8.9 cd A−1 (Fig. 3), respectively, accompanied with a brightness of 622 cd m−2. The performances of the green -emitting device based on FTO-BT5 are summarized in Table 1. We notice that this green-emitting device possesses significantly improved EL efficiency relative to the device based on a copolymer derived from fluorene and BT units.6b We attribute the improved device performances to the more facile charge injection and the more-efficient charge recombination within FTO-BT5 due to the presence of the electron-rich TPA and electron-deficient OXD pendent groups. In addition, the introduction of an electron injecting and transporting TPBI layer, which also functions as the hole blocking and exciton confinement layer, may contribute to the high performance, even though we used a high-work-function alloy of Mg : Ag, rather than Ba, as the cathode.
 |
| | Fig. 2 Current density–voltage–luminance characteristics of ITO/PEDOT/FTO-BT5/TPBI/Mg : Ag. Inset: solid state PL spectra of the FTO-BT5 film and EL spectra of the ITO/PEDOT/FTO-BT5/TPBI/Mg : Ag devices, recorded at various voltages. | |
 |
| | Fig. 3 Plots of the luminance efficiency and external quantum efficiency as a function of the current density for the green-emitting device. | |
Table 1 The device performances of devices with structures ITO/PEDOT/polymer/TPBI/Mg : Ag
| Device |
FTO-BT5
c
|
Double-doped blendc |
|
At 1 cd m−2.
At 20 mA cm−2.
The data in the parentheses were taken at 100 mA cm−2.
At 15 V.
|
| Turn-on voltage/Va |
7.2 |
9.9 |
| Brightness/cd m−2b |
1702 (6539) |
1492 (5783) |
| Luminance efficiency/cd A−1b |
8.5 (6.6) |
7.5 (5.8) |
| External quantum efficiency (%)b |
2.3 (1.8) |
3.7 (2.8) |
| Maximum brightness/cd m−2 |
10232 |
8698 |
| Maximum luminance efficiency/cd A−1 |
8.9 |
8.3 |
| Maximum external quantum efficiency (%) |
2.4 |
4.1 |
| EL peak positions/nmd |
531 |
426, 523, 618 |
| CIE coordinates, x and yd |
0.32 and 0.61 |
0.34 and 0.32 |
White polymer light-emitting diode from a double-doped blend
Previously, we described white-emitting devices that were based on the dispersion of green and red fluorene-derived fluorescent dyes in PF-TPA-OXD. Through a balance between charge injection and transportation within the host polymer, the resulting device displayed a relatively high EQE of 0.82%;2e clearly there remains some room for improvement. One approach to raising the device's efficiency is to incorporate phosphorescent dopants into the blends to harvest luminescence from both singlet and triplet excitons.13 However, due to the relative low triplet state energy of PF (ca. 2.15 eV), doping green phosphor into PF would lead to a reverse triplet-energy transfer from the green phosphor back to the non-phosphorescent PF, which competes with the radiative decay from the triplet state of the green emitter and subsequently reduces the device efficiency.4b,14 Thus, we chose to use the green fluorophore FTO-BT5 and a red phosphor, Os(fppz), as dopants in the PF-TPA-OXD host to realize white light that covered the entire visible region. The osmium complex, which contains both pyridine pyrazolate chelates and phosphine ligands, was selected in this study considering its highly efficient red electrophosphorescence that can be achieved through physical doping of Os(fppz) into PF-TPA-OXD.4f
Fig. 4 displays the EL spectra of the devices fabricated using PF-TPA-OXD, FTO-BT5, or the Os(fppz)-doped PF-TPA-OXD as the emitting layer. The EL spectrum of PF-TPA-OXD exhibits its maximum intensity at 426 nm, and we determined its maximum value of EQE to be 1.2%.3h The device prepared from FTO-BT5 copolymer emits exclusively green light, which is characteristic of the BT unit (vide supra). In the red-emitting device prepared by doping Os(fppz) into PF-TPA-OXD at a concentration of 1.7 wt%, we obtained a saturated red emission—its peak at 620 nm is characteristic of Os(fppz) triplet emission—and the corresponding value of EQE reached as high as 8.37%.4f Because of the high energy level of its highest occupied molecular orbital (HOMO), Os(fppz) serves as an effective trap site for holes in the blend; hence, direct hole trapping on Os(fppz), followed by recombination with electrons, results in the red emission from the triplet state of Os(fppz), and this process was mainly responsible for the EL process.4f From the performances of these devices, it is clear that PF-TPA-OXD is not only an efficient blue emitter but also an ideal host material for lower-energy dopants. As depicted in the inset of Fig. 5, the CIE color coordinates corresponding to the EL spectra of the blue-, green-, and red-emitting devices in Fig. 5 are (0.18, 0.12), (0.32, 0.61), and (0.65, 0.34), respectively. The emission color within the triangle drawn by connecting these three positions in the CIE chromaticity diagram, including the equal energy white point at (0.33, 0.33), can be generated by combining the three emission colors with appropriate control of their relative emission intensities. Therefore, we anticipated that we could achieve white electrophosphorescence from a blend of FTO-BT5 and Os(fppz) as low-energy dopants in PF-TPA-OXD as the host.
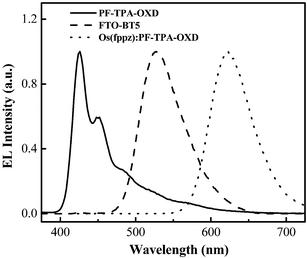 |
| | Fig. 4 EL spectra of devices formed using PF-TPA-OXD, FTO-BT5, or Os(fppz) (1.1 mol% or 1.7 wt%) : PF-TPA-OXD as the emitting layer in the configuration ITO/PEDOT/emitting layer/TPBI/Mg : Ag. | |
Fig. 5 displays the white-light EL spectrum of a blend comprising 4 wt% FTO-BT5 and 0.3 wt% Os(fppz) in PF-TPA-OXD. It is obvious that the EL spectrum of this double-doped device covers the whole visible region with a full-width at half-maximum (FWHM) of 221 nm, which is composed of three distinct emission bands—peak maxima located at 426, 523, and 618 nm—lying within the ranges of the three primary colors. The peak positions of these three individual bands are very close to the positions shown in Fig. 4 for the blue-, green-, and red-emitting devices; accordingly, we conclude that the green and red emissions arose purely from the FTO-BT5 and Os(fppz) components, respectively, and no exciplex emission contributed to the resulting white light emission. Table 1 summarizes the performances of the white-emitting device. The main drawback for display or illumination applications of white-light OLEDs formed using dye-doped polymers as an active layer is typically their color instability. Gratifyingly, we found that the CIE coordinates of our white-light OLED are quite insensitive to the current density and brightness. For instance, when we increased the current density from 0.39 mA cm−2 (26 cd m−2) to 88.2 mA cm−2 (5258 cd m−2), the CIE color coordinates underwent only a minor shift from (0.36, 0.32) to (0.33, 0.31); these values remain well within the white-light region and very close to those of ideal white light (0.33, 0.33). One possible reason for the instability in the CIE color coordinates has been identified as phase separation within the blend.2c,g,13d In our case, the chemical structures of the FTO-BT5 copolymer and the host are very similar, and the amount of the Os(fppz) phosphor required to achieve white light was only 0.3 wt%; thus, phase separation or dye aggregation is efficiently suppressed in such a blend. Furthermore, the polar moieties of the host may also help to provide a stabilizing environment for the osmium dopant.15
 |
| | Fig. 5 EL spectrum of the doubly doped device formed using PF-TPA-OXD as the host matrix at an applied voltage of 15 V. Inset: Variation in the CIE color coordinates of the doubly doped device as the brightness increased from 26 to 5258 cd m−2; the CIE color coordinates corresponding to the EL spectra in Fig. 4 are marked with crosses. | |
To better understand the operating mechanism behind this emission of white light, we fabricated a control device that had the same device geometry, but was doped with only FTO-BT5 (4 wt%) in the PF-TPA-OXD host. In this control device, we observed only blue and green emission bands at 426 and 523 nm, respectively, in the EL spectrum; the corresponding CIE coordinates were (0.23, 0.36). Fig. 6 presents a comparison between the device characteristics of the white-light and control PLEDs. The current density–voltage (I–V) characteristics shifted slightly to higher voltages upon introduction of Os(fppz) into the host matrix (Fig. 6), because the Os complexes serve as effective trapping sites for holes as discussed above. The introduction of the red phosphor Os(fppz) improved the device efficiency. As Fig. 7 indicates, the value of EQE of the doubly doped device reached 4.1% at a current density of 4.84 mA cm−2 and a brightness of 402 cd m−2; in contrast, the value of EQE of the control device was only 1.47% at a current density of 31.0 mA cm−2 and a brightness of 1069 cd m−2. We notice that the EL efficiency obtained from our device using FTO-BT5 as the green-emitting dopant is comparable to those of other white-emitting devices employed green phosphor as a dopant in a polyfluorene host.16 Furthermore, the observed exciton lifetime of Os(fppz) was 0.7 µs, which is considerably shorter than that of Ir(btp)2(acac) (5.8 µs), a commonly used red phosphor.17 This shorter lifetime, which may suppress both TT and PT annihilations,18 leads to an improvement in the device's quantum efficiency, especially at a high current density. This property is an important one for illumination purposes because such sources of white light are usually operated at higher luminance levels (i.e., a higher current density is required in the case of white-light OLEDs). When we increased the luminance of our white-light-emitting device up to the order of 1 × 103 cd m−2 (at ca. 13.5 V), nearly 95% of the maximum value of EQE was maintained; we calculated it to be 3.81% (7.76 cd A−1). When the brightness was increased further to near 5 × 103 cd m−2—the emission remained in the white-light region—and the corresponding EL efficiency remained above 3%. We attribute the high performances of this three-band white-light OLED to the balanced charge injection and transportation provided by the PF-TPA-OXD host and to the direct formation of excitons at the Os(fppz) dopants. Effective exciton confinement within the emitting layer, caused by the hole/exciton blocking (TPBI) layer, may also account for these properties.
 |
| | Fig. 6 Current density–voltage–luminance characteristics for both the white-light-emitting and control devices. | |
 |
| | Fig. 7 Curves of the luminance efficiency and external quantum efficiency as a function of the current density for both the white-light-emitting and control devices. | |
Conclusions
We have synthesized a green-emitting copolymer (FTO-BT5) by incorporating low-bandgap BT moieties into a blue-light-emitting polyfluorene copolymer (PF-TPA-OXD). A highly efficient and color-stable white-electrophosphorescent device based on a single-layer polymer blend was fabricated readily through spin-casting a solution containing PF-TPA-OXD, FTO-BT5 and a red-emitting Os(fppz) complex. In addition to being the host matrix, PF-TPA-OXD plays the role of an emitter: it offers its efficient fluorescence to the blue emission region of the resulting white-light emission; this situation not only reduces the number of dopants required but also simplifies the fabrication process. This approach should be valuable for solid-state lighting applications because of the simple device architecture and the promise of low-cost manufacturability. Without physically codoping any charge-transporting molecules into the host matrix, we obtained a device efficiency of 4.1% (8.3 cd A−1) at 402 cd m−2 for the resulting white light, the spectrum of which covered the entire visible region. When we increased the luminance from 30 to 5 × 103 cd m−2, the corresponding CIE coordinates exhibited only a slight shift—from (0.36, 0.32) to (0.33, 0.31)—and the value of EQE remained above 3%. More recently, white-electrophosphorescent devices based on a single polyfluorene emitter, chemically doped with BT units and red phosphor, have been demonstrated with a maximum LE of 6.1 cd A−1;19 in comparison with them, our polymer-blend-based device shows comparably decent color stability with relatively higher EL efficiency.
Acknowledgements
We thank MOE ATU Program and the National Science Council for financial support. Our special thanks go to Professor C.-H. Cheng for his support and cooperation during the preparation and characterization of the light-emitting devices.
References
-
(a) J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332 CAS;
(b) S. Tasch, E. J. W. List, O. Ekström, W. Graupner, G. Leising, P. Schlichting, U. Rohr, Y. Geerts, U. Scherf and K. Müllen, Appl. Phys. Lett., 1997, 71, 2883 CrossRef CAS.
-
(a) E. J. W. List, G. Leising, N. Schulte, D. A. Schlüter, U. Scherf and W. Graupner, Jpn. J. Appl. Phys., 2000, 39, L760 CrossRef CAS;
(b) R. W. T. Higgins, A. P. Monkman, H.-G. Nothofer and U. Scherf, Appl. Phys. Lett., 2001, 79, 857 CrossRef CAS;
(c) Y.-Z. Lee, X. Chen, M.-C. Chen, S.-A. Chen, J.-H. Hsu and W. Fann, Appl. Phys. Lett., 2001, 79, 308 CrossRef CAS;
(d) B. Hu and F. E. Karasz, J. Appl. Phys., 2003, 93, 1995 CrossRef CAS;
(e) J. H. Kim, P. Herguth, M.-S. Kang, A. K.-Y. Jen, Y.-H. Tseng and C.-F. Shu, Appl. Phys. Lett., 2004, 85, 1116 CrossRef CAS;
(f) Q. Xu, H. M. Duong, F. Wud and Y. Yang, Appl. Phys. Lett., 2004, 85, 3357 CrossRef CAS;
(g) G. Tu, Q. Zhou, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, Appl. Phys. Lett., 2004, 85, 2172 CrossRef CAS;
(h) Y. Xu, J. Peng, Y. Mo, Q. Hou and Y. Cao, Appl. Phys. Lett., 2005, 86, 163502 CrossRef;
(i) P.-I. Shih, Y.-H. Tseng, F.-I. Wu, A. K. Dixit and C.-F. Shu, Adv. Funct. Mater., 2006, 16, 1582 CrossRef CAS;
(j) P.-I. Shih, C.-F. Shu, Y.-L. Tung and Y. Chi, Appl. Phys. Lett., 2006, 88, 251110 CrossRef.
-
(a) M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867 CrossRef CAS;
(b) D. Neher, Macromol. Rapid Commun., 2001, 22, 1365 CrossRef CAS;
(c) U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS;
(d) S. Becker, C. Ego, A. C. Grimsdale, E. J. W. List, D. Marsitzky, A. Pogantsch, S. Setayesh, G. Leising and K. Müllen, Synth. Met., 2002, 125, 73 CrossRef CAS;
(e) C. D. Müller, A. Falcou, N. Reckefuss, M. Rojahn, V. Wiederhirn, P. Rudati, H. Frohne, O. Nuyken, H. Becker and K. Meerholz, Nature, 2003, 421, 829 CrossRef;
(f) C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. C. Grimsdale, K. Müllen, J. D. MacKenzie, C. Silva and R. H. Friend, J. Am. Chem. Soc., 2003, 125, 437 CrossRef CAS;
(g) R. Yang, R. Tian, J. Yan, Y. Zhang, J. Yang, Q. Hou, W. Yang, C. Zhang and Y. Cao, Macromolecules, 2005, 38, 244 CrossRef CAS;
(h) H.-J. Su, F.-I. Wu, Y.-H. Tseng and C.-F. Shu, Adv. Funct. Mater., 2005, 15, 1209 CrossRef CAS;
(i) Q. Peng, E. T. Kang, K. G. Neoh, D. Xiao and D. Zou, J. Mater. Chem., 2006, 16, 376 RSC.
-
(a) A. Charas, J. Morgado, J. M. G. Martinho, A. Fedorov, L. Alcácer and F. Cacialli, J. Mater. Chem., 2002, 12, 3523 RSC;
(b) F.-C. Chen, G. He and Y. Yang, Appl. Phys. Lett., 2003, 82, 1006 CrossRef CAS;
(c) H.-J. Su, F.-I. Wu and C.-F. Shu, Macromolecules, 2004, 37, 7197 CrossRef CAS;
(d) X. Gong, W. Ma, J. C. Ostrowski, G. C. Bazan, D. Moses and A. J. Heeger, Adv. Mater., 2004, 16, 615 CrossRef CAS;
(e) C. Jiang, W. Yang, J. Peng, S. Xiao and Y. Cao, Adv. Mater., 2004, 16, 537 CrossRef CAS;
(f) F.-I. Wu, P.-I. Shih, Y.-H. Tseng, G.-Y. Chen, C.-H. Chien, C.-F. Shu, Y.-L. Tung, Y. Chi and A. K.-Y. Jen, J. Phys. Chem. B, 2005, 109, 14000 CrossRef CAS.
- C.-F. Shu, R. Dodda, F.-I. Wu, M. S. Liu and A. K.-Y. Jen, Macromolecules, 2003, 36, 6698 CrossRef CAS.
-
(a) J. Huang, Y. Niu, W. Yang, Y. Mo, M. Yuan and Y. Cao, Macromolecules, 2002, 35, 6080 CrossRef CAS;
(b) F. Huang, L. Hou, H. Wu, X. Wang, H. Shen, W. Cao, W. Yang and Y. Cao, J. Am. Chem. Soc., 2004, 126, 9845 CrossRef CAS.
- Y.-L. Tung, P.-C. Wu, C.-S. Liu, Y. Chi, J.-K. Yu, Y.-H. Hu, P.-T. Chou, S.-M. Peng, G.-H. Lee, Y. Tao, A. J. Carty, C.-F. Shu and F.-I. Wu, Organometallics, 2004, 23, 3745 CrossRef CAS.
-
J. Shi, C. W. Tang and C. H. Chen, US Pat. 5,645,948, 1997 Search PubMed.
- S. W. Culligan, Y. Geng, S. H. Chen, K. Klubek, K. M. Vaeth and C. W. Tang, Adv. Mater., 2003, 15, 1176 CrossRef CAS.
- F.-I. Wu, D. S. Reddy, C.-F. Shu, M. S. Liu and A. K.-Y. Jen, Chem. Mater., 2003, 15, 269 CrossRef CAS.
-
(a) R. Yang, R. Tian, Y. Yang, Q. Hou and Y. Cao, Macromolecules, 2003, 36, 7453 CrossRef CAS;
(b) J. Yang, C. Jiang, Y. Zhang, R. Yang, W. Yang, Q. Hou and Y. Cao, Macromolecules, 2004, 37, 1211 CrossRef CAS.
- S. Tokito, H. Tanaka, K. Noda, A. Okada and Y. Taga, Appl. Phys. Lett., 1997, 70, 1929 CrossRef CAS.
-
(a) Y. Kawamura, S. Yanagida and S. R. Forrest, J. Appl. Phys., 2002, 92, 87 CrossRef CAS;
(b) I. Tanaka, M. Suzuki and S. Tokito, Jpn. J. Appl. Phys., 2003, 42, 2737 CrossRef;
(c) M. Suzuki, T. Hatakeyama, S. Tokito and F. Sato, IEEE J. Sel. Top. Quantum Electron., 2004, 10, 115 CrossRef CAS;
(d) H. A. Al
Attar, A. P. Monkman, M. Tavasli, S. Bettington and M. R. Bryce, Appl. Phys. Lett., 2005, 86, 121101 CrossRef.
- F.-C. Chen, S. C. Chang, G. He, S. Pyo, Y. Yang, M. Kurotaki and J. Kido, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2681 CrossRef CAS.
- F.-I. Wu, P.-I. Shih, C.-F. Shu, Y.-L. Tung and Y. Chi, Macromolecules, 2005, 38, 9028 CrossRef CAS.
-
(a) Y. Xu, J. Peng, J. Jiang, W. Xu, W. Yang and Y. Cao, Appl. Phys. Lett., 2005, 87, 193502 CrossRef;
(b) T.-H. Kim, H. K. Lee, O. Park O, B. D. Chin, S.-H. Lee and J. K. Kim, Adv. Funct. Mater., 2006, 16, 611 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS.
-
(a) F.-C. Chen, Y. Yang, M. E. Thompson and J. Kido, Appl. Phys. Lett., 2002, 80, 2308 CrossRef CAS;
(b) M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B, 2000, 62, 10967 CrossRef CAS.
- J. Jiang, Y. Xu, W. Yang, R. Guan, Z. Liu, H. Zhen and Y. Cao, Adv. Mater., 2006, 18, 1769 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: characterization data of PF-TPA-OXD and Os(fppz). See DOI: 10.1039/b610111a |
|
| This journal is © The Royal Society of Chemistry 2007 |
Click here to see how this site uses Cookies. View our privacy policy here. 