Atom economic synthesis of amides via transition metal catalyzed rearrangement of oxaziridines†
Chin Hin
Leung
a,
Adelina M.
Voutchkova
a,
Robert H.
Crabtree
*a,
David
Balcells
b and
Odile
Eisenstein
*b
aDepartment of Chemistry, Yale University, 225 Prospect Street, New Haven, CT 06520-8017, USA. E-mail: robert.crabtree@yale.edu; Fax: +1 (203) 432 6344; Tel: +1 (203) 432 3925
bInstitut Charles Gerhardt, Université Montpellier 2, CNRS – cc-1501 Place Eugène Bataillon, 34095, Montpellier, France. E-mail: odile.eisenstein@univ-montp2.fr; Fax: +33 467144839; Tel: +33 467143306
First published on 6th June 2007
Abstract
A mild synthetic route to amides involves imine oxidation to an oxaziridine followed by a transition-metal catalyzed rearrangement to the amide . This route shows potential for a greener pathway to amides . DFT studies showed a possible rearrangement pathway in the case of the Rh catalyst.
Introduction
Amide groups appear in a vast range of useful compounds, for example in pharmaceuticals. Traditional methods for synthesis of amides from carboxylic acids and amines have poor atom economy since they necessitate stoichiometric amounts of dehydrating agents such as carbodiimides, which consequently generate stoichiometric amounts of the corresponding urea as byproducts (eqn (1)).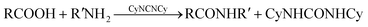 | (1) |
Although acyl halides and esters can also react directly with amines to form amides , their preparation also generates undesirable waste (eqn (2)).
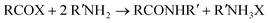 | (2) |
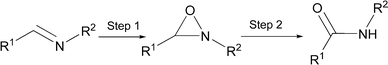
Atom-economic and environmentally more benign synthetic methods are thus highly desirable. One approach involves formation of imine by condensation of aldehyde and amine , oxidation of the imine to the oxaziridine, and subsequent isomerization of the oxaziridine to the amide , catalyzed by transition metal complexes (Scheme 1). This approach was proposed by Duhamal and Plaquevent1 for peptide synthesis but using stoichiometric reagents. Using Mn, W, Ir and Rh catalysts, we now explore the potential of the oxaziridine route for a general synthesis of amides .
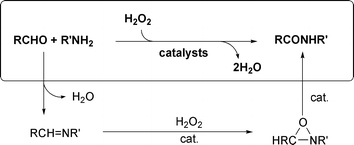 | ||
| Scheme 1 | ||
Oxaziridines were first synthesized independently in the 1950s by Krimm,2 Emmons3 and Horner and Jurgens.4 Their unusual reactivity pattern is a consequence of the strained three-membered ring and of the weak N–O bond. Oxaziridines are well known as both aminating and oxygenating reagents.5
The principal synthetic routes to oxaziridines involve the oxidation of imines with peracids.6 Other methods to oxidize imines include oxone,7O2/CoCl2 (cat.),8H2O2/benzonitrile9 or H2O2/urea10 and H2O2/Na2WO4 (cat.).11 These alternatives to the harsh peracid methods usually generate stoichiometric amounts of undesirable waste. For example, in the O2/Co(II) system, an aldehyde has to be used as coreductant. The atom economic method of Rao and coworkers,11 using the catalyst Na2WO4 and the green oxidant H2O2,12 goes at room temperature in moderate yield.
Rearrangement of oxaziridines to amides usually requires harsh conditions. Treatment of certain oxaziridines with a strong base such as NaH in HMPA13 or LDA14 gives amides , but can also lead to decomposition.15 Both thermal and photochemical isomerization of oxaziridines are known, but nitrones are sometimes formed instead of amides .16
Oxaziridine rearrangements to amides with transition metal complexes are mostly stoichiometric.17 Apart from an early example by Emmons using FeSO4,3 catalytic oxaziridine rearrangements have been found with iron or manganese porphyrin catalysts with high (10–20%) catalyst loadings.18
While direct oxidation of imines to amides is sometimes possible, this usually requires stoichiometric oxidants such as KMnO419 or mCPBA/BF3.OEt2.20
Results and discussion
Catalytic synthesis of oxaziridines
Imines were synthesized from the corresponding amines and aldehydes by direct condensation. In some cases, this even occurred in water, following the prior work by Tashiro et al.21 Otherwise Al2O3, a potentially recyclable reagent, was used as the reaction medium in a solvent-free reaction. Oxaziridines were then prepared from the corresponding imines by the known mild method using Na2WO4 (cat., 2 mol%) and H2O2 in MeCN (eqn (3): Ar = p-NO2C6H4, p-MeOC6H4, p-ClC6H4, R = Cy; Ar = Ph, R = Cy, nBu, tBu, iPr, C2H4Ph).22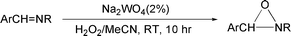 | (3) |
Catalytic rearrangement to amides
The rearrangement of the 2-cyclohexyl-3-phenyloxaziridine (1; Ar = Ph, R = Cy) to the corresponding amide was used for screening catalysts and conditions (Table 1).23Common heterogeneous catalysts (Rh/alumina, Pd/C) showed no activity. The commercially available [Rh(cod)Cl]2 and [Ir(cod)Cl]2 and some simple cationic derivatives all showed good activity (entries 3–6, 9, 10), but yields were lowered if air was not excluded. The catalyst Rh2(octanoate)4 was less active but unaffected by air.| Entry | Catalystb | Loading (mol%) | Solvent | % Yield amide c |
|---|---|---|---|---|
| a Reaction time 12 h in refluxing solvent. b NHC = 1,3-dimethyl-imidiazole-2-ylidene; cod = 1,5-cyclooctadiene; TPPMnCl = tetraphenylporphyrin chloro manganese(III); L = 4′-phenyl-2,2′:6,2″-terpyridine. c Yields based on 1H NMR integrations using 1,3,5-trimethoxybenzene as internal standard. | ||||
| 1 | Pd(OAc)2 | 5 | MeCN | 14 |
| 2 | Pd(PPh3)2Cl2 | 5 | MeCN | 30 |
| 3 | [Rh(cod)Cl]2 | 2.5 | Toluene | 57 |
| 4 | [Rh(PPh3)2(cod)]BF4 | 5 | MeCN | 12 |
| 5 | [Rh(NHC)2(cod)]PF6 | 5 | MeCN | 56 |
| 6 | [Rh(py)2cod] PF6 | 5 | Toluene | 61 |
| 7 | Rh2(octanoate)4 | 2.5 | Toluene | 50 |
| 8 | [IrCp*Cl2]2 | 2.5 | Toluene | 36 |
| 9 | [Ir(cod)Cl]2 | 2.5 | Toluene | 58 |
| 10 | [Ir(py)2cod] PF6 | 5 | Toluene | 58 |
| 11 | TPPMnCl (2) | 5 | MeCN | 83 |
| 12 | [(L)2Mn2(µ-O)2(H2O)2] (ClO4)3 (3) | 2.5 | MeCN | 74 |
| 13 | Pd/C | 5 | Toluene | 0 |
| 14 | Rh/alumina | 5 | Toluene | 0 |
| 15 | MnO2 | 10 | Toluene | 0 |
Manganese complexes were tested based on the previously reported activity.19 Indeed, the two homogeneous manganese species showed the best activities for the rearrangement step (entries 11 and 12), and exclusion of air was not necessary. Manganese dioxide, on the other hand, was inactive.
The Mn catalyzed rearrangement showed some generality (Table 2) but yields of 2-t-butyl-3-phenyloxaziridine (entry 3) and 2-cyclohexyl-3-(p-nitrophenyl)oxaziridine (entry 7) were poor. TPPMnCl (2) did not catalyze the rearrangement of 2-t-butyl-3-phenyloxaziridine at all, but the sterically less hindered Mn dimer (3) achieved a low yield (36%, entry 3).
DFT study of the Rh-catalyzed oxaziridine rearrangement
DFT studies24 have been carried out using Gaussian 0325 for [Rh(cod)Cl]2 where the catalyst is most likely the mononuclear Rh(cod)Cl. Although the Mn catalysts are somewhat more active, the mechanism in these cases has proved to be much harder to tackle, so we defer discussion of this point until the full paper. The substrate was modelled by replacing Cy and Ph by Me. A reasonable reaction mechanism for this catalyst consists of the oxidative addition of the oxaziridine N–O bond to [Rh(cod)Cl] followed by β-H elimination and, finally, reductive elimination. The potential energy surface and a schematic representation of the intermediates and transition states are shown in Fig. 1.![DFT potential energy surface for the Rh-catalyzed rearrangement. Energies are given in kcal mol–1and [Rh] = Rh(cod).](/image/article/2007/GC/b706164a/b706164a-f1.gif) | ||
| Fig. 1 DFT potential energy surface for the Rh-catalyzed rearrangement. Energies are given in kcal mol–1and [Rh] = Rh(cod). | ||
The coordination of the substrate to [Rh(cod)Cl] leads to the square-planar Rh(I) intermediate I1, with the oxaziridine coordinated to Rh via N. This process is exothermic with a ΔE of –27.9 kcal mol–1. Only potential energy (E) needs to be considered because the oxaziridine rearrangement is a unimolecular process.
TS1 is the transition state for oxidative addition of the oxaziridine. At TS1, O is coplanar with the plane defined by Rh, Cl and cod C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond mid-points, and the N–O bond is perpendicular to that plane. TS1 is connected to the intermediate I2, on the reactant side of the reaction, and to I3, on the product side. In I2, the oxaziridine is bound to Rh via O and I2 is less stable than I1 by 14.7 kcal mol–1. I3 is a square pyramidal Rh(III) species with a four-membered metallacycle in which N occupies the apical site. The step I1 → TS1 → I3 has a ΔE of +7.9 kcal mol–1 and a ΔE‡ of +28.3 kcal mol–1, indicating that the oxidative addition is relatively slow and moderately endothermic.
C bond mid-points, and the N–O bond is perpendicular to that plane. TS1 is connected to the intermediate I2, on the reactant side of the reaction, and to I3, on the product side. In I2, the oxaziridine is bound to Rh via O and I2 is less stable than I1 by 14.7 kcal mol–1. I3 is a square pyramidal Rh(III) species with a four-membered metallacycle in which N occupies the apical site. The step I1 → TS1 → I3 has a ΔE of +7.9 kcal mol–1 and a ΔE‡ of +28.3 kcal mol–1, indicating that the oxidative addition is relatively slow and moderately endothermic.
It is remarkable that the N–O cleaving step starts from the O-bound complex I2 and not from the more stable N-bound complex I1. This is consistent with the fact that the N–O bond, with a bondlength of 1.486 Å in the free substrate, shortens to 1.477 Å in I1 and lengthens to 1.500 Å in I2. To cleave the N–O bond, Rh needs to transfer electron density into the σ*NO orbital, while avoiding the repulsive interaction from the heteroatom lone pairs. This situation is only obtained when Rh is coplanar with the three-membered ring, which is best achieved by coordination of Rh to the in-plane sp2 lone pair on oxygen.
The metallacycle I3 undergoes β-H elimination through the transition state TS2 leading to the formation of the hydride I4. The Rh–O bond is weakened on going from C–O to CO and the resulting hydride complex (I4) is octahedral, N and Cl being mutually trans. The exothermic and lower energy barrier β-H elimination, with ΔE = –28.4 kcal mol–1and ΔE‡ = +15.9 kcal mol–1, is more favourable than the endothermic and higher energy barrier oxidative addition. β-H elimination may be assisted by the N lone pair, via hyperconjugation of the lone pair into σ*CH and delocalization of the lone pair into the developing π system of the C–O bond.
At the transition state TS3 for the reductive elimination, the apical N bends over the Rh,O,H plane in order to approach the hydride while the Rh–N and Rh–H bonds elongate. TS3 connects to the square-planar intermediate I5 in which the amide is coordinated to Rh via O. At I5, the Rh–N bond is broken and the new N–H bond forms a hydrogen-bond with the Cl.
The I4 → TS3 → I5 step has a ΔE of –44.6 kcal mol–1 and a ΔE‡ of +21.3 kcal mol–1. These values indicate that the N–H reductive elimination is more exothermic than the β-H elimination but involves a moderately higher energy barrier. N–H reductive elimination has been relatively rarely observed.26–28 However, the energy released by the overall reaction (ΔE = –65.1 kcal mol–1) is a driving force for going over moderately high energy barriers.
The final amide decoordination from I5 releases the product and recycles the catalyst. The bond dissociation energy of the amide (+30.3 kcal mol–1) is slightly larger than that of the oxaziridine maybe because of the H-bond between N–H and Cl. However, the bond dissociation energies of the reactant and product are sufficiently similar to keep the catalyst active.
Conclusions
Our data suggests that an atom economic route to amides from imines may be possible using the oxidation/rearrangement strategy of Scheme 1. This would entail a redesign of the overall synthesis versus the conventional routes. Better catalysts are desirable for both steps, however, because generality is still not optimal. DFT studies could not provide a pathway for the metal-free oxaziridine rearrangement in agreement with the need for a catalyst. In contrast with this, in the case of the Rh-catalyzed reaction, the calculations show that the reaction pathway consisting of substrate coordination, oxidative addition (rate-determining), β-H elimination, reductive elimination and then product decoordination is reasonable.Acknowledgements
R.H.C, C.H.L., and A.V. thank the U.S. DOE and Johnson Matthey Co. for support. O.E. and D.B. thank the CNRS and the Ministère de l'Education Nationale for funding. D.B. thanks SANOFI-Aventis for a post-doctoral fellowship and R.H.C. also thanks them for funding.Notes and references
- J. C. Plaquevent, D. Benard and B. Goument, New J. Chem., 1991, 15, 579–585 Search PubMed and references therein.
- For reviews on early oxaziridine work, see: (a) H. Krimm, Chem. Ber., 1958, 91, 1057–1068 CrossRef CAS; (b) J. F. Dupin, Bull. Soc. Chim. Fr., 1967, 3085–3092 CAS.
- W. D. Emmons, J. Am. Chem. Soc., 1957, 79, 5739–5754 CrossRef CAS.
- L. Horner and E. Jurgens, Chem. Ber., 1957, 90, 2184–2189 CrossRef CAS.
- For select examples, see: (a) P. C. B. Page, C. Limousin and V. L. Murrell, J. Org. Chem., 2002, 67, 7787–7796 CrossRef CAS; (b) L. Bohe, M. Lusinchi and X. Lusinchi, Tetrahedron, 1999, 55, 155–166 CrossRef CAS; (c) Y. Hata and M. Watanabe, J. Am. Chem. Soc., 1979, 101, 6671–6676 CrossRef CAS.
- K. Kloc, E. Kubicz, J. Mlochowski and L. Syper, Synthesis, 1987, 1084–1087 CrossRef CAS.
- D. Mohajer, N. Iranpoor and A. Rezaeifard, Tetrahedron Lett., 2004, 45, 631–634 CrossRef CAS.
- (a) L. Martiny and K. A. Jorgensen, J. Chem. Soc., Perkin Trans. 1, 1995, 699–704 RSC; (b) B. J. Auret, D. R. Boyd and P. B. Coulter, J. Chem. Soc., Chem. Commun., 1984, 463–464 RSC.
- J. Kraiem, Y. Kacem, J. Khiari and B. Ben Hassine, Synth. Commun., 2001, 31, 263–271 CrossRef CAS.
- J. A. Damavandi, B. Karami and M. A. Zolfigol, Synlett, 2002, 933–934 CrossRef CAS.
- M. Shailaja, A. Manjula and B. V. Rao, Synlett, 2005, 1176–1178.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2473 CrossRef CAS.
- G. M. Rubottom, Tetrahedron Lett., 1969, 3887–3889 CrossRef CAS.
- (a) S. E. Dinizo and D. S. Watt, J. Am. Chem. Soc., 1975, 97, 6900–6901 CrossRef CAS.
- (a) M. Newcomb and R. A. Reeder, J. Org. Chem., 1980, 45, 1489–1493 CrossRef CAS; (b) D. R. Boyd, K. M. McCombe and N. D. Sharma, J. Chem. Soc., Perkin Trans. 1, 1986, 867–872 RSC.
- (a) L. H. Sternbach, E. Reeder and B. A. Koechlin, J. Org. Chem., 1962, 27, 4671–4672 CrossRef CAS; (b) D. R. Boyd, P. B. Coulter, W. J. Hamilton, W. B. Jennings and V. E. Wilson, Tetrahedron Lett., 1984, 25, 2287–2288 CrossRef CAS.
- J. Aube, X. Peng, Y. G. Wang and F. Takusagawa, J. Am. Chem. Soc., 1992, 114, 5466–5467 CrossRef CAS and references therein.
- K. Suda, T. Umehara and F. Hino, Chem. Pharm. Bull., 1990, 38, 839–841 CAS.
- J. Larsen, K. A. Jorgensen and D. Christensen, J. Chem. Soc., Perkin Trans. 1, 1991, 1187–1190 RSC.
- S. Y. Kim, G. I. An and H. Rhee, Synlett, 2003, 112–114.
- A. Simion, C. Simion, T. Kanda, S. Nagashima, Y. Mitoma, T. Yamada, K. Mimura and M. Tashiro, J. Chem. Soc., Perkin Trans. 1, 2001, 2071–2078 RSC.
- In slight modification of the published procedure,9 2 mol% of Na2WO4 (instead of 10 mol%) was found to be adequate. Further experimental details are described in the ESI†.
- A mixture of oxaziridine (0.25 mmol) 1,3,5-trimethoxybenzene (10 mg) and metal in solvent (5 ml) was heated at reflux for 12 h. For Pd/Rh/Ir cases, air was excluded. Yields were determined by 1H NMR peak integration 1,3,5-trimethoxybenzene as internal standard.
- DFT calculations were carried out using the hybrid B3PW91 functional. The basis set was the ECP-adapted SDDALL with a set of polarization functions for Rh and Cl and the all-electron 6–31G(d,p) for all other atoms. Geometry optimizations were carried out without any geometrical constraints. The analytical calculation of frequencies was performed in order to classify each stationary point as a minimum or a transition state. Each transition state was relaxed towards reactants and products using the vibrational data to confirm its nature. All energies in the text are potential energies. The zero-point, thermal and entropy corrections were evaluated to compute enthalpies and Gibbs free energies, which are given in the ESI†. It was verified that the free energy profiles and the potential energy profiles are similar. The references for the level of calculations are given in the ESI†.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision D.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- M. Kanzelberger, X. Zhang, T. J. Emge, A. S. Goldman, J. Zhao, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 13644–13645 CrossRef CAS.
- D. M. Roundhill, Chem. Rev., 1992, 92, 1–27 CrossRef CAS.
- D. S. Glueck, L. J. N. Winslow and R. G. Bergman, Organometallics, 1991, 10, 1462–1479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, computational references, optimized structures with E, H and G. See DOI: 10.1039/b706164a |
| This journal is © The Royal Society of Chemistry 2007 |