 Open Access Article
Open Access ArticleCopper(II) cyclam-based complexes for radiopharmaceutical applications: synthesis and structural analysis†‡
Jon D.
Silversides
,
Cheryll C.
Allan
and
Stephen J.
Archibald
*
Department of Chemistry and Institute of Clinical Biosciences, The University of Hull, Cottingham Road, Hull, UK HU6 7RX. E-mail: s.j.archibald@hull.ac.uk; Fax: +44(0)1482 466410; Tel: +44(0)1482 465488
First published on 1st February 2007
Abstract
Two molecular structures of the copper(II) complex, Cu(H2TETA), have been determined by X-ray crystallography. The Jahn–Teller distortion differs between the two structures; occurring either along the axis of the pendant acetate arms or across the macrocyclic ring. An analysis of deposited data from over one hundred copper(II) cyclam X-ray structures in the Cambridge Structural Database (CSD) reveals that Jahn–Teller distortion across the ring is highly unusual for such compounds in the solid state. Novel chelators based on the piperazino/side-bridged cyclam have been prepared and copper(II) complexes formed. The single crystal X-ray structures of two copper(II) complexes, with either an ester or acid N-pendant arm, have been determined and in both cases the pendant arm is bound to the metal centre.
Introduction
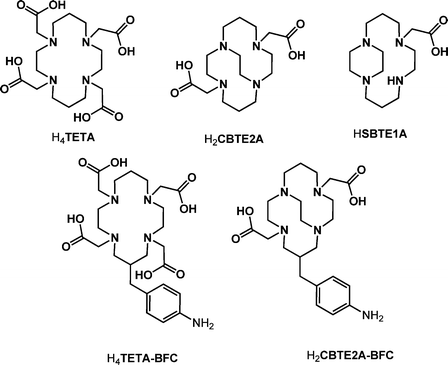
1,4,8,11-Tetraazacyclotetradecane (cyclam)-based macrocycles form complexes of high thermodynamic and kinetic stability with transition metals.1–3 N-functionalised derivatives are selective metal ion chelators and are commonly used as the chelating component (known as a bifunctional chelator or BFC) of targeted radiopharmaceuticals in nuclear medicine.4–6 A bifunctional chelator contains a reactive site for attachment to a targeting group and, ideally, will fully retain the radiolabel under physiological conditions to allow localisation at the target site without transchelation occurring.7,8 We are particularly interested in copper radioisotopes 60Cu, 61Cu, 62Cu and 64Cu, which are positron emitters with half lives varying from 0.16 to 12.7 h that have been investigated for applications in positron emission tomography (PET) imaging.6,9–1167Cu is a longer lived β emitter (t1/2 = 61.9 h) that has been used in targeted radiotherapy.12,13
The 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid (H4TETA) chelator, where cyclam has been functionalised with four N-pendant acetate groups, is one of the most common chelator components in BFCs for copper(II) e.g. H4TETA–BFC.14,15 The addition of pendant carboxylic acid groups further increases stability, forming six-coordinate copper(II) complexes where two of the pendant arms are coordinated to the metal centre. The charge on the resultant complex can vary with deprotonation of the remaining carboxylic acid pendant arms. We report X-ray crystal structures of the neutral copper(II) complex in which only the two coordinating arms are deprotonated (H2TETA2−).
However, despite the relatively high stability of these complexes, in vivo retention of the radiolabel is still not optimum for medical imaging and therapeutic applications. Hence, we are interested in modified chelators based on the cyclam framework that also incorporate acetate arms but may have increased in vivo stability.
Complexes of topologically-constrained aza-macrocycles have improved kinetic stability. Weisman, Wong and Anderson have designed and characterised novel cross-bridged tetraza-macrocyclic chelators (CBTE2A and CBDO2A) and demonstrated increased in vivo stability of the radiolabelled complexes in comparison to TETA.16,17 For 64CuCB-TE2A, initial transchelation in vivo to ceruloplasmin, followed by transfer to Cu–Zn superoxide dismutase, is significantly reduced.17,18 We have recently reported the first BFC based on the CBTE2A framework, H2CBTE2A–BFC.19,20 Other structurally reinforced macrocycles such as the side-bridged cyclam, with an ethylene bridge between adjacent nitrogens, are also of interest.21 In this work, we describe the synthesis and structure of copper(II) complexes of the first side-bridged macrocycle incorporating a coordinating acetate pendant arm.
Results and discussion
Copper TETA structures
For such an important chelator in biomedicine, TETA is relatively underrepresented in the Cambridge Structural Database (CSD). The solid state structure of neutral copper TETA complexes has not been investigated. Our main interest in TETA is to gather structural information about the flexibility of this chelator from the X-ray structural parameters and then to correlate this with the known in vivo kinetic stability issues.The two copper TETA structures, reported by Kaden and co-workers, deposited in the CSD are [CuTETA]2− and Cu2TETA; both have fully deprotonated TETA4− chelators.22–24 The charge of the [CuTETA]2− anion is balanced by barium ions and extensive H-bonding is observed with lattice-incorporated water molecules. Two forms of this compound are known with an identical copper-containing anion but varying numbers of water molecules of crystallisation.§ The Cu2TETA structure is of less interest as neither of the two metal ions is incorporated into the four nitrogen donor macrocyclic cavity. The structure of the neutral Cu(H2TETA) complex has not been determined previously, although Kaden and co-workers report the isolation of this complex. To produce crystals for structural characterisation of the neutral compound, our initial approach was to follow Kaden's synthetic procedure and grow crystals from water at an appropriate acidic pH to ensure protonation of the two unbound carboxylates. Large blue crystals were produced by dissolving the compound in distilled water and adjusting the pH to 2.0 with hydrochloric acid. These crystals were of suitable quality for X-ray diffraction studies and revealed the expected neutral complex, Cu(H2TETA) (A).
An alternative crystallisation technique was attempted by redissolving the isolated blue crystalline compound in acetonitrile. Slow evaporation of the acetonitrile solution resulted in formation of violet crystals that were of sufficient quality for X-ray crystallographic analysis. Once again, a neutral complex of Cu(H2TETA) (B) was present in the unit cell. Some interesting structural differences were observed, with significant variation observed in the copper–ligand bond lengths. Both structures show the expected Jahn–Teller distortion around the d9 copper(II) centre. However, the orientation of the elongated axis varies; for one complex the increased bond lengths are observed across the macrocycle (N–Cu–N axis) and for the other along the O–Cu–O axis of the coordinated pendant arms, see Fig. 1. As expected, the axial elongation also results in compression of the four equatorial bonds. This demonstrates significant flexibility of the cyclam ring in the TETA ligand complex with copper(II).
 | ||
| Fig. 1 Ball and stick representations of the X-ray structures of two Jahn–Teller distorted Cu(H2TETA) structures from this work (A and B) and Weisman and co-workers X-ray structure of CuCBTE2A (C). The elongated axis is indicated by the dashed bonds in all cases. | ||
The Cu(H2TETA) (A) complex crystallised from water at pH 2.0 exhibits a distorted octahedral geometry with two weakly coordinating axial carboxylates and the four macrocyclic nitrogens in the equatorial positions, Fig 2. This structure has both six-membered CuN2C3 chelate rings in the chair conformation and the four nitrogens coordinated in a planar arrangement. There is crystallographic symmetry within the molecule and the copper atom lies on an inversion centre. The Cu–N1 bond lengths are 2.06(1) Å and Cu–N2 bond lengths are 2.16(1) Å, Jahn–Teller distortion is observed through the O–Cu–O axis with bond lengths of 2.27(1) Å, Table 1.
| Cu(H2TETA) (A)1 | Cu(H2TETA) (B)a | [Cu3]CuCl3 | [Cu(HSBTE1A)](ClO4)2 | |
|---|---|---|---|---|
| a Angles given for the symmetric macrocyclic units are equivalent to those with N3 and N4 labels. | ||||
| Cu–N1 | 2.057(1) | 2.002(2) | 1.995(6) | 2.049(8) |
| Cu–N2 | 2.164(1) | 2.378(2) | 2.063(6) | 2.028(5) |
| Cu–N3 | — | — | 1.980(6) | 2.015(8) |
| Cu–N4 | — | — | 2.021(6) | 2.003(7) |
| Cu–O1 | 2.269(1) | 2.020(2) | 2.414(5) | 2.225(5) |
| N1–Cu–N2 | 93.16(5) | 93.31(6) | 102.2(3) | 101.4(3) |
| N1–Cu–N3 | — | — | 163.8(3) | 151.7(3) |
| N1–Cu–N4 | 86.84(5) | 86.69(6) | 74.8(3) | 75.0(4) |
| N2–Cu–N3 | — | — | 87.1(3) | 87.1(3) |
| N2–Cu–N4 | — | — | 163.1(3) | 174.3(4) |
| N3–Cu–N4 | — | — | 100.0(3) | 98.1(4) |
| O1–Cu–N1 | 79.75(4) | 84.68(6) | 107.8(2) | 100.1(3) |
| O1–Cu–N2 | 100.25(4) | 95.32(6) | 77.1(2) | 81.1(2) |
| O1–Cu–N3 | 92.43(4) | 90.45 (6) | 87.1(2) | 108.0(3) |
| O1–Cu–N4 | 87.57(4) | 89.55(6) | 87.9(2) | 95.2(3) |
![ORTEP plots (50% probability ellipsoids) of the single crystal X-ray structures of Cu(H2TETA) (A) and (B) [symmetry transformation used to generate equivalent atoms (A) −x + 1, −y + 1, −z; (B) −x, −y, −z + 1].](/image/article/2007/DT/b615329a/b615329a-f2.gif) | ||
| Fig. 2 ORTEP plots (50% probability ellipsoids) of the single crystal X-ray structures of Cu(H2TETA) (A) and (B) [symmetry transformation used to generate equivalent atoms (A) −x + 1, −y + 1, −z; (B) −x, −y, −z + 1]. | ||
Similarly, the ‘ring-distorted’ compound, Cu(H2TETA) (B), has the two six membered CuN2C3 chelate rings in the chair conformation and the four nitrogens coordinated in a planar arrangement, see Fig. 2. Again, crystallographic symmetry is present within the neutral complex and the copper atom lies on an inversion centre. The Cu–O bond lengths are 2.020(2) Å and Cu–N1 bond lengths 2.002(2) Å. Jahn–Teller distortion is observed through the N2–Cu–N2 axis with bond lengths of 2.378(2) Å. Comparison with the published isomorphous structure of Zn(H2TETA) is of interest as Jahn–Teller distortion will not be observed for a d10 complex.25 The Zn–O1 bond lengths are 2.13(1) Å and the Zn–N1 bond lengths are 2.11(2) Å. However, a smaller elongation is observed in the Zn–N2 bond lengths at 2.25(2) Å, probably due to a mismatch between the cavity size and the radius of the zinc(II) ion.
Although the two samples used in these X-ray studies were crystallised from different solvent systems, only water molecules are observed in the crystal lattice in both structures.
A linked 3D network of hydrogen bonded species involving both the water molecules of crystallisation and the uncoordinated pendant arms is observed for each structure (see Fig. S2 in the ESI‡). Structure (A) has four water molecules and structure (B) two water molecules present for every complete copper macrocyclic unit. Linear and bifurcated H-bonds are present in both structures.
There is a considerable amount of published data for copper(II) cyclam complexes going back over more than 30 years. We believe the distortion of the cyclam macrocycle to accommodate the ring-based Jahn–Teller distortion demonstrates flexibility that explains the increased kinetic lability of such complexes in vivo. Over a hundred X-ray structures are known for copper(II) cyclam-based complexes and an analysis of all the deposited structures in the CSD was carried out using CONQUEST and VISTA to determine how common this phenomenon was for six-coordinate cyclam complexes with copper(II).26–28 A parameter was specified for each bond length and the Cu–N ring bond lengths forming the square plane plotted in histogram Fig. 3. A greater range of bond lengths was observed for the non-ring donors as the nature of the donor atom (i.e. N, S, O, P) varied.‡ The distribution of bond lengths from the 118 X-ray crystal structures in the CSD¶ shows only one other example is known where the elongated axis from the Jahn Teller distortion is across the macrocyclic ring, see Fig. 3.
 | ||
| Fig. 3 Histogram representing the Cu–N cyclam ring bond length data from all six coordinate copper(II) cyclam type X-ray structures deposited in the CSD (for further details, see ESI‡). The frequency values for bond lengths 2.02 to 2.04 Å and 2.04 to 2.06 Å are truncated with actual values of 199 and 132, respectively. | ||
This other ring-distorted complex is Cu(H2TETA–BFC), a neutral complex of the C-functionalised nitrobenzyl derivative,14 giving only two examples out of 120 structures. The energy difference between the two Jahn Teller distorted variations must be relatively low, as both structures are observed. However, the much higher frequency of occurrence of the non-macrocyclic Jahn–Teller distortion suggests it is the more stable arrangement. It may be that N–Cu–N elongation is only observed in structures where crystal packing forces and the optimisation of H-bonded networks compensate for the energy penalty of adopting this arrangement.
The existence of these two arrangements does suggest that the TETA conformation will be dynamic in solution and this flexibility may be the key factor that increases kinetic lability, accounting for the overall inadequate chelating ability of TETA in vivo. It is certainly not valid to assume that the Jahn–Teller distortion is static along the axis between the two acetate arms. Any chelator system reducing this flexibility may consequently increase the kinetic stability of the six- coordinate copper(II) complex in vivo.
The X-ray structure of the configurationally restricted and higher stability CuCBTE2A is known. In this case, the carboxylate oxygens are cis rather than trans due to the topological constraint of the macrocycle giving a folded conformation.29 The axial elongated bonds are present along one of the O–Cu–N axes, Fig. 1, with average values of Cu–O 2.314(2) and Cu–N 2.235(2). The more rigid backbone will prevent the macrocycle flexing and it is highly unlikely a cross macrocycle N–Cu–N Jahn–Teller distortion would be observed for this system.
Side-bridged chelators
Reduced flexibility is one feature of configurational restraint in bridged cyclam chelators. Due to the potential increase in kinetic stability and favourable results already observed for cross-bridged chelators, side-bridged chelators may also be of use in 64Cu radiopharmaceutical applications. We have produced the first example of a side-bridged cyclam chelators with a pendant acetate ‘arm’.The side-bridged cyclam ligand has two adjacent nitrogen atoms linked by an ethylene bridge, forming a piperazine fragment within the macrocyclic ring, Scheme 1.30 The constraint imparted by the bicyclic structure results in restriction of the metal complex configuration solely to the trans type configuration (only the trans-II or trans-IV forms, as described by Bosnich and co-workers, have been observed).31–33 Although interest has been expressed in this chelator class for use in radiopharmaceuticals, it has not been extensively studied.21
 | ||
| Scheme 1 Synthesis of the N-carboxymethyl side-bridged cyclam (a) tert-butyl bromoacetate, CH3CN, RT, 16 h, 93%; (b) NaBH4, EtOH, reflux, 3 h, 93%; (c) HCl, reflux, 6 h, 95% (HCl salt). | ||
The first syntheses of side-bridged cyclam ligands by Wainwright produced symmetrical, rigidified macrocycles with two secondary amine sites.34 The synthetic procedures reported in 1995 by Kolinski enable the mono-functionalisation of these macrocycles to produce asymmetrically-substituted analogues.35 A modification of this strategy has been used to produce a mono-substituted side-bridged cyclam with a tert-butyl ester-protected carboxymethyl arm, 3, Scheme 1. The substitution of tert-butyl bromoacetate by a tetracyclic bisaminal intermediate 1 occurs efficiently to produce the mono-quaternary ammonium salt 2. Reductive ring opening with sodium borohydride produces the ethylene bridged cyclam, in total yield from 1 of 85%. Acidic hydrolysis, using either trifluoroacetic acid (TFA) or hydrochloric acid, of the tert-butyl ester group in 3 gives a carboxymethyl arm in chelator HSBTE1A, analogous to the pendant arms of TETA. It was expected that the carboxymethyl arm would deprotonate and coordinate to a copper(II) centre bound in the macrocyclic cavity. Complexes of 3 and HSBTE1A with copper(II) have been produced by reaction with copper(II) salts at reflux in methanol. Single crystal X-ray structures were determined and show that the pendant arm is coordinated through an oxygen atom in both cases.
Complexes with the ester pendant ligand, 3, were prepared with copper(II) chloride or perchlorate. Single crystals of the sample prepared with the chloride counter anion were grown by diffusion of diethyl ether into a methanolic solution. On solving the structure, the counter anion present was found to be copper(I) trichloride, [CuCl3]2−. The copper(I) trichloride anion must have been formed during the complexation of compound 3 with copper(II) dichloride. [CuX3]2− (where X = Cl, Br) has previously been observed in several crystal structures, and has been produced from CuX2 during complexation.36–38
The [Cu3]2+ cation has a five coordinate copper centre in a distorted square-based pyramidal geometry (Addison and Reedijk's τ parameter shows no distortion towards trigonal bipyramidal, with a value of 0.01),39Fig. 4(a). The four ring nitrogens form a distorted square plane with Cu–N bond lengths in the range 1.980(6) to 2.063(6) Å. The side-bridged cyclam ring is in the trans-II configuration. There is a longer bond to the ester carbonyl oxygen in the apical position at 2.414(5) Å. The Cu–O bond seems to be longer than expected for a five-coordinate square pyramidal species, for example, a mono-armed methyl ester cyclam complex has a Cu–O distance of 2.285(5) Å.40 The increased Cu–O bond distance in [Cu3]2+ is required to accommodate an additional long range interaction between the copper(II) and a chloride from the copper(I) trichloride anion at 3.048(2) Å. This results in the metal ion moving further into the macrocyclic cavity. This structure contains no solvent or water molecules within the crystal lattice and no H-bonded networks are present. The copper(I) trichloride counterions are packed into channels within the lattice.
![ORTEP plots (50% probability ellipsoids) of the single crystal X-ray structures of (a) [Cu3]CuCl3 and (b) [Cu(HSBTE1A)]2+.](/image/article/2007/DT/b615329a/b615329a-f4.gif) | ||
| Fig. 4 ORTEP plots (50% probability ellipsoids) of the single crystal X-ray structures of (a) [Cu3]CuCl3 and (b) [Cu(HSBTE1A)]2+. | ||
A single crystal sample of [Cu(HSBTE1A)](ClO4)2 was grown by diffusion of diethyl ether into a methanolic solution of [Cu3](ClO4)2 that hydrolysed over three weeks. Analysis of the methanolic mother liquor by mass spectrometry showed complete hydrolysis had occurred. The complex could also be formed by direct reaction with chelator HSBTE1A. The complex cation has a charge of 2+, with the pendant acetic acid arm having unexpectedly retained the proton. The proton on the carboxylate group was located on the difference map and the bond lengths within the carboxylic acid group are consistent with this assignment. Other examples are known of cyclam-type ligands with pendant acetic acid arms that coordinate to copper(II) through the carbonyl oxygen.41–45 IR and UV/vis. studies by Kang and co-workers show coordination of a protonated acetate pendant arm in acetonitrile solution and deprotonation to form a carboxylate complex when dissolved in water.44
[Cu(HSBTE1A)]2+ contains a five-coordinate copper(II) ion bound in a distorted square based pyramidal geometry with a considerable distortion towards trigonal bipyramidal (a τ parameter value of 0.37). The apical bond is between the copper ion and the carbonyl oxygen atom, with a Cu–O bond length of 2.224(5) Å, which is within the expected range for such an interaction and comparable to the value observed for the methyl ester compound.44 The secondary amine N–H forms an H-bond with the perchlorate counterion, which in turn H-bonds to a lattice water molecule forming a 1D H-bonded chain. The observed cyclam ring configuration is again trans-II. Comparison of [Cu(HSBTE1A)]2+ with [Cu3]2+ shows that the additional long-range interaction with [CuCl3]2− forces [Cu3]2+ into a less-distorted square-based pyramidal structure with an unusually elongated Cu–O distance.
Although the side-bridged copper complexes have not been extensively investigated previously, data on six X-ray structures of copper(II) side-bridged cyclams are currently available in the CSD.26 All of these chelators are tetradentate with four N-donors with no intramolecularly-coordinating pendant arm donor atoms.46,47 Fabbrizzi and coworkers report a side-bridged cyclam complex formed with copper(II) perchlorate, which exhibits a distorted square-planar geometry that is attributed to the constraint of the ring geometry.48 Distortion from planarity of the four ring N-donors is also observed in our two structures. Fabbrizzi has already demonstrated that the non-functionalised side-bridged macrocycle does not benefit from a reinforced kinetic macrocyclic effect.48 Further investigations are required to determine whether this is also the case for functionalised ligands with coordinating pendant ‘arms’ and higher denticity.
The other key properties of radiopharmaceutical chelators, in addition to stability, should also be considered. Another important issue is rapid complex formation under mild conditions. Increased rigidity will slow the complex formation kinetics. This means that a compound with optimised stability properties may still be unusable as a radiopharmaceutical component if a high yield of radiolabelling cannot be achieved under suitable conditions and on an appropriate timescale.
The synthetic methodology used to produce the complexes [Cu3]2+ and [Cu(HSBTE1A)]2+ (2 h reflux in MeOH) is not compatible with conditions required for radiolabelling. However, the colour changes observed suggest that the final complex is formed much more rapidly. For example, the addition of copper(II) perchlorate to ligand 3 results in a blue-coloured solution immediately on addition of the metal salt, and within a few minutes of stirring at room temperature this has changed to a purple-blue colour. No further colour change was observed during the course of the reaction, suggesting any intermediate complex that is formed rapidly reacts to give the final product and that complexation is complete long before the two hour reflux. Further investigation of complexation under radiolabelling conditions and in aqueous solution is required.
Conclusions
The neutral copper(II) complex of the TETA ligand was synthesised and two different X-ray crystal structures obtained, showing Jahn–Teller distortion through either across the macrocycle or along the axis where the pendant arms were coordinated bonds. This reveals the flexible nature of the TETA chelator and provides evidence to explain the more rapid in vivo exchange kinetics observed for copper complexes of TETA in comparison to chelators with configurational constraint and reduced flexibility. Chelator flexibility, cavity size match and complexation/decomplexation kinetics are all key factors in determining the utility of these chelators as radiopharmaceutical components.Side-bridged chelators have also been of interest as radiopharmaceutical chelators. We have reported a facile synthesis of the first derivative of this type to incorporate a coordinating acetate pendant arm. The X-ray structures show that the pendant donor atom is coordinated intramolecularly to give five-coordinate copper(II) complexes with the novel pentadentate chelators. Our further investigations of such chelators are underway, including attempts to synthesise side-bridged derivatives with two coordinating pendant arms. Radiolabelling conditions and the stability of the copper complexes of both one- and two-armed derivatives will also be studied.
Experimental
All reagents and solvents were purchased from Sigma-Aldrich, Lancaster or Fisher and were used as supplied unless otherwise stated. CAUTION: Perchlorate salts are potentially explosive when dry. Although no problems were encountered during this study, caution must be exercised when handling such substances.All 1H NMR and proton decoupled 13C NMR spectra were collected on a Jeol JNM-LA400 spectrometer at 400 and 100 MHz respectively, and referenced against residual solvent signals. Mass spectrometry was carried out by electrospray ionisation with the data collected on a Finnigan LCQ spectrometer, or by the EPSRC National Mass Spectrometry Service at the University of Swansea on a Waters ZQ4000 spectrometer. UV/visible spectra were obtained using an HP-Agilent 8453 diode array spectrometer. Elemental analyses were carried out at the University of Hull.
X-Ray crystallography
Diffraction data sets were collected on a Stöe IPDS-II imaging plate diffractometer using Moκα radiation (λ = 0.71073 Å). The temperature of each crystal was kept at 150 K during data collection and controlled using the Oxford Cryosystems Cryostream Cooler.49 All structures were solved using direct methods (SHELXS) and refined against F2 (SHELXL).50,51 H atoms were either located on the difference map or placed in idealised positions and refined using a riding model with C–H = 0.97 Å, N–H = 0.91 Å and Uiso(H) = 1.2 (or 1.5 for some H atoms) times Ueq of the carrier atom. Some of the water protons were located using calc-OH as part of the WinGX package. The WinGX package was used for refinement and production of data tables, and ORTEP-3 used for structure visualisation.52,53 All ORTEP representations show ellipsoids at the 50% probability level. Crystal data are summarized in Table 2.CCDC reference numbers 624741–626744.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b615329a
| Cu(H2TETA) (A) | Cu(H2TETA) (B) | [Cu3]CuCl3 | [Cu(HSBTE1A)](ClO4)2 | |
|---|---|---|---|---|
| Formula | C18H38 N4O12Cu | C18H34 N4O10Cu | C18N4H36O2Cl3Cu2 | C14N4H32O12Cl2Cu |
| M r | 566.06 | 530.03 | 573.94 | 582.88 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P 21/c | P 21/a | C c | P 212121 |
| a/Å | 8.5304(15) | 7.946(2) | 8.3937(16) | 14.0640(15) |
| b/Å | 15.337(3) | 16.644(3) | 27.970(6) | 9.4730(11) |
| c/Å | 9.010(2) | 8.775(2) | 10.273(2) | 17.359(2) |
| β/° | 90.472(18) | 95.49(2) | 102.069(15) | — |
| Volume/Å3 | 1178.8(4) | 1155.2(5) | 2358.5(8) | 2312.7(5) |
| Z | 2 | 2 | 4 | 4 |
| Density (calc.)/Mg m−3 | 1.595 | 1.518 | 1.616 | 1.674 |
| µMoKα/mm−1 | 0.997 | 1.006 | 2.165 | 1.243 |
| T/K | 150 | 150 | 150 | 150 |
| θ range/° | 2.62 to 34.78 | 2.85 to 34.63 | 2.91 to 32.50 | 2.59 to 32.54 |
| Measured reflections | 17146 | 16962 | 23231 | 27974 |
| Unique reflections [Rint] | 5072 [0.0318] | 4900 [0.0644] | 7665 [0.1083] | 8246 [0.1298] |
| Completeness (θ/°) | 99.3% (34.78) | 99.2% (34.63) | 99.9% (32.5) | 99.4% (32.5) |
| Goodness-of-fit on F2 | 1.127 | 0.975 | 0.933 | 1.022 |
| R1, wR2 (I > 2σ(I)) | 0.0380, 0.1065 | 0.0498, 01319 | 0.0681, 0.1662 | 0.1132, 0.2791 |
| R1, wR2 (all data) | 0.0471, 0.1115 | 0.0726, 0.1428 | 0.1278, 0.1972 | 0.1521, 0.3102 |
| Extinction coefficient | 0.030(3) | 0.033(4) | 0.0031(5) | 0.007(3) |
| Largest diff. peak and hole/e Å−3 | 0.791 and −0.791 | 0.957 and −1.212 | 1.566 and −1.187 | 1.194 and −0.680 |
Synthetic procedures
Cyclam was synthesised by the Barefield method.54 Glyoxal-cyclam, (1), was synthesised in accordance with literature procedures.55 H4TETA·6H2O was synthesised following the literature procedure.56 Cu(H2TETA) was synthesised according to the methods of Kaden and co-workers.57Cu(H2TETA)
m/z: 502 [M+(ClO4)]+, 100%; 446 [M+(ClO4)–C(CH3)3]+, 12%; 402, M+, 23%. HRMS: calcd. for C18H36O6N4ClCu: 502.1614; found 502.1620. λmax(MeOH)/nm 583 (ε/dm3 mol−1 cm−1 150). νmax/cm−1 1680, 1607, 1110.
The compound was also synthesised directly from HSBTE1A. HSBTE1A.4HCl.2H2O (128.0 mg, 281 µmol) was dissolved in methanol (10 mL) and a solution of copper(II) perchlorate hexahydrate (104 mg, 281 µmol) in methanol (3 mL) was added dropwise to the stirred solution. This was followed by addition of a 2M aqueous sodium hydroxide solution (0.70 mL, 1.40 mmol). The mixture was heated to reflux for 2 h. After cooling, solids were removed by filtration through Hyflo filter-aid, and solvents were removed from the filtrate to yield a deep blue solid (145 mg, 95%). ES-MS, m/z: 346 [M]+. HRMS: calcd. for C14H28O6N4ClCu: 446.0988; found 446.0986. λmax(MeOH)/nm 624 (ε/dm3 mol−1 cm−1 60). νmax/cm−1 1625, 1145, 1110, 1090.
Acknowledgements
C. C. A. is grateful to the EPSRC for studentship funding. J. D. S. is grateful to the EPSRC and the Wellcome Trust (Grant 069719) for funding. We thank the EPSRC mass spectrometry service. We wish to acknowledge the use of the EPSRC's Chemical Database Service at Daresbury.Notes and references
- M. Meyer, V. Dahaoui-Gindrey, C. Lecomte and R. Guilard, Coord. Chem. Rev., 1998, 180, 1313–1405 CrossRef.
- H. Elias, Coord. Chem. Rev., 1999, 187, 37–73 CrossRef CAS.
- X. Y. Liang and P. J. Sadler, Chem. Soc. Rev., 2004, 33, 246–266 RSC.
- J. R. Morphy, D. Parker, R. Kataky, M. A. W. Eaton, A. T. Millican, R. Alexander, A. Harrison and C. Walker, J. Chem. Soc., Perkin Trans. 2, 1990, 573–585 RSC.
- D. Parker, Chem. Soc. Rev., 1990, 19, 271–291 RSC.
- C. J. Anderson and M. J. Welch, Chem. Rev., 1999, 99, 2219–2234 CrossRef CAS.
- C. F. Meares and T. G. Wensel, Acc. Chem. Res., 1984, 17, 202–209 CrossRef CAS.
- G. R. Mirick, R. T. O'Donnell, S. J. DeNardo, S. Shen, C. F. Meares and G. L. DeNardo, Nucl. Med. Biol., 1999, 26, 841–845 CrossRef CAS.
- D. W. McCarthy, L. A. Bass, P. D. Cutler, R. E. Shefer, R. E. Klinkowstein, P. Herrero, J. S. Lewis, C. S. Cutler, C. J. Anderson and M. J. Welch, Nucl. Med. Biol., 1999, 26, 351–358 CrossRef CAS.
- P. J. Blower, Dalton Trans., 2006, 1705–1711 RSC.
- P. McQuade, K. E. Martin, T. C. Castle, M. J. Went, P. J. Blower, M. J. Welch and J. S. Lewis, Nucl. Med. Biol., 2005, 32, 147–156 CrossRef CAS.
- D. S. Ma, F. Lu, T. Overstreet, D. E. Milenic and M. W. Brechbiel, Nucl. Med. Biol., 2002, 29, 91–105 CrossRef CAS.
- J. S. Lewis, A. Srinivasan, M. A. Schmidt and C. J. Anderson, Nucl. Med. Biol., 1999, 26, 267–273 CrossRef CAS.
- M. K. Moi, M. Yanuck, S. V. Deshpande, H. Hope, S. J. Denardo and C. F. Meares, Inorg. Chem., 1987, 26, 3458–3463 CrossRef CAS.
- C. J. Anderson, F. Dehdashti, P. D. Cutler, S. W. Schwarz, R. Laforest, L. A. Bass, J. S. Lewis and D. W. McCarthy, J. Nucl. Med., 2001, 42, 213–221 CAS.
- X. K. Sun, M. Wuest, G. R. Weisman, E. H. Wong, D. P. Reed, C. A. Boswell, R. Motekaitis, A. E. Martell, M. J. Welch and C. J. Anderson, J. Med. Chem., 2002, 45, 469–477 CrossRef CAS.
- C. A. Boswell, X. K. Sun, W. J. Niu, G. R. Weisman, E. H. Wong, A. L. Rheingold and C. J. Anderson, J. Med. Chem., 2004, 47, 1465–1474 CrossRef CAS.
- L. A. Bass, M. Wang, M. J. Welch and C. J. Anderson, Bioconjugate Chem., 2000, 11, 527–532 CrossRef CAS.
- E. A. Lewis, C. C. Allan, R. W. Boyle and S. J. Archibald, Tetrahedron Lett., 2004, 45, 3059–3062 CrossRef CAS.
- E. A. Lewis, R. W. Boyle and S. J. Archibald, Chem. Commun., 2004, 2212–2213 RSC.
- T. M. Jones-Wilson, K. A. Deal, C. J. Anderson, D. W. McCarthy, Z. Kovacs, R. J. Motekaitis, A. D. Sherry, A. E. Martell and M. J. Welch, Nucl. Med. Biol., 1998, 25, 523–530 CrossRef CAS.
- A. Riesen, M. Zehnder and T. A. Kaden, Helv. Chim. Acta, 1986, 69, 2074–2080 CrossRef CAS.
- A. Riesen, M. Zehnder and T. A. Kaden, Acta Crystallogr., Sect. C, 1988, 44, 1740–1742 CrossRef.
- A. Riesen, M. Zehnder and T. A. Kaden, J. Chem. Soc., Chem. Commun., 1985, 1336–1338 RSC.
- A. Riesen, M. Zehnder and T. A. Kaden, Acta Crystallogr., Sect. C, 1991, 47, 531–533 CrossRef.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746–749 CrossRef.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B, 2002, B58, 389–397 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380–388 CrossRef.
- E. H. Wong, G. R. Weisman, D. C. Hill, D. P. Reed, M. E. Rogers, J. S. Condon, M. A. Fagan, J. C. Calabrese, K. C. Lam, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 2000, 122, 10561–10572 CrossRef CAS.
- R. D. Hancock, G. Pattrick, P. W. Wade and G. D. Hosken, Pure Appl. Chem., 1993, 65, 473–476 CrossRef CAS.
- G. C. Valks, G. McRobbie, E. A. Lewis, T. J. Hubin, T. M. Hunter, P. J. Sadler, C. Pannecouque, E. De Clercq and S. J. Archibald, J. Med. Chem., 2006, 49, 6162–6165 CrossRef CAS.
- A. Khan, J. D. Silversides, L. Madden, J. Greenman and S. J. Archibald, Chem. Commun., 2007, 416–418 RSC.
- B. Bosnich, C. K. Poon and M. L. Tobe, Inorg. Chem., 1965, 4, 1102–1108 CrossRef CAS.
- K. P. Wainwright, Inorg. Chem., 1980, 19, 1396–1398 CrossRef CAS.
- R. A. Kolinski, Pol. J. Chem., 1995, 69, 1039–1045 CAS.
- W. Clegg, J. R. Nicholson, D. Collison and C. D. Garner, Acta Crystallogr., Sect. C, 1988, 44, 453–461 CrossRef.
- R. D. Willett and A. Vij, J. Chem. Crystallogr., 2000, 30, 399–404 CrossRef CAS.
- R. D. Kohn, G. Seifert, Z. D. Pan, M. F. Mahon and G. Kociok-Kohn, Angew. Chem., Int. Ed., 2003, 42, 793–796 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Vanrijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- S. G. Kang, K. S. Ryu, K. Nam and J. Kim, Inorg. Chim. Acta, 2005, 358, 2224–2230 CrossRef CAS.
- D. M. Tonei, D. C. Ware, P. J. Brothers, P. G. Plieger and G. R. Clark, Dalton Trans., 2006, 152–158 RSC.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. J. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Rheingold, Eur. J. Inorg. Chem., 2005, 4829–4833 CrossRef CAS.
- H. Aneetha, Y. H. Lai, S. C. Lin, K. Panneerselvam, T. H. Lu and C. S. Chung, J. Chem. Soc., Dalton Trans., 1999, 2885–2892 RSC.
- S. G. Kang, S. J. Kim, K. Ryu and J. Kim, Inorg. Chim. Acta, 1998, 274, 24–31 CrossRef CAS.
- J. Chapman, G. Ferguson, J. F. Gallagher, M. C. Jennings and D. Parker, J. Chem. Soc., Dalton Trans., 1992, 345–353 RSC.
- L. Siegfried, R. Kowallick and T. A. Kaden, Supramol. Chem., 2001, 13, 357–367 CrossRef CAS.
- R. Kowallick, M. Neuburger, M. Zehnder and T. A. Kaden, Helv. Chim. Acta, 1997, 80, 948–959 CAS.
- M. Boiocchi, M. Bonizzoni, L. Fabbrizzi, F. Foti, M. Licchelli, A. Poggi, A. Taglietti and M. Zema, Chem.–Eur. J., 2004, 10, 3209–3216 CrossRef CAS.
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105–107 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97 Program for Crystal Structure Refinement, ( 1997), Institüt für Anorganische Chemie der Universität: Tammanstrasse 4, D-3400 Gottingen, Germany Search PubMed.
- G. M. Sheldrick, SHELXS-97 Program for Crystal Structure Solution, ( 1997), Institüt für Anorganische Chemie der Universität: Tammanstrasse 4, D-3400 Gottingen, Germany Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- E. K. Barefield, F. Wagner, A. W. Herlinger and A. R. Dahl, Inorg. Synth., 1976, 16, 220 CAS.
- G. R. Weisman, S. C. H. Ho and V. Johnson, Tetrahedron Lett., 1980, 21, 335–338 CrossRef CAS.
- H. Stetter, W. Frank and R. Mertens, Tetrahedron, 1981, 37, 767–772 CrossRef CAS.
- A. Riesen, M. Zehnder and T. A. Kaden, Helv. Chim. Acta, 1986, 69, 2067–2073 CrossRef CAS.
Footnotes |
| † The HTML version of this article has been enhanced with colour images. |
| ‡ Electronic supplementary information (ESI) available: Packing diagrams for the Cu(H2TETA) structures and further details of the compounds used in the structural distortion analysis. See DOI: 10.1039/b615329a |
| § CSD identification codes EFENEI and VUYSOX. |
| ¶ At the time of searching there were 132 hits for six-coordinate copper complexes incorporating the 14-membered tetraaza macrocycle. Of these, 14 were excluded as either duplicates, constrained systems (such as cross-bridged macrocycles or cryptands), or amides. |
| This journal is © The Royal Society of Chemistry 2007 |