‘Magic bullets’ for bone diseases: progress in rational design of bone-seeking medicinal agents
Sufeng Zhanga, Geeti Gangal†a and Hasan Uludağ*ab
a#526, Department of Chemical & Materials Engineering, Faculty of Engineering, University of Alberta, Edmonton, Alberta, Canada T6G 2G6. E-mail: hasan.uludag@ualberta.ca; Fax: +1 (780) 492-2881; Tel: +1 (780) 492-0988
bFaculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada
First published on 14th September 2006
Abstract
An ideal therapeutic agent for bone diseases should act solely on bone tissue with no pharmacological activity at other anatomical sites. Current therapeutic agents, however, do not usually display a preferential affinity to bones and non-specifically distribute throughout the body after administration. Attempts to design bone-specific agents have relied on engineering a desired therapeutic agent with bone-seeking molecules so that the latter delivers the therapeutic agents specifically to bones. In this critical review, we summarize the latest attempts to engineer bone-seeking therapeutic agents based on formulating therapeutic agents with bisphosphonates, a class of compounds with high affinity to biological apatite. We first provide a relevant summary of the structure of bone mineral and bisphosphonates, highlighting the mode of interaction between these two entities. The use of bisphosphonates in the diagnosis of bone diseases is then presented, since this application helps us to understand the bone-carrier properties of bisphosphonates under physiological conditions. A summary of recent attempts to formulate bisphosphonates with traditional therapeutic agents to restrict their activities to bone tissues is then provided, with special emphasis on the structure–function relationships of the engineered compounds. Finally, attempts to use bisphosphonates to deliver macromolecular therapeutics (i.e., proteins) are summarized, based on recent data from the authors' lab. The collective research into bone-seeking medicinal agents is progressively laying the foundation for next-generation ‘magic bullets’ that display desirable activities at the disease sites with no undesirable activity on other organ systems. (164 references.)
 Sufeng Zhang | Sufeng Zhang, born in Pingdingshan (Henan, China) in 1978, obtained her MSc in Pharmaceutical Chemistry from Tianjin University (Tianjin, China) in 2003. She is currently a PhD student at the Department of Chemical & Materials Engineering, University of Alberta (Edmonton, AB, Canada). Her research interests are focused on designing bone targeting systems for protein therapeutics, especially modifying proteins with bisphosphonate ligands for increased bone affinity. |
 Geeti Gangal | Geeti Gangal completed her BS, MS and PhD in Organic Chemistry at the University of Delhi, India. After finishing her PhD in 2002, she joined the Department of Chemical & Materials Engineering at the University of Alberta (Edmonton, AB, Canada), and worked on the design and synthesis of bone targeting conjugates. She is currently a postdoctoral associate at the Massachusetts Institute of Technology, Cambridge, MA. She has been honored twice with a Science Meritorious Award for her academic excellence. She was also awarded a research fellowship and eligibility for lectureship after qualifying in the National Eligibility Test conducted by the Council of Scientific and Industrial Research (CSIR), India. |
 Hasan Uludağ | Hasan Uludag, a native of Turkish Republic of Northern Cyprus, obtained dual BSc degrees in Biomedical Engineering and Biology from the Brown University (Providence, RI, USA) in 1989. He then completed his PhD degree in 1993 from the Department of Chemical Engineering & Applied Chemistry at the University of Toronto (Toronto, ON, Canada). After spending four years at Genetics Institute Inc. (now part of Wyeth Pharma, Andover, MA, USA), he joined the University of Alberta (Edmonton, AB, Canada), holding joint appointments at the Departments of Chemical & Materials Engineering, and Biomedical Engineering, and the Faculty of Pharmacy & Pharmaceutical Sciences. Dr Uludag is currently directing interdisciplinary research programs into designing bone targeting systems for protein therapeutics, and non-viral delivery systems for transgene expression in primary cells. |
I. Introduction
Bone tissue constitutes our bodily scaffold around which our organs are compartmentalized. It is a dynamic tissue that maintains the mineral balance in an organism, as well as providing an environment for cellular machinery involved in different physiological functions.1 Bone tissue undergoes constant remodeling, where tightly regulated anabolic and catabolic processes enable bone adaptation during the lifespan of an organism. In addition to the cells involved in regulating bone tissue mass, i.e., bone-depositing osteoblasts, bone-resorbing osteoclasts and regulatory osteocytes, bone tissue provides a home for a diverse array of cells involved in systemic functions. Immune regulatory cells involved in host defense, mesenchymal stem cells involved in tissue healing/repair, and hematopoietic precursors destined for systemic gas transport, are distinct cell populations residing in the bone tissue. Bone tissue is distinguished from the rest of our tissues by the presence of a massive mineral phase, i.e., biological apatite. Approximately 3–4 kg of mineral mass is present in our bodies, and two-thirds of this mineral mass is estimated to be present in the bone tissue.2 More than 99% of bodily calcium deposits are located in bone. With the exception of dental tissue and pathological calcifications, such as kidney stones and calcified atherosclerotic plaques, no other tissue systems contain such a concentrated mineral phase. It is the mineral phase in bones that can serve as a unique receptacle for absorption of molecules from the systemic circulation, and molecules in circulation that display a preferential affinity to biological apatite have the potential to seek and concentrate in the bone tissue. This provides a unique opportunity for developing magic bullets for bone diseases, following on from Paul Ehrlich's idea that an ideal drug will act specifically on a disease-causing agent, in this case in bones, without affecting other tissues in an organism.Only a limited number of molecules exhibit a strong affinity to bone. These include heavy metals, such as strontium, rhenium and lead, and the well-known antibacterial agent tetracycline.3 The conventional therapeutic agents, except one class of molecules (bisphosphonates, the subject of this critical review), do not exhibit any particular affinity to bone. The systemic administration of these molecules accordingly results in non-specific distribution throughout an organism. For developing bone-specific therapeutic agents, the critical challenge becomes the design of molecules that display a preferential affinity to biological apatite with no affinity to other tissues. Systemic administration of such molecules will result in specific deposition to bone tissue with no accumulation at other tissues. This goal is likely to be difficult to achieve, since all therapeutic molecules will display a certain degree of affinity to other tissues, given the diverse array of functional groups (e.g., hydrophobic, polar, charged, etc.) found in biological membranes and surfaces. However, a step towards this goal is to engineer the currently utilized therapeutic agents for an apatite affinity. The benefits of this endeavor will be two-fold. First, the molecules that are currently acceptable for treatment of bone diseases (i.e., where the therapeutic action overshadows the undesired activities) will be more effective, since bone targeting will concentrate the pharmacological agents at the desired site of activity. This will allow a more potent activity without increasing the administered dose, which is not always possible due to undesirable activities of the therapeutic agents at extra-skeletal sites. Secondly, promising molecules not previously tested for bone diseases due to unacceptable side effects (i.e., where the undesired activities outweigh the therapeutic action) may become effective on bone diseases after being concentrated in the bone tissue. Modifying the therapeutic agents for bone affinity, of course, should not alter the inherent pharmacological activity of the agents. In this way, a given therapeutic agent can be tailored to have a higher specificity by concentrating it to bone sites and, possibly, to display lower toxicity by reducing its exposure to extra-skeletal sites.
Efforts in this direction were set into motion in the early 1960s while probing the physiological function of an endogenous molecule, pyrophosphate (Fig. 1). Pyrophosphate is localized throughout an organism, and displays a dual activity on both the formation and dissolution of biological apatite, a carbonated form of the stoichiometric hydroxyapatite [HA; Ca10(PO4)6(OH)2].4,5 The strong affinity of the pyrophosphate to nucleating HA crystals was considered to be the underlying basis of this dual activity.6 On one hand, the pyrophosphate appeared to become localized on the growing crystal surfaces, preventing the growth of the HA (i.e., ‘poisoning’ the fledgling crystal growth). On the other hand, this pyrophosphate coating on HA surfaces provided a protective layer against the dissolution of the already nucleated crystals. The ability of the pyrophosphate to suppress crystal growth is put into constant use in our bodies where preventing aberrant calcification from the supercritical solutions found in the tissues is an enduring process. Indeed, pyrophosphate administration was found early on to be beneficial in an animal model of aberrant calcification, namely the rat aortic calcification model.7 The ability of the pyrophosphate to suppress apatite dissolution, on the other hand, suggested a means to prevent the loss of tissues already mineralized (i.e., deposited bone). Unlike its beneficial effect in suppressing aortic calcification, pyrophosphate was not beneficial in suppressing bone loss, and this lack of activity in bone resulted in a search for pyrophosphate analogues that displayed superior stability in the bone milieu, the presumed shortcoming of the pyrophosphate in this environment. The search led to identification of phosphonate-based molecules, where the hydrolysis-resistant –C–P(O)–(OH)2 moieties replaced the labile –O–P(O)–(OH)2 moieties in the pyrophosphate.8–11 Such diphosphonates were shown to be capable of controlling HA dissolution,8,9 as well as preventing bone loss induced by immobilization10 and parathyroid extract injection in animal models.9 The diphosphonates were also active in preventing pathological aorta calcification,11 similar to the first beneficial use of the pyrophosphates. The diphosphonates used in these early studies were dichloromethylene diphosphonate,9,10 methylene diphosphonate9 and 1-hydroxyethylene-1,1-diphosphonate11 (Fig. 2). The two phosphonate moieties in these compounds were located on the same carbon (α-carbon), in fact forming the basis of the bisphosphonate (BP) class of compounds. This promising work spurred intense research activity where the end-goal was to identify pharmacologically active analogues of BPs, i.e., potent compounds where a predictable inhibition of bone loss could be obtained when delivered in a convenient, clinically acceptable fashion without significant side-effects. Human use of the BPs immediately followed with almost no lag time for clinical entry.12 Some of the successful BPs emanating from this research activity are summarized in Fig. 2. As with the first generation of BPs, contemporary BPs display an exceptional affinity to HA; once localized to the bone tissue, however, they exert their respective pharmacological activities primarily by modulating local cellular activities, rather than affecting the physicochemical properties of the apatite (see Section III for a more detailed discussion of the latter issue). It must be pointed out that Fleisch's early work also recognized the possibility of cellular effects by the early BPs, in addition to their effects on inhibition of HA dissolution.9
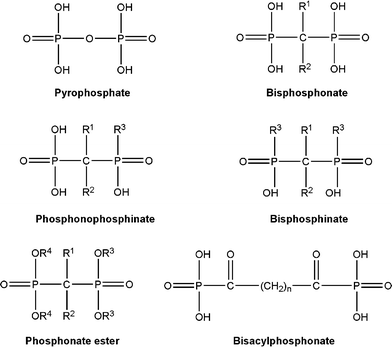 | ||
| Fig. 1 Structure of the endogenous pyrophosphonate, and its synthetic analogue, bisphosphonate (BP), which exhibit a strong bone affinity. The geminal (α) carbon in BPs typically contains two separate substituents, R1 and R2, which may significantly affect both the mineral affinity and the pharmacological activity. Other BP-related compounds are also shown, but the latter compounds either lack or exhibit a reduced affinity to the bone apatite. | ||
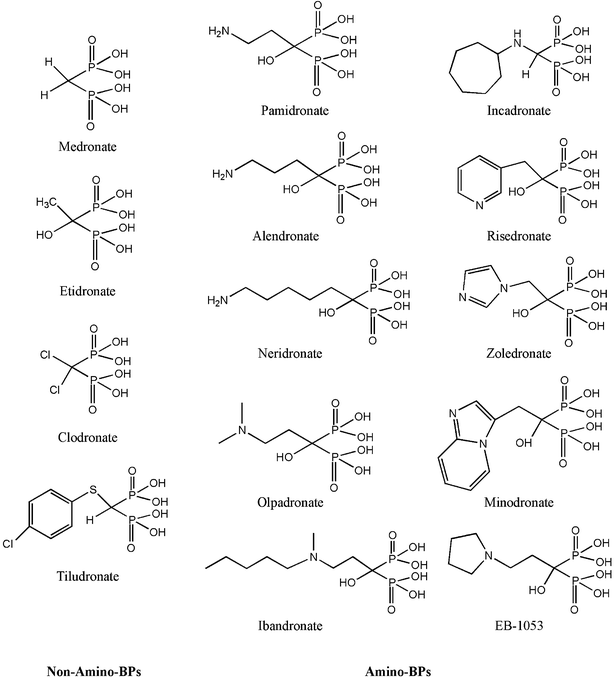 | ||
| Fig. 2 Examples of BP class of compounds currently used in a clinical setting. The compounds have been categorized into two classes, based on the presence of an amino group in the R2 side-chain. The amino-BPs typically exhibit a higher potency in antiresorptive effects, the primary clinical utility of BPs. Most of the BPs contain a geminal –OH group that enhances the mineral affinity of the compound. | ||
It was within a few years of realization of the pharmacological activities of BPs that their utility as bone carriers was also demonstrated. The initial use of BPs for bone targeting was for delivering the radionucleotide 99mTc to skeletal tissues for imaging purposes.13,14 Complexes formed between a BP and 99mTc did not compromise the bone-seeking capability of the compounds, providing a means to visualize skeletal tissues via the γ-emitting isotopes. Several critical observations were immediately noted from this collective activity: heterogeneity in bone uptake of the labeled complexes, ability to detect osteolytic metastasis in bones, as well as locating neoplastic tissues extra-skeletally in soft tissues (presumably due to local spots of calcification) spurred a diagnosis-centered BP research.15 These studies initially established the existence of a structure–function relationship for BPs, and inspired subsequent studies to further elucidate this relationship. It was not until 1986, however, when the development of a bone targeted therapeutic (i.e. the synthesis of BP-incorporating molecules with pharmacological activities distinctly different from the BP action) was first reported. Two of the earliest examples of bone-seeking therapeutics, which relied on a BP moiety for bone targeting and a distinct moiety for pharmacological activity, were an 131I-containing BP,16 and the antineoplastic drug 1,2,4-triglycidylurazol chemically linked to a BP.17 A wide spectrum of bone-seeking therapeutic agents has subsequently been pursued.
Scope
This review is intended to update the recent advances in the development of bone-specific medicinal agents and, in particular, design of bone-seeking proteins. It is a continuation of a previous communication from the authors' group that summarized advances in the bone targeting field until 2001,18 and a recent communication that specifically focused on protein delivery.19 Other reviews on this topic have since appeared in the literature.20,21 The foundation of this review is based on the premise that the exceptional affinity of BPs to bone makes them useful candidates as carriers of molecules to bone tissue. Although other molecules with high affinity to bone exist,18 the versatility of the BP structure facilitates the design of a diverse range of bone delivery systems. We therefore focused on the BP class of molecules in this manuscript. This review first provides a summary of biological apatite structure and the structural basis for BP affinity to the apatite, since it is the structural features of BPs that form the foundation of bone targeting efforts. Note that this review does not explore the structural features responsible for the pharmacological actions of BPs (see ref. 22 on this topic). Recent attempts on the delivery of BP-based diagnostic and therapeutic agents are then presented. We then focus on bone delivery of protein-based agents, first investigating the structural basis of protein affinity to bone, then focusing on functional BPs that are desirable for protein targeting efforts. The current attempts to deliver proteins to bones are then summarized, with a critical analysis of recently published data from the authors' lab. Finally, avenues of future research into the design of bone-seeking medicinal agents are discussed.II. Biological apatite
Bone mineral was recognized as an analog of naturally-occurring geological HA in the 1920s.23 The unit cell of crystalline HA has the chemical formula Ca10(PO4)6(OH)2 with an ideal Ca : P stoichiometric ratio of 1.67 : 1 (Fig. 3A). However, the analysis of bone mineral shows a Ca : P ratio ranging from 1.3 : 1 to 1.9 : 1.24 This deviation is mainly attributed to the carbonated groups present distorting the crystal structure of most biological apatites.25,26 The kinetic factors arise in solid–solution equilibrium during mineralization, which may give rise to the creation of vacancies at Ca2+, PO43−, and OH− sites, substitution of other cations (such as Mg2+ for Ca2+), and protonation of PO43− to substitute HPO42− in the crystal lattice (Fig. 3B and 3C).27 Moreover, the organic phosphate in the bone matrix might also partly contribute to this deviation.24 Two primary classes of binding sites in such a structure are provided by the superficial Ca2+ for anions and PO43− for cations. The zeta (ζ) potential of a synthetic HA (“HA” refers to synthetic HA herein till the end of this paragraph) surface increases from negative to positive, with the Ca : P ratio increasing from 1.55 to 1.70.28 This effect is likely due to excess Ca2+ neutralizing surface anions such as CO32− or OH− located in the PO43− defects. The surface charge of HA was recently probed by titration to determine the ability of the surface to adsorb H+ (and/or OH−) in an indifferent (non-interacting) electrolyte solution.29 The pH at which the point of zero charge occurs for synthetic HA was found to be ∼7.3. HA accumulated positive charge more readily below the point of zero charge than it accumulated negative charge above the point of zero charge, consistent with previous observations.28 The Ca2+ ions in solution were readily exchanged during deprotonation of HPO42−, indicating the dynamic nature of HA surfaces.29![Schematic representation of biological apatite. A. The synthetic hydroxyapatite [HA; Ca10(PO4)6(OH)2] is represented by the stoichiometric ratio of Ca2+, PO43− and OH− ions. B. Substitution of other cations (e.g., Mg2+) and anions (e.g., HPO42−) are typical for the physiological apatite. C. Non-equilibrium formation conditions also create defects in the apatite structure, leading to ionic gaps in the crystal structure. D. Recent investigations of physiological apatite crystals have suggested charge densities organized in larger scales (>10 nm) in the form of ‘bands’ (as revealed by probing surfaces with charged atomic force microscope tips).](/image/article/2007/CS/b512310k/b512310k-f3.gif) | ||
| Fig. 3 Schematic representation of biological apatite. A. The synthetic hydroxyapatite [HA; Ca10(PO4)6(OH)2] is represented by the stoichiometric ratio of Ca2+, PO43− and OH− ions. B. Substitution of other cations (e.g., Mg2+) and anions (e.g., HPO42−) are typical for the physiological apatite. C. Non-equilibrium formation conditions also create defects in the apatite structure, leading to ionic gaps in the crystal structure. D. Recent investigations of physiological apatite crystals have suggested charge densities organized in larger scales (>10 nm) in the form of ‘bands’ (as revealed by probing surfaces with charged atomic force microscope tips). | ||
Unique insights into HA surfaces are beginning to be acquired with the atomic force microscope (AFM).30,31 By virtue of functionalizing the AFM tip with specific chemical groups, the nature of surface charge on HA crystals could be probed in addition to the surface topography. To obtain precise and positionally-sensitive measurements of the surface properties of HA, Vandiver et al.32 measured the electrostatic forces between AFM tips derivatized with –COO− and –NH3+, and the synthetic polycrystalline HA at nm-resolution. The results revealed that HA has a net negative surface charge per unit area with an average value of −0.019 C m−2, consistent with previous measurements33,34 and the given pH (6.0) of the system. More importantly, HA did not present a uniform charge density over its surface; the surface charges varied greatly as a function of distance within a grain boundary (a ∼7-fold variation over a distance of ∼400 nm within the boundaries of a grain). This variation presumably resulted from a variation of surface PO43− groups, and suggests a heterogeneous template for molecular binding within a single grain. The same approach (with –COO− and –NH3+ functionalized tips) was also adopted for probing protein-free apatite enamel surfaces.35 These surfaces exhibited a net positive surface charge at neutral pH due to Ca2+ rich surface layers on biologically derived crystals.36,37 Within each crystal (approximately 90 nm in width, 50 nm in thickness and >1000 nm long) the surfaces exhibited a ‘striated’ pattern of alternative surface charge, perpendicular to the crystal “c”-axis in the absence of any topographical changes on the surfaces. Using enamel surfaces where the endogenous proteins were extracted under neutral conditions, the striated pattern of the surface became more pronounced under low pH (4–5.5) conditions;38i.e., the functionalized tips exhibited significant frictional variations while sliding on the etched surface. The striated pattern was readily observed with AFM tips derivatized with –COO−, but not with –NH3+ derivatized tips. Therefore, it is likely that this striated pattern corresponds to the distribution of Ca2+ on the HA surface (Fig. 3D). Other mechanisms, such as protonation of surface moieties, mobility of charges in and out of a buffer environment and/or dissolution of crystal interfaces, are also likely reasons for the appearance of such striated patterns.
An important consideration for the design of bone-seeking agents is the relationship between the observed heterogeneity in charge distribution and molecular binding; do such variations lead to differences in quantitative and/or selective molecular binding? A well-studied molecule in this context is amelogenin, a peptide macromolecule known to play a physiological role in mediating mineralization in enamel formation. Recent studies have indicated that the –COOH rich C-terminus of the protein binds and orients the molecule on HA surfaces.39 The protein forms 10–20 nm aggregates under physiological conditions,40 so that its HA binding is not likely to involve specific secondary motifs. However, the aggregated protein still binds to the discrete regions on enamel surfaces, consistent with the cationic banding pattern.31,41 Even with synthetic HA, a recombinant amelogenin exhibited ∼64% surface coverage, selectively interacting with some of the crystal faces but not uniformly with all available surfaces.42 The binding pattern is not unique to the amelogenin, and heterogeneous binding patterns were also observed by two other anionic proteins: (i) bovine serum albumin, which has no significant role in biomineralization,41 and; (ii) phosphophoryn, which is also involved in modulating biomineralization.42 Whereas albumin desorption was complete with a high molar (500 mM) phosphate buffer, a significant fraction of amelogenin nanospheres remained bound to enamel crystals under these conditions, indicating also a variation in the strength of binding among the surface sites.
The striated pattern of biological apatite (enamel) was also revealed after investigating the surface binding of a totally synthetic macromolecule, a –COOH-terminated 7th-generation polyamidoamine (PAMAM) dendrimer.43,44 However, an –NH2-terminated PAMAM dendrimer exhibited a diffuse, as opposed to a striated, binding pattern. Both functional groups were >95% ionized under the utilized experimental conditions (in distilled water, at pH 7.4), so that electrostatic interactions were considered to be the main contributor to the surface binding. In fact, a –CH3-terminated PAMAM, unlike its –COOH and –NH2-terminated analogs, was readily removed from the enamel surfaces (the desorption buffer: 100 mM and 200 mM phosphate buffer at pH 7.4),44 indicating the terminal groups of the PAMAM, rather than the internal hydrophobic moieties or polar amide linkages, to be the participants in the enamel matrix interactions. These results were indicative of a diffuse pattern of anionic charges (presumably PO43−) on enamel surfaces, unlike the ‘clustered’ cationic charges (primarily Ca2+). Although the exact structural features responsible for these variations are not known, the substitution of H-bonding for Ca2+, or variations in apatite solubility (due to carbonate or Mg2+ substitutions) could be likely reasons.45,46 These results might also suggest a stronger interaction between the basic groups and the surface, whose variations in ionic composition do not necessarily influence the conducive binding of basic groups.
III. Structural basis of bisphosphonate affinity to bone
The exceptional selectivity of BPs to bone mineral rather than other tissues lacking a mineral phase is the basis for their value in clinical practice. Due to the affinity of spatially-optimized, deprotonated –O− to the bone apatite, the bisphosphonic acid form of the compound is the bone-seeking entity. Monophosphonates, phosphonate esters, chemically-modified phosphonate groups (e.g., methylated phosphonates, phosphonophosphinates or bisphosphinates), or compounds with P–N–P and P–C–C–P backbones all display reduced affinity to mineral to make them not useful for bone therapy.47 Though non-geminal diphosphonates are biologically inactive as anticalcification agents, introduction of a keto group at the α-carbon makes bisacylphosphonates active as antiresorptive agents with lower potencies than the BPs48 (Fig. 1). The pharmacological activity of BPs varies a great deal from compound to compound,22 in line with variations in the R1 and R2 substituents shown in Fig. 1. Whereas an increased affinity to HA is desirable to further increase the concentrations of the active agents in bone, it is important for the BP levels not to reach inhibitory concentrations on mineralization of osteoid in bones.49There are two main considerations in the design of BP-based drugs: (a) the relative affinity for bone mineral, and (b) the inhibitory effect on cellular mechanisms responsible for bone resorption. These aspects are fulfilled by different parts of the BP molecules and early studies suggested that they did not necessarily rely on each other. The P–C–P linkage, along with the 3D configuration of the R2 substituent, determines the interaction with specific cellular targets essential for pharmacological activity. The (HO)2–(O)P–C–P(O)–(OH)2 moiety, on the other hand, is responsible for chelation of Ca2+ in HA; the bite distance of deprotonated oxygens, –O−⋯−O–, between the two phosphonates is 2.9–3.1 Å, and this separation is within the range found for the oxygens in HA.50,51 This is presumably the ideal distance for the chelation of Ca2+ ions in HA. The affinity of BPs for bone mineral was proposed to bear no relationship to their marked differences in antiresorptive potencies.52 BPs with an –OH group in the R1 position have increased affinity for bone mineral (the so called ‘bone-hook’ effect, as suggested by Russell et al.53) and the R2 substituent was believed to play a nominal role in the bone affinity; BPs with variable R2 substituents but a common –OH in the R1 position bound with equal affinity to bone mineral; For example, olpadronate and etidronate (see Fig. 2), two BPs with considerably different R2 substituents, could displace [14C]-3-dimethylamino-1-hydroxypropylidene-1,1-bisphopshonate (dimethyl-pamidronate) from mouse fetal bones with equal potency, but clodronate, which lacked the –OH moiety on the R1 position, was ∼10 times less potent in such a displacement.52 The differences in binding can be explained with the mode of binding,49i.e., bidentate binding, involving two deprotonated –O− from each phosphonate moiety binding to HA Ca2+ (as in the case of clodronate) vs. tridentate binding, involving participation of the –OH at the R1 position. Although tridentate binding is generally accepted, the amino-BPs pamidronate and alendronate contain –OH groups in a gauche configuration, which enable formation of an intramolecular N–H⋯O(hydroxyl) H-bond and impairs tridentate binding under some conditions.50,51 Tridentate binding can also be obtained with an –NH2 group in the R1 position.54,55 The amino-substituted BPs and their –OH analogs (etidronate, pamidronate and olpadronate, in this case) bound with similar affinity to mouse bones in vitro, and inhibited the growth of calcium oxalate crystals to the same extent.55
Recent studies, however, indicated that the R2 substituent may also influence the HA affinity of BPs.56,57 Nancollas et al. compared the binding affinities of several BPs to HA, all of which bore the same R1 substituent (–OH), but differing R2 substituents.57 Different HA binding affinities were observed among the BPs, with a decreasing rank order of zoledronate > alendronate > ibandronate ≈ risedronate > etidronate. The corresponding affinity constants KLi, which were calculated from the kinetics of HA crystal growth, were 3.47, 2.94, 2.36, 2.19, and 1.19 × 106 M−1, respectively. These results were attributed to the differences in the protonation of the R2 substituent, dictated by the pKa of the ionizable moieties. In addition, measurement of the ζ-potential of the BP-treated HA suggested that there were changes in the overall charge of the crystal surface, which were best explained by molecular charges on the R2 substituent. The latter affected HA binding of additional BPs by enhancing or suppressing further binding on the surface. Under physiological conditions, higher binding was obtained when the pKa of the R2 substituent was ≥7. Consistent with the HA growth studies, the BP affinity to HA was determined by a direct method under chromatographic conditions; a decreasing order of HA affinity was obtained with zoledronate, risedronate and a risedronate analogue in which one of the phosphonate groups was replaced by a –C(O)–OH. Moreover, a recent computational model (3D) demonstrated the differences in HA affinities of different BPs, with consideration of the interactions of the N side chain conformations with the [001] surface of HA.58 This model also showed a similar rank order in HA affinity to the above studies (note that ibandronate was not included in the simulation study). Along the same lines, a sulfate-bearing BP, whose R1 and R2 substituents were –H and –CH2–SO3H, respectively, was compared in vivo to the –OH-bearing etidronate's capacity to target to bone.59 Equivalent or better bone targeting was achieved (at 1 h and at 24 h, respectively) with this non-hydroxyl bearing BP as compared to etidronate. Therefore, increasing evidence is accumulating to confirm the subtle contribution of the R2 substituent to the mineral affinity, which could be utilized to further enhance the ability of BPs to seek bone.
Finally, the overall hydrophobicity of BPs has been shown to influence their bone targeting ability.60 This observation was derived from studies where BP conjugates from a diverse group of compounds and drugs (e.g., 17ß-estradiol, diclofenac, and benzene) were constructed. As expected, the hydrophobicity of the conjugates was significantly different among BPs with different side-groups (the R2 substituent being the major variable), based on the calculated octanol–water partition coefficient (Clog P) of the acid form of the compounds. The use of the acid form is a simplification, but it is justifiable considering that the exact nature of the BP complexes with metal ions such as Ca2+ and Na+in vivo is difficult to predict. The Clog P of the acid form will presumably be closely related to the Clog P of the ion-chelated form. After systemic administration, an inverse correlation was obtained between the Clog P and the ability of the compounds to deposit to bones, which ranged from 6 to 70% of the administered dose. All compounds used in that study contained an –H on the α-carbon, and lacked either an –OH or –NH2 at this position. Using such a uniform population of BP conjugates has likely contributed to the relatively good correlation between the Clog P and the skeletal deposition. The authors expected the hydrophobicity of BPs to determine the propensity of the BP–metal chelates, formed between the endogenous ions in circulation and the exogenous BPs through the interaction between negatively charged phosphate groups on the BPs and positively charged metal ions, to precipitate in systemic circulation; i.e. chelates with more hydrophobic character will presumably be removed faster by the clearance organs. These results highlight the need to use smaller, hydrophilic BPs as the basis of bone-specific carriers.
IV. Bisphosphonates in the diagnosis of bone diseases
The ability of BPs to chelate radioactive isotopes, such as 99mTechnetium (99mTc) and 186Rhenium (186Re), while preserving their HA affinity made them useful as skeletal radiodiagnostics. 99mTc-labeled skeletal imaging agents were first introduced in 1971 with long chain polyphosphates,61 and then superseded by the pyrophosphate and subsequently the current bone-seeking BPs. 99mTc–pyrophosphates and 99mTc–BP are known as the second and third generation diagnostics, respectively, following the initial use of 99mTc–polyphosphate. Normal uptake of the radiolabeled BPs by osseous tissues typically shows the area of physiological bone turnover, whereas abnormally high uptake at specific osseous sites corresponds to regions of injury. Various tumors at bone sites, whether primary or metastatic, are characterized by rapid bone deposition, and/or osteolytic lesions, and scans with 99mTc–methylene diphosphonate (MDP) are very sensitive for visualization of tumor burden at osseous sites.62 More importantly, the localization of 99mTc-labeled BPs at extra-skeletal sites could highlight pathological tissues, and recent experiences broaden the utility of 99mTc–BP chelates to diagnose meningiomas,63 cirrhosis of the liver,64 cerebral infarction,65 myocardial infarction,66 osteosarcoma,67 neurofibroma,68 methotrexate osteopathy,69 relapsing polychondritis,70 and breast cancer.71 As an example, 99mTc–MDP detected calcifications in meningiomas arise from calvarial erosion, the formation of reactive bones, and tumor-induced calcification.72 The latter is postulated to arise from low levels of intracellular adenosine triphosphate, leading to metabolic impairment and weakening of cell membrane, and followed by accumulation of calcium in the mitochondria. Presumably this intracellular nucleation provides a template for the uptake of radiotracers reminiscent of BP absorption onto the HA crystal of osseous tissue, whose nucleation is mediated in part by phosphorylated membrane components during normal matrix mineralization. It is difficult for the computed tomography (CT) scan, the most precise non-invasive test for diagnosis of meningiomas, to detect lesions close to the vertex or the base of the skull because of the low level of calcification in tumor tissue, and its proximity to high-mineral density skull. BP scintigraphy provides an excellent complementary test for this clinical syndrome.The chromatographic analysis of commercially available 99mTc–BP preparations has resolved multiple 99mTc species, indicating that 99mTc does not form a single complex with BPs. The only 99mTc–MDP structure reported 73 shows a 1 : 1 complex, where each 99mTc atom chelates with two BPs, and each BP chelates with two 99mTc atoms (Scheme 1). This polymeric 99mTc–BP complex plays a central role in the chemistry of this bone-scanning agent. 99mTc bridges to the HA by the deprotonated –O− of BP, because metals like 99mTc contain acidic oxides that have no affinity for HA. In addition to the bidentate ligand MDP, the tridentate ligands 1-hydroxyethylidene-1,1-diphosphonate (HEDP; –OH on α-carbon; Fig. 2) and 1,3-dicarboxypropane-1,1-diphosphonate (DPD; –COOH on α-carbon) have also been utilized74–76 due to the enhanced mineral binding of the respective 99mTc-bearing radiopharmaceuticals.
![The portions of the Tc–MDP polymer [Tc(MDP)(OH)−]∞ showing (A) one Tc center binds to two MDP ligands, and (B) one MDP ligand bridging two Tc centers. (Adopted with permission from ref. 73. Copyright 1980 American Chemical Society.)](/image/article/2007/CS/b512310k/b512310k-s1.gif) | ||
| Scheme 1 The portions of the Tc–MDP polymer [Tc(MDP)(OH)−]∞ showing (A) one Tc center binds to two MDP ligands, and (B) one MDP ligand bridging two Tc centers. (Adopted with permission from ref. 73. Copyright 1980 American Chemical Society.) | ||
Bone scintigraphy with 99mTc–MDP has recently been used with single photon emission computer tomography (SPECT), which enables 3D reconstruction of tissues along with a thin sectioning technique. Images obtained from MDP-SPECT analysis have been helpful in the diagnosis of mandibular condylar hyperplasia,77 carcinoma of the nasopharynx,78 ovarian cancer,79 and ossification of the ligamentum flavum.80 HEDP has been also used to deliver 186Re and 188Re for the diagnosis of metastatic cancers within the bone.74–76 These radiopharmaceuticals are not only γ-emitters (suitable for diagnostic use), but also emit β-particles of 1.07 MeV energy (maximum) in the case of 186Re–HEDP and of 2.1 MeV in the case of 188Re–HEDP, both of which are suitable for therapeutic use. The clinical studies have confirmed that even small doses of these compounds were palliative for treatment of painful bone metastases. With Re–HEDP complexes, the –OH groups of HEDP was found to coordinate to some extent with the Re, resulting in reduced accumulation of HEDP in bones.81 Ogawa et al. have recently synthesized a bifunctional radiopharmaceutical, one composed of mercaptoacetylglycylglycylglycine, and an α-OH-bearing BP (186Re–MAG3–HBP; Fig. 4, 1).81 Whereas the latter group acted as the bone-seeking component, the former group chelated the 186Re, preventing the interaction of the α-OH group with the radionucleotide. This compound was more stable in vitro, and showed better accumulation in bones as compared to other tissues; 2-fold increased selectivity to bones was evident with this new reagent as compared to 186Re–HEDP.
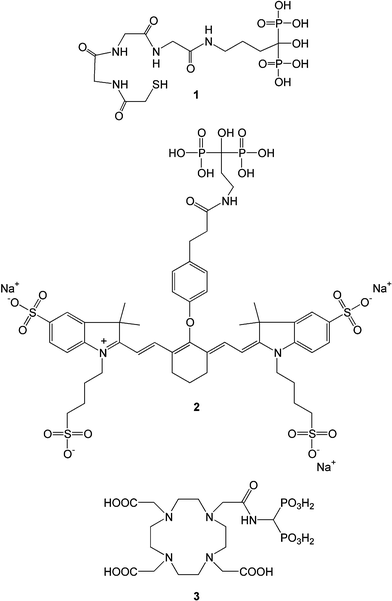 | ||
| Fig. 4 Recent diagnostic agents being pursued for bone diseases. The agents were constructed based on a compound useful for a desired detection modality (e.g., γ-emission in 1, fluorescence in 2 and nuclear magnetic resonance in 3) and a bone-seeking BP. More information about the compounds 1–3 is in the body of the manuscript. | ||
A significant shortcoming of all of the above diagnostics is their reliance on radioactive emitters for detection. Considering the health hazards related to exposure to such isotopes, a fluorescence-based imaging agent was constructed based on the linkage of a BP (pamidronate) and a near-infrared (NIR) emitting dye (IRDye78) (Fig. 4, 2).82 Using a hairless mouse model, it was possible to visualize the osseous tissues, such as the spine, skull, and pelvis, under NIR fluorescence imaging. The construct's amide linkage appeared to be stable for a short duration (∼6 hours), permitting rapid visualization of skeletal tissues (<500 ms) at high resolution (<1 mm). This was unmatched by 99mTc–MDP imaging in the same animal model, but the latter appeared to be better suited for imaging deep tissues, since the NIR fluorescence decayed as a function of tissue depth.82 It was possible to visualize HA implanted at extra-skeletal sites by the newly synthesized probe,83 suggesting that detecting extra-skeletal calcification is also feasibile using the newly synthesized probe.
Finally, a BP-derivative of gadolinium (Gd)-binding tetraazacyclododecanetetraacetic acid (DOTA) was prepared as a carrier of Gd(III) as an MRI contrast agent.84 The compound 3 (Fig. 4) bound the Ln(III) ion in the macrocyclic cavity, after the initial contact of the ion with the BP moiety. The construct was found to bind to HA in a reversible manner, and exhibited a mineral affinity that was far superior to the HA affinity of tridentate HEDP and bidentate MDP. Presumably, the additional –COOH groups on 3 contributed to HA binding, even though the BP on DOTA was a bidentate ligand. In vivo evaluation of this compound remains to be performed.
V. Bisphophonates in the delivery of therapeutic agents to bones
The exceptional affinity of BPs to HA has previously been used to transport several classes of therapeutic agents to bone tissue. These included: (i) antineoplastic agents intended to control cancerous cell growth; (ii) antibacterial agents to inhibit bacterial colonization, and; (iii) anti-osteoporosis agents acting to protect against excessive bone loss with advancing age. In addition, BPs are beginning to be utilized as the building blocks of generic carriers that can transport a spectrum of molecules to bone, rather than linking them directly to a given molecule for its delivery to bone. Below are the recent developments in the use of BPs to deliver therapeutic agents to bones.A. Anticancer agents
Combining anticancer agents with BPs is intended to concentrate the antineoplastic activity of therapeutic agents in bones after systemic administration. Towards this end, a new compound was prepared by linking 1-aminomethylene-1,1-bisphosphonate with 5-fluorouracil (Fig. 5, 4).85 5-Fluorouracil is a broad-spectrum anticancer agent, and it has been utilized against solid tumors, such as breast and colorectal cancers, but with a low (<30%) success rate.86 The bone localization of 4 after IV injection in normal mice was similar to that of an equivalent BP (MDP), confirming previous observations that conjugating small molecular drugs to BPs did not alter the bone affinity of BPs. The individual compounds in 4 were linked together with a urea linkage, which was suggested to be labile in situ, and possibly releases the anticancer drug from its BP counterpart (remains to be determined). The anticancer efficacy of 4 has not been reported and, ideally, 4 is expected to display an anticancer effect at bone sites at significantly lower doses than the parent 5-fluorouracil. The same group also reported a BP conjugate of the metal-chelator diethylenetriaminepentaacetic acid (DTPA; Fig. 5, 5), which afforded bone targeting of 99mTc equivalent to that of MDP. However, DTPA might exhibit some osteotropic character due to multiple –COOH groups, and no studies on the ability of the parent DTPA to seek bone were reported in this paper (hence, the benefit of BP conjugation to DTPA could not be assessed). Such a BP–DTPA conjugate was intended as a carrier of radioisotopes beneficial in cancer therapy, such as 153Sm and 186Re, and to target the radioisotopes to bone. In a follow-up study, the same group reported the ability of 4 and 5 to deliver 188Re to bones.87 Both compounds were able to deliver the radioisotope to the bone, but the extent of delivery was similar or slightly lower than the reference carrier, HEDP. The bidentate binding by HEDP was a likely reason for better targeting efficiency. Presumably, the 188Re–HEDP mixture exhibited some dissociation in vivo,81 and it appears that having a separate radioisotope binding center (DTPA) in these constructs did not improve non-specific distribution to kidneys and stomach. Given the similar level of bone delivery among these compounds, the simpler BP will obviously be preferable in a clinical setting. However, it must be stated that the significant delivery of a radioisotope to bones with 4 in this preliminary study offers the possibility of a single bone-seeking agent that incorporates a chemotherapeutic as well as a radioisotope component for combating malignancies at bone tissue.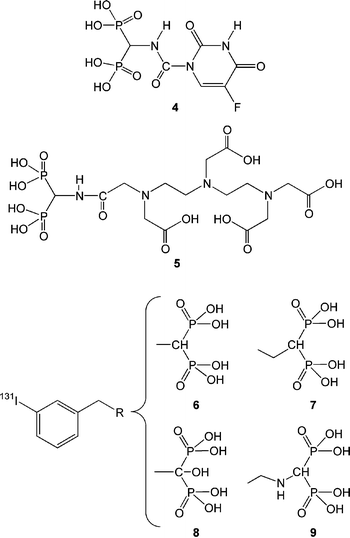 | ||
| Fig. 5 Recently reported anticancer agents developed specifically for bone tumors. Compound 4 is a BP conjugate of the wide spectrum anticancer agent, 5-fluorouracil. Compound 5 is a BP conjugate of the radioisotope chelator diethylenetriaminepentaacetic acid (DTPA). Compounds 6–9 are BPs with a benzene ring in the R2 side-chain that is capable of 131I addition. | ||
In contrast to BP-based carriers of radioisotopes that hold their cargo by physical complexation, BPs with the ability to covalently couple radionucleotides could be desirable for constructing more stable reagents. BPs chemically coupled to 125I and 131I were recently reported towards this end.88 The new compounds offer the possibility of a more stable linkage between the bone carrier and the radioisotope, as well as the possibility of benefiting from the unique emission patterns of 125I and 131I. The 131I–BP derivatives tested in animals (Fig. 5, 6–9) exhibited rapid (<30 minutes) localization to bones, which did not change over a 24-hour period. There was little accumulation of the radioisotope in the thyroid, indicating the expected stability of the iodinated compounds. Among the four compounds tested in mice, 8, with an –OH moiety on the α-carbon, was most effective in targeting the radioisotopes to bone, and 9 was surprisingly not bone-seeking (Fig. 5). The reason(s) for the failure of the latter compound was not known. We utilized a similar BP, 1-aminomethylene-1,1-bisphosphonate, for coupling the –NH2 group to proteins, but this BP linkage afforded successful targeting of proteins to bone (see Section VI). The 5-fluorouracil conjugate in 4 was also constructed from such a BP linkage, but this compound also displayed the expected bone targeting. It remains to be seen whether 9 contains unique structural features that led to its lack of expected bone targeting. In efficacy studies, 131I-labeled 8 significantly increased the survival times in rats inoculated with a model of human breast cancer, as well as an osteosarcoma.88 While an increase in efficiency was seen with increasing 131I dose, this animal study did not evaluate the efficacy of non-labeled 8. In the absence of this control, it is not known whether the beneficial effect was due to the radioisotope or to the BP component, which may retard tumor growth per se.
At this point, it must be pointed out that several studies in recent years have yielded important clues about the possible effects of BPs on cancerous tissues. Unlike the initial use of BPs to reduce the osteolytic burden on bone tissue, it is now recognized that BPs can display direct effects on cancerous cell mass, such as the induction of cellular apoptosis, and inhibition of cellular invasiveness.89,90 This has led to the preparation of novel BPs specifically to inhibit cancer-related events, including inhibition of angiogenesis necessary for the growth of solid tumors.91,92 In the same way a wide spectrum of BPs has been evaluated for their antiresorptive activity, such an effort is beginning to be directed towards anticancer activity based on the purely BP class of compounds, rather than relying on BPs to target currently accepted antineoplastic agents to bone.
B. Antibacterial agents
A new class of BP conjugates recently reported is the BP conjugates of antibacterial agents, and a BP conjugate of ciprofloxacin in particular93,94 (Fig. 6, 10). A bone-seeking antibacterial agent will be beneficial for osteomyelitis where the bacterial infection poses difficulty in treatment once colonization has occurred in the bone tissue. The prepared conjugate 10 retained the antibacterial activity of the parent compound ciprofloxacin, albeit at a slightly lower level (based on the minimum inhibitory concentration for bacterial growth in vitro).93 Ethyl esters of BPs were equally effective antibacterial agents as the bone-seeking bisphosphonic acidic form. This was an indication of the BP component not directly participating in the antibacterial effects. An in vivo study where the conjugate was delivered locally with a sintered HA (Skelite™) was effective in preventing bacterial burden in a rat fracture model of osteomyelitis.94 The mineral carrier presumably bound and maintained a high concentration of the antibacterial agent at the fracture site (no data was provided on in situ release kinetics of the compounds). Although the proposed therapy was superior against no treatment (i.e., HA implantation alone), it remains to be seen whether the BP conjugate will be superior to the standard therapy, i.e., systemic administration of antibacterial agents which are not specifically targeted to bone.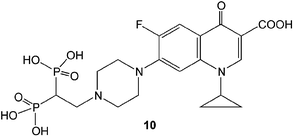 | ||
| Fig. 6 Bone-seeking antibacterial agent constructed by conjugating a BP to ciprofloxacin. | ||
C. Antiresorptive agents
Src tyrosine kinase in osteoclasts has been recently utilized as a potential target to develop antiresorptive therapeutics. Src tyrosine kinase is a critical enzyme for the development of osteolytic activity.95,96 Such inhibitors will be useful in inhibiting aberrant osteoclastic activity observed in several diseases, including postmenopausal osteoporosis, arthritis and osteolytic bone tumors. Since Src tyrosine kinase is involved in regulating a multitude of cellular processes, targeting the inhibitors to bone is critical to reduce the undesired activity at other tissues. Several compounds are available that can inhibit the kinase activity at nM concentrations, and these compounds can serve as the initial molecular scaffolds for further design of bone specific inhibitors.97 Early work with a Src kinase inhibitor indicated successful inhibition of bone resorption following its systemic administration, providing the initial proof-of-principle of this approach in an animal model.98 Bone targeting inhibitors were designed based on conformational information of the catalytically active domain of the enzyme, and by incorporating bone targeting moieties to the base molecules.99 An obvious precaution in this effort is ensuring that the interaction of the inhibitors with the active site of the enzyme is not compromised due to the presence of the conjugated bone targeting moiety. Several bone targeting moieties have been used towards this end,100–102 which included a conventional geminal BP, as well as structurally modified geminal BPs and other diphosphonates (Fig. 7A). As expected, addition of a bone targeting moiety sometimes reduced the ability of the starting scaffold to inhibit the enzyme (10∼100-fold lower specific inhibitory activity; given by concentration required for 50% inhibition of Src tyrosine kinase activity, IC50), or did not affect the inhibitory activity by yielding a specific inhibitory activity that was equivalent to the starting scaffold. However, one particular bone targeting moiety (phosphinomethyl phosphonic acid) was found to increase the specific inhibitory activity of multiple active molecules. This was also the case in an in vitro cell-based anti-osteolytic activity assay using osteoclasts, where the most promising compounds 11a, 12a and 13a (Fig. 7B) inhibited the extent of osteolytic lesions. The cell viability was not altered in the in vitro assay, so that the obtained anti-osteolytic activities were not due to general toxicity on the osteoclastic cells. It was interesting to note that the higher potency of phosphinomethyl phosphonic acid derivatives 11avs. 11, and 12avs. 12 observed in the enzymatic assay did not readily transfer to higher potency in cell-based assays.101,102 This might have been due to the reduced penetration of the modified molecules through the cell membrane. As expected, the synthesized compounds displayed a higher mineral affinity in an in vitro HA binding assay.100–102 It is not straightforward to extrapolate the observed HA affinity to actual bone targeting in vivo, and the extent of bone targeting remains to be investigated in animal models. Considering some of the starting molecular scaffolds are relatively bulky and hydrophobic, the bone targeting might not be as strong as anticipated, as observed with certain hydrophobic derivatives of BP-conjugates that were found to be located to fatty tissue.60 Ability to target bone (directly proportional to BP substitution) and to penetrate cell membranes (inversely proportional to BP substitution) may need to be further optimized for this class of compounds.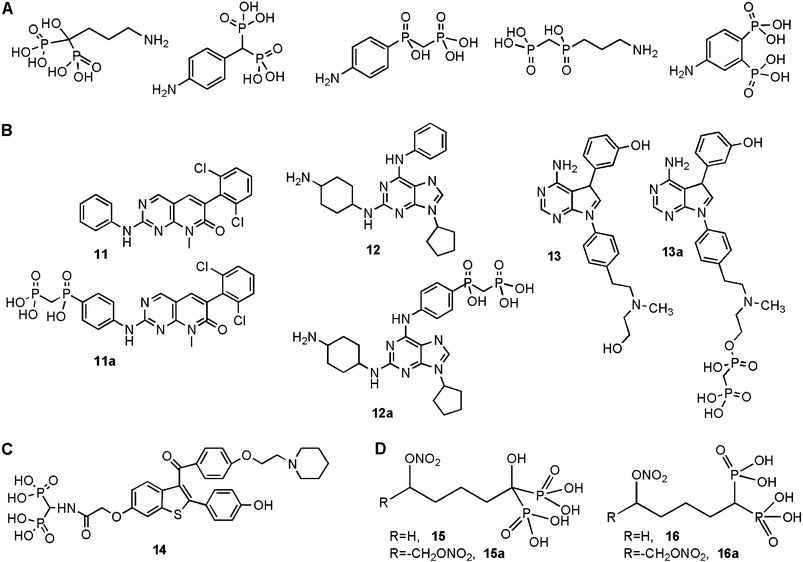 | ||
| Fig. 7 Summary of antiresorptive agents specifically designed for bone targeting. A. Several novel phosphonate-based moieties, in addition to a BP, were explored for bone targeting. B. Most successful antiresorptive agents that displayed both a strong mineral affinity and anti-osteoclastic activity. 11, 12 and 13 are the anti-osteoclastic agents, and 11a, 12a and 13a are their bone-seeking analogues. C. BP-conjugate of selective estrogen receptor modulator raloxifene (14). D. New BPs designed as nitric-oxide donors. The compounds were either single NO donors (15 and 16) or double NO donors (15a and 16a), and contained either a geminal –OH (15 and 15a) or an –H (16 and 16a). | ||
Several groups previously reported different bone-seeking estrogenic agents as a way to reduce non-skeletal exposure of systemically administered estrogens,18 since undesirable activities at these sites have limited the usefulness of this hormone against the rapid bone loss associated with menopause. A recent report also contributed to this field by synthesizing a bone-seeking raloxifene,103 a clinically-approved selective estrogen receptor modulator (SERM). Using 1-aminomethylene-1,1-bisphosphonate, raloxifene was linked to the BP via a short amide linkage (Fig. 7C, 14), and its pharmacological activity was evaluated in ovariectomized rats, which undergo rapid bone loss due to systemic estrogen depletion after the removal of the ovaries. The synthesized compound protected against the ovariectomized-induced bone loss, with a potency similar to that of BP alendronate.103 No pharmacokinetics data was provided to demonstrate the bone targeting ability of this compound, and the efficacy assessment was evaluated without a raloxifene control group. Hence, this preliminary study did not indicate (i) whether the BP conjugation provides a higher bone targeting, or (ii) whether the conjugate provided any benefit over that of the standard therapy, i.e., raloxifene alone.
Finally, a unique class of BPs was recently reported where the compounds acted as bone-selective nitric-oxide (NO˙) donors.104 The NO˙ was capable of inhibiting osteoclastic activity, and based on this activity, –O–NO2 bearing moieties were incorporated into BPs (Fig. 7D). Consistent with previous experience, bone targeting of BPs with an α-carbon –OH group (15 and 15a) was better than that of the BPs lacking this –OH group (16 and 16a), but both types of compounds were equally effective in vitro in inhibiting development of osteoclastic phenotype from the RAW264.7 cells in vitro. BPs lacking –O–NO2 groups were not active in this assay, and compounds with a single –O–NO2 group (15, 16) were less potent than the disubstituted analogues (15a, 16a), indicating the NO˙ delivery to be the primary determinant of the observed anti-osteoclastic activity. These unique compounds were also active in inhibiting osteoclastic development from rat bone marrow cells. Preclinical testing in an animal model is the next step for these unique compounds.
D. Drug carriers
An alternative to direct chemical linkage of BPs to therapeutic agents is to assemble the BPs into generic drug carriers. In this way, the bone-specific carriers serve as a general platform for delivery of a range of medicinal agents to bone. Three independent efforts in this direction have been pursued by preparing drug carriers that are based on synthetic polymers,105 heparin106 and a fullerene.107The use of polymeric carriers in controlled delivery of therapeutic agents, especially anticancer agents, has been pioneered by the group of Kopecek and, based on this approach, poly[N-(2-hydroxypropyl)methacrylamide] polymers with alendronate and oligo(aspartic acid)8 moieties were prepared. Oligo(aspartic acid)n with n > 6 is known to display significant HA binding (see Section VI) and this group reported a strong HA affinity of both types of polymers with no obvious major differences between the two constructs in vitro.105 Using fluorescein isothiocyanate (FITC)-labeled conjugates, both types of carriers were shown to be targeted to bone after intravenous injection in mice (based on FITC fluorescence obtained at the bone tissue). As expected, the polymeric carriers were localized at the sites of high bone turnover and/or high vascular supply, consistent with such sites receiving the first line of exposure to systemic agents. Unlike the similarity in the in vitro HA affinities, however, the oligo(aspartic acid)8 conjugate appeared to exhibit a lower bone targeting than the alendronate conjugate for reasons not yet clear. A critical issue, namely the ability of these carriers to deliver a cargo to bones in an animal model, remains to be reported.
In an analogous approach, BPs were conjugated to the endogenous proteoglycan heparin as a means to target heparin-binding molecules to bone.106 Protein growth factors, such as basic fibroblast growth factor and bone morphogenetic protein-2, display a preferential affinity to heparin via electrostatic interactions involving cationic amino acid residues, and these proteins will be particularly suitable for heparin–BP based bone delivery, since direct modification of the proteins may compromise their bioactivity (see Section VI). As with other molecules, BP substitution on heparin imparted a strong HA affinity, and the heparin–BP conjugate was shown to facilitate binding of the growth factors to HA in vitro. The ability of the heparin–BP conjugates to deliver the growth factors to bone in vivo remains to be investigated.
A BP conjugate of fullerene (C60) was prepared as a carrier, in principle, of metallic radionucleotides, as well as other molecules that can be entrapped in/on fullerenes.107 The conjugate was constructed from 3-amino-3-carboxylpropane-1,1-bisphosphonate, and it was shown to exhibit an inhibitory activity on HA formation in vitro, suggesting an affinity of the construct to the mineral. Characterization of actual HA binding in vitro, and bone targeting in vivo remain to be reported with this class of carriers.
E. Photodynamic therapy
The bone targeting approach is likely to provide a beneficial option in photodynamic therapy at bone tissues, for example in the therapy of osteomyelitis,108 tumors at bone sites109 and periodontitis.110 Attempts at clinical application of this approach are in their early stages and are limited where surgical entry is justified,111 due to the low penetration capability of the activating illumination though the soft tissues. No BP-based targeting has been utilized for photodynamic therapy at bones to date. Preclinical research with conventional photoreactive molecules has indicated the feasibility of this approach after transcutaneous activation of the photosensitizer. The conventional photosensitizers, such as methylene blue and acridine orange, do have some bone-seeking capability since they accumulate sufficiently at bones for effective activation,108–110 but their targeting efficiency as compared to the BPs is not known. Concentrating the photosensitizers at bones after BP-mediated targeting should allow a more effective therapeutic effect after photoactivation.VI. Design of proteins for bone delivery
Several endogenous proteins exhibit a significant affinity to biological apatite, so that their interactions with mineralizing surfaces serve to facilitate or inhibit the mineral formation process in vivo. The protein structural features responsible for mineral affinity are being continually elucidated. Below we review these structural features, since these features will help us to understand the interactions of exogenous molecules with bone, and might ultimately be utilized as a means for bone targeting (i.e., replace BPs in targeting efforts). However, clinically-useful recombinant proteins do not effectively target bone, and BP substitution on proteins has been the preferred method for protein targeting to bone. We review BPs suitable for chemical conjugation to proteins in this section, and summarize the experience accumulated with BP-mediated targeting of proteins to bone.A. Bone affinity of proteins
The amphoteric nature of bone mineral presents a complex mosaic of charges to proteins due to Ca2+, PO43−, OH− and other ill-defined substituents formed under physiological conditions. Such ionic species provide ample sites for binding of proteins via electrostatic interactions. The affinity of specific proteins for HA is the basis for influencing the nucleation, orientation, and growth of bone apatite under physiological conditions,112 ultimately regulating mineralization to suit the needs of an organism. The interactions of proteins with HA were first probed in 1956 under chromatographic conditions by Tiselius et al.,113 revealing the electrostatic nature of the underlying interactions. Gorbunoff and Timasheff114–116 subsequently conducted a comprehensive investigation of HA binding using an extensive set of proteins whose isoelectric points (pI) ranged from 3.5 to 11.0. Basic proteins (high pI) were proposed to bind to HA primarily via non-specific electrostatic interactions between the –NH3+ groups and anionic moieties on the HA surface, while acidic proteins (low pI) were bound specifically by complexation of their –C(O)–O− moieties to Ca2+ sites on HA. A concentrated charge density was necessary for a significant protein binding, and diffuse distribution of the charged groups on a protein generally weakened the HA binding (see below). The binding mediated by the –C(O)–O− groups was proposed to be rather specific, since replacement of –C(O)–OH by –S(O)(O)–OH obliterated the binding.105 Both the ionic composition and the pH of the immediate environment influenced the interactions of proteins with HA. For acidic proteins, HA binding is enhanced in the presence of Ca2+ ions in the medium,117 and elution of the acidic proteins is difficult to achieve even with the Ca2+ concentration as high as 3 M. In addition to direct effects on HA binding, Ca2+ in solution was proposed to facilitate HA binding by inducing an optimal configuration of protein α-helixes most suitable for HA binding,118 which was observed in circular dichroism spectra for an analog of matrix Gla protein.119 The α-helical conformation was proposed to act as a scaffold, aligning the charged residues of proteins in a complementary pattern to the Ca2+ ions in a hydroxyapatite crystal lattice. The PO43− in solution suppresses the HA binding of acidic proteins, possibly due to competitive binding of the phosphate ions with the Ca2+ on HA. On the other hand, the basic proteins are eluted by cationic ions Na+, K+, and especially Mg2+ and Ca2+.120 Acidic proteins generally display increased HA binding with decreasing pH. Since protein –C(O)–OH groups will be less ionized at lower pH, and, hence, display a lower propensity towards cationic moieties in HA, this pH effect was likely due to an increased hydrogen bonding interaction between acidic proteins and the HA surface and/or an overall decrease in the collective surface charge of HA (i.e., protonation of PO43− to HPO42− at lower pH).121 Also the possible change in the conformation of the proteins (i.e., more favorable binding configuration at lower pH),122,123 and stronger HA interactions mediated by the protein –NH3+ groups (i.e., due to the higher extent of amine protonation at lower pH) are all likely factors contributing to enhanced protein binding at lower pH.An active area of mineralization research has focused on elucidating protein structural motifs responsible for HA binding, with the expectation that the optimal distribution of “point” interactions imparts a unique, and stronger mineral affinity as compared to the simple additive effect of the interacting components.124–129 Consecutive aspartic acid residues found in osteopontin, and consecutive glutamic acid residues found in osteonectin and bone sialoprotein are two specific protein motifs shown to concentrate anionic groups for a superior mineral affinity.124 A minimum length of six anionic residues was necessary, and poly(aspartic acid) was shown to be a superior for HA binding.130 This simple sequence of consecutive amino acids has been utilized to facilitate binding of RGD-peptides (arginine–glycine–aspartic acid peptides involved in cell-surface integrin binding) to HA,131,132 as well as in drug delivery efforts to design model drugs with high affinities to bone mineral.133
In the absence of consecutive aspartic/glutamic acid residues, post-translational modification of proteins with acidic moieties provides an alternative means for imparting mineral affinity. Phosphorylation of serine/tyrosine imparts a high affinity to proteins by virtue of phosphate binding to the mineral, analogous to pyrophosphate binding to HA. Phosphophoryn binding to bone is a well-known example where dephosphorylation of specific protein residues reduces its mineral binding.134 Partial dephosphorylation of bovine osteopontin with alkaline phosphatase also reduced its ability to bind HA.135 Phosphorylated sequences may act in concert with other acidic residues for HA binding; using several isoforms of salivary statherins, the N-terminal sequence Asp–pSer–pSer–Glu–Glu136,137 was found to be responsible for the HA affinity. The phosphorylated serines in this case provided the appropriate high-charge density in the absence of long acidic amino acid repeats, and induced a helical conformation conducive for HA binding. Several uncharged polar residues (e.g., glutamine, tyrosine and threonine) adjacent to the Asp–pSer–pSer–Glu–Glu sequence additionally participated in H-bonding interactions with the HA surface. γ-Carboxylation of glutamic acid residues also enhances mineral affinity. In a recent model of HA binding,138 osteocalcin was found to display 3 α-helical regions, where 3 glutamic acid residues in helix α-1 were γ-carboxylated to create an extensively anionic surface, and in conjunction with an adjacent aspartic acid, coordinated 5 Ca2+ ions on HA surfaces. The spatial arrangement of the Ca2+ binding moieties on osteocalcin was well-aligned with the Ca2+ arrangement in a synthetic analogue of HA (note that the exact arrangement of Ca2+ ions in bone mineral might be significantly different from this HA model). Other post-translation modifications, such as carbohydrate moieties, sulfation and sialic acid addition, may also contribute to increased bone binding (as observed after comparison of native bone sialoprotein, and unmodified bone sialoprotein expressed in Escherichia coli without post-translational modification123,139). Gericke et al.140 suggested that osteopontin, whose binding propensity was usually ascribed to aspartic acid-rich sequences, incorporates phosphorylated sequences that caused alternations in protein conformation (detected by Fourier transform-infrared spectroscopy, FT-IR) to influence the interaction of osteopontin with HA. Finally, transglutaminase-sensitive sequences present in osteopontin can be crosslinked to form an osteopontin-network, and provide another avenue for post-translational control of mineral affinity.141 Upon polymerization, a conformational change in osteopontin structure was observed with circular dichroism spectroscopy, suggesting a more-ordered structure for the osteopontin polymer as compared to the monomeric form of the protein.141 A more ordered structure was more likely to provide the HA binding motif by the spatial arrangement well-aligned with the HA lattice. It was suggested that the initiation of HA crystallization required protein aggregates rather than the single osteopontin monomers.142
The structural studies in recent years have clearly indicated the availability of several distinct acidic protein motifs for HA binding. It is likely that the strength and the specific protein binding sites on HA will depend on the details of the interacting motif. No information is available about the differences in the binding mode of the different motifs on biological apatite. A recent study, which utilized octacalcium phosphate as a model matrix, indicated poly(aspartic acid) interacted preferentially with Ca2+ ions exposed on the hydrated {100} surface parallel to the c-axis of the crystals.143 The phosphorylated proteins, on the other hand, interacted specially with the apatite-like motifs on the {010} surface through occupation of the exact lattice site of the crystal phosphate groups. The availability of both motifs in a single protein, such as osteopontin, is likely to enhance the affinity due to additive binding of the two motifs to different sites on apatite.
Relatively little is known about the structural motifs responsible for HA binding of basic proteins. It is generally accepted that the binding arises due to interactions between the protein –NH3+ and the anionic moieties of HA. Using site-directed mutagenesis of lysozyme (pI ∼ 11), where individual amino acids were replaced with alanine, Lys-1, Lys-13, Arg-14 and Arg-10 residues were found critical for HA contact, which were all located on the protein surface away from the active site of the enzyme.144,145 This preliminary study also suggested that cationic charges on a protein may form ‘clustered’ patterns on a protein site, possibly enhancing the strength of HA interactions reminiscent of anionic protein motifs.
Unfortunately, these mineral-interacting proteins do not have a medicinal value at the present time. On the other hand, the current recombinant proteins useful for medicinal purposes do not express a strong affinity to bone due to lack of structural motifs or lack of post-translational modification. One, then, is faced with engineering the proteins to impart a mineral affinity to make them useful in bone diseases. This is most effectively achieved with BP substitution.
B. Functional bisphosphonates for protein targeting to bone
To impart a mineral affinity to a protein, one needs to couple the BPs to proteins in aqueous media. Unlike the majority of BP constructs summarized here and in previous reviews,18–20 this consideration rules out the possibility of using BP esters that are not soluble under aqueous conditions, and whose de-esterification after protein coupling is likely to abolish the protein activity. High-temperature conditions, pH extremes, and exposure to highly reactive, non-specific reagents (e.g., free radicals) are not possible if one wishes to retain the activity of the proteins. Water-soluble BPs with readily-reactive functional groups need to be employed for this purpose. The three most common moieties for convenient protein chemistry are –NH2, –SH, and –C(O)OH groups; hence, BPs with these groups will be ideal for chemical schemes designed for protein substitution. The current schemes for synthesis of such BPs were previously provided,19 and below we summarize the strategies for modifying such groups for protein attachment.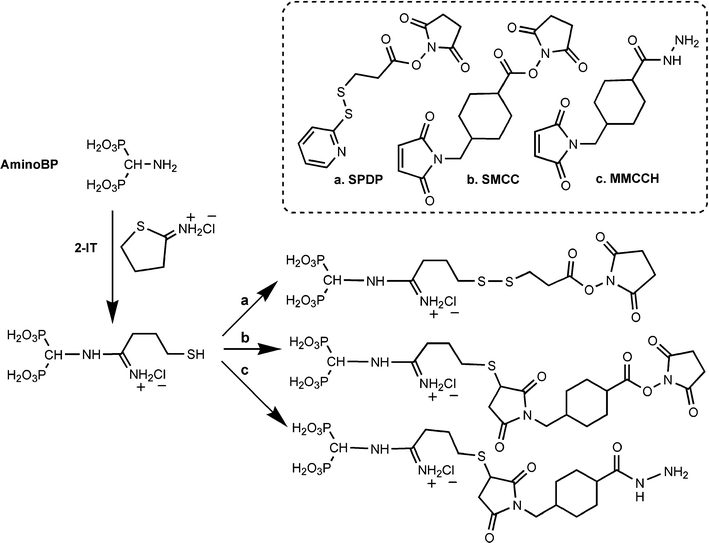 | ||
| Scheme 2 | ||
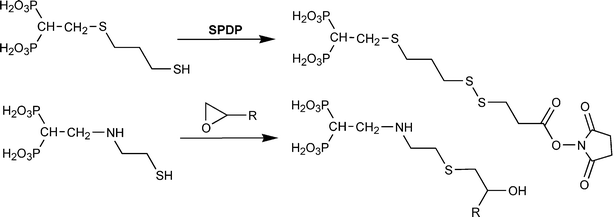
Unlike our experience, others were able to utilize the –NH2 groups of BPs for direct coupling to target molecules under aqueous conditions. 3-Amino-1-hydroxypropylidene-1,1-bisphosphonate (pamidronate) was utilized to prepare 2 (Fig. 4) after reaction with an NHS ester of a fluorescent dye.82 Similarly, 4-aminobutylene-1,1-bisphosphonate (alendronate) was reacted with an NHS ester of poly(ethylene glycol) before conjugation to a polymeric carrier.105 In both cases, (i) the BPs contained a methylene spacer between the –NH2 group and the α-carbon; (ii) the target compounds for substitution were much smaller than the proteins used in our studies, and; (iii) a relatively high pH (∼9) was needed for BP coupling. Relatively large proteins will favor hydrolysis of NHS esters before aminolysis (a ∼100-fold less reactivity towards protein amines is typical for NHS-esters149), and such a high hydrolysis rate could have been the reason for lack of protein coupling in our hands. As an alternative to NHS esters, 4-aminobutylene-1,1-bisphosphonate was capable of coupling to a tetrafluorophenol ester of a short peptide, triglycine, leading to a BP–(glycine)3 conjugate with an amide linkage.82 The feasibility of –NH2 coupling to aldehyde groups has also been reported,150 but the same reaction (using an oxidized protein and 1-aminomethylene-1,1-bisphosphonate) was not effective in our hands.
 | ||
| Scheme 3 | ||
The thiol groups of BPs are also reactive with epoxides under neutral pH conditions (Scheme 3).151 An amine group at the β-position was found necessary for this reaction, since an –SH linked to the α-carbon of BPs with an aliphatic chain was unreactive with the epoxide moieties. The conjugates will yield a thioether in the tether, which is expected to be more stable than the disulfide linkages. This chemistry was successfully used to graft thiol-BPs onto biological matrices (i.e., heart valves151), and it should be straightforward to adopt it with epoxide-containing linkers for modification of soluble proteins.
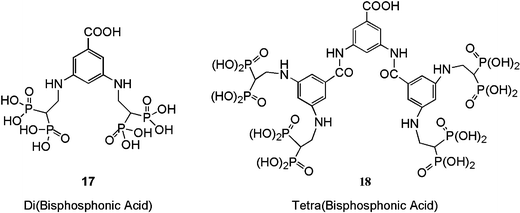 | ||
| Fig. 8 Novel dendritic BP-containing compounds. The compounds contained a single –COOH moiety for protein coupling, and either two (17) or four (18) BPs. | ||
C. Current attempts for protein delivery to bone with BPs
Proteins produced in large quantities by recombinant techniques have provided unique possibilities for treatment of bone diseases.19 As with most drugs, proteins do not seek bone once introduced into the blood stream. Bone targeting is especially vital for therapeutic proteins since they are ubiquitous modulators of cellular activity, and non-specific distribution of the proteins extra-skeletally will inevitably lead to undesirable activities in these tissues. The feasibility of protein targeting to bone was demonstrated in 2002,155 and recent studies are beginning to yield important information about the effectiveness of various bone targeting approaches, as well as the challenges ahead. Unlike conventional drugs, which are relatively small in size (<1 kDa), proteins pose a special challenge for BP-based delivery since bioactive proteins are relatively large (>5 kDa, and as large as 150 kDa for antibody-based therapeutics). A one-to-one construct (i.e., a single therapeutic molecule linked to a single BP), typical of small molecular therapeutics, is not expected to yield effective bone targeting for proteins. Hence, all proteins used in the bone targeting studies were substituted with multiple copies of BPs.| Ref. | Protein | Linker | Substitution | Route | 3–6 Hours | 1 Day | 2–4 Days | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Tibia | Femur | Tibia | Femur | Tibia | Femur | |||||
| a The results from the animal studies were grouped into 3 time points (initial 3–6 h, 1 day, and 2–4 days post-injection), and summarized as the ‘targeting efficiency’ (defined as the bone delivery of the BP-conjugate divided by the bone delivery of the native protein) at two select bone sites (tibia and femur). The proteins studied were bovine serum albumin (BSA), lysozyme (LYZ), bovine fetuin, bovine non-specific IgG, and E. coli-derived human bone morphogenetic protein-2 (BMP-2). The crosslinkers used for BP conjugations were N-succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC), 4-(maleimidomethyl)cyclohexane-1-carboxyl hydrazide (MMCCH), or 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide–N-hydroxysuccinimide (EDC–NHS). The amino-BP (aBP) was used after thiolation with 2-IT, as shown in Scheme 2. The conjugates were administered by either intravenous (IV), subcutaneous (SC), or intraperitoneal (IP) routes. * indicates testing in ovariectomized (osteopenic) rats. UP: Unpublished. | ||||||||||
| 155 | BSA-aBP | SMCC | 11.0 | IV | 2.2 | 2.0 | 3.7 | 3.7 | ||
| 155 | BSA-aBP* | SMCC | 11.0 | IV | 2.2 | 3.0 | 7.5 | 5.9 | ||
| 155 | BSA-aBP | SMCC | 11.0 | SC | 2.5 | 1.9 | ||||
| 155 | LTZ-aBP | SMCC | 3.9 | IV | 4.1 | 5.3 | 5.6 | 3.7 | ||
| 155 | LTZ-aBP | SMCC | 3.9 | SC | 3.1 | 4.5 | ||||
| 158 | Fetuin-aBP | SMCC | 6.1 | IV | 0.9 | 1.0 | 0.9 | 0.6 | 0.6 | 0.8 |
| 158 | Fetuin-aBP | SMCC | 6.1 | IP | 0.7 | 0.6 | ||||
| 158 | Fetuin-aBP | MMCCH | 7.1 | IV | 1.0 | 1.0 | ||||
| 158 | Fetuin-diBP | EDC–NHS | 1.2 | IV | 1.8 | 2.1 | ||||
| 153 | BSA-diBP | EDC–NHS | 2.6 | IV | 3.7 | 2.9 | ||||
| 154 | BSA-tetraBP | EDC–NHS | 3.6 | IV | 4.1 | 4.7 | ||||
| 154 | BSA-tetraBP | EDC–NHS | 3.6 | IP | 3.4 | 3.7 | ||||
| 153 | IgG-diBP | EDC–NHS | 6.3 | IV | 2.6 | 1.5 | ||||
| UP | BMP-2-aBP | SMCC | ∼3 | IV | 2.8 | 4.3 | ||||
The targeting efficiency appeared to increase with the assessment time after systemic administration (Table 1; except for conjugates that did not yield bone targeting at the initial time point). The reason behind this observation was the better retention of the BP-conjugate at the bone tissue as compared to the native protein. Presumably, the substituted BPs ensured a longer duration of binding between the administered conjugates and the bone. If this is the case, a direct correlation between the extent of substitution and the in situ residence time was expected. This concept was recently put to the test in an implant model where albumins with variable degrees of BP substitution were implanted with two types of mineral matrices, a synthetic HA and a silicone-substituted/sintered HA. No relationship between the rate of protein loss from the matrices and the extent of BP substitution was evident in this study (Fig. 9). This result suggests that the rate of protein loss from an implanted mineral matrix is independent of the extent of BP substitution. If the extent of BP substitution does not contribute to the rate of protein loss, interactions between the protein molecule and the mineral matrix might be alternatively responsible for controlling the conjugate loss from the mineral implants. This study was performed at an extra-skeletal (subcutaneous) site, and it will be important to determine whether the protein loss is still independent on the extent of BP substitution at bone sites. This study, in addition to the studies outlined in Table 1, clearly highlighted steady loss of proteins from the bound mineral matrix even after BP conjugation; it appears that the proteins do not simply get ‘glued’ to the mineral matrix after BP conjugation, but rather slowly released from such a matrix with a half-life in the order of weeks. The observed desorption half-lives were significantly shorter than the loss kinetics exhibited by the BPs at bone tissues in similar experimental models, where terminal half lives in the order of months to years are typical (see ref. 157 for a review).
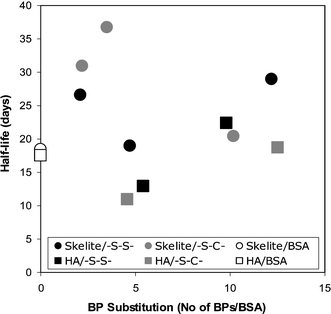 | ||
| Fig. 9 In situ half-life of two closely related BP conjugates of BSA in a rat subcutaneous implant model. The conjugates were prepared with thiolated amino-BP, and by using either thioether (–S–C–; via SMCC) or disulfide (–S–S–; via SPDP) linkages. Several conjugates with variable BP substitution numbers were implanted with synthetic HA and Skelite™ (a silicon rich, high-temperature sintered HA), and explanted over a 10-day period, as described in ref. 156. The loss of each protein from the implants was fitted with a one-compartmental decay model (y = Ae−mt), and the half-life of protein loss (t½) was determined by t½ = 0.691 m−1. No clear relationship between the t½ and the nature of the protein was evident. The t½ was also not affected by the extent of BP substitution on BSA. | ||
The natural consequence of increased protein delivery to bones after systemic administration should be a reduced delivery to extra-skeletal tissues. This is an important consideration that currently limits the clinical progress of a range of recombinant proteins. Reduced extra-skeletal delivery of BP-conjugates is shown to be the case in some of our studies,155 but not all.158 Undesirable changes in the physicochemical properties of proteins as a result of BP conjugation might be one reason for this observation. For example, reduced solubility in biological fluids, propensity for aggregation especially with cationic ions, or association with specific extracellular matrix molecules may result in non-specific delivery of conjugates to other tissues. A more rapid clearance of BP-conjugates, or a newly acquired cellular interaction/uptake characteristic might be other factors that might have contributed to increased delivery to other sites. It is likely that the desired inverse relationship between bone and extra-skeletal delivery is going to be protein specific, and the current experience is simply not extensive enough to elucidate this issue. Even if a reduced extra-skeletal delivery is achieved, the extent of this reduction might not be significant. Using hypothetical targeting efficiencies of 5–20 for BP conjugates (i.e., 5–20-fold increased delivery to bones as a result of BP conjugation), the reduction in extra-skeletal delivery will be ∼10% when bone delivery of the native protein is ∼1% (Fig. 10). The reduction in extra-skeletal delivery becomes significant (i.e., ∼50%) for proteins (i) that are already targeted to bones without the need for BP substitution, or (ii) that display a substantially high targeting efficiency (>10) as a result of BP conjugation. Our unpublished data in rodent models indicated a bone delivery in the range of 0.1–0.5% of the administered dose for the proteins used in Table 1, so that expecting an extra-skeletal benefit at this level of targeting might not be realistic. This analysis indicates that if the therapeutic administration of a recombinant protein calls for a single dose, the increased targeting to bones is not likely to cause a reduction in extra-skeletal delivery significant enough (say >50%) to minimize the protein activity at these sites. Alternatively, if the increased delivery can cause a corresponding reduction in the efficacious dose (e.g., 10-fold increased delivery causes 10-fold reduction in efficacious dose—assuming the therapeutic activity is linearly dependent on local protein concentration), the benefit of bone targeting will manifest itself in the possibility of reducing the administered dose of the protein. In this case, the reduction in the extent of extra-skeletal delivery will be directly proportional to the reduction in the administered protein dose.
 | ||
| Fig. 10 Hypothetical relationship between improved targeting efficiency (TE) as a result of BP conjugation and reduction in extra-skeletal delivery of a protein. The analysis assumes a simple mass balance where the improved delivery leads to reduced delivery to extra-skeletal sites. | ||
| R1SH → R1S− + H+ |
| ●–SS–BP + R1S− → {●–SS–R1 + −S–BP} or {●–S− + R1–SS–BP} |
| {−S–BP + H+ → HS–BP} or {●–S− + H+ → ●–SH} |
Such a cleavage mechanism may lead to either a thiol-adduct of the protein (●–SS–R1), or preferably liberation of the original protein (●–SH) if a protein cysteine thiol is utilized in the conjugate construction. In vitro studies did not indicate a strong dependence of cleavage rate on the nature of the cleaving thiol at equimolar concentrations of the major thiols, glutathione, cysteine, and homocysteine.159 Hence, cysteine, by virtue of its high concentration in serum, is likely to be the major cleaving thiol. Our recent studies on cysteine cleavage of albumin–SS–BP indicated the cysteine adduct of the protein to be the primary species (unpublished). Cysteine is a relatively small molecule, so that it is not likely to interfere with receptor binding of the proteins once an adduct is formed; nevertheless, this needs to be confirmed on an individual protein basis.
A special consideration for the BP conjugates is the need for successful cleavage after the conjugates are bound to a mineral surface.‡ Whereas cleavage in solution involves freely diffusable species, a disulfide linkage in the vicinity of a mineral surface must be adequately accessible to small thiols. This was shown to be the case for protein–BP conjugates,159 suggesting sufficient accessibility of the disulfide linkage for the thiolate (R1S−) attack. However, values for the relative cleavage rate of HA-bound conjugates were conflicting; whereas one study indicated a slower cleavage rate after the HA adsorption,147 a follow up study indicated a similar cleavage rate for the HA-bound and the soluble conjugates.159 This issue was further evaluated in a subcutaneous implant model in rats, by using HA matrices for protein implantation. Unlike the in vitro observations, the conjugate cleavage was not apparent in this model, in fact yielding no obvious differences in the implant binding of conjugates constructed with thioether and disulfide linkages.156 The actual disulfide cleavage, or loss of substituted BPs per se, could not be evaluated in vivo due to interference from endogenous compounds. However, two critical parameters dependent on the presence of BPs on proteins were used as an indirect measure of cleavage: first, the rate of protein loss from the implants was not affected whether the conjugates were disulfide-linked or thioether-linked, and secondly; a correlation between the original (i.e., pre-implantation) extent of BP substitution and implant binding was retained at different post-implantation times (1, 4, 10 days post-implantation). These two observations suggested that the disulfide cleavage rate in vivo might be slower than desired, and certainly slower than the in vitro conditions.
(1) A BP-conjugate encountering a surface is likely to experience additional surface interactions due to specific motifs and/or charged species on the protein surface. AFM measurements of protein interactions with specific ligands on a surface recently revealed multiple ‘clusters’ of interactions, whose strength varied depending on the distance between the ligand and the surface.160 These clusters represented different modes of interaction, including non-specific interactions of the ligand with the surface, as well as a specific affinity of the ligand to receptors on the surface. Our unpublished data indicated protein –NH2 groups participate in HA binding in our in vitro binding assay; e.g., modification of –NH2 groups of albumin with FITC resulted in a weaker affinity to HA and abolished the protein binding at high FITC substitutions (Fig. 12). Such protein–surface interactions secondary to BP binding to a mineral surface are not likely to take place when a long tether separates the protein from the surface. If this is the mechanism responsible for lower affinity of long-tethered BPs, then the secondary interactions between the protein and the mineral surfaces seem to have a stabilizing influence on the mineral binding of BP–protein conjugates.
(2) An alternative explanation is the increased affinity of the protein-attached BPs to the mineral due to a higher local concentration of the BPs arising from the spatial restriction of shorter tether lengths. It is possible to estimate the local concentration of a molecule tethered to a fixed location (i.e., an attachment site on a protein) by molecular dynamics simulations, taking into account the length as well as the rigidity of a tether.161 This analysis assumes a lack of steric effects, so that the BPs present in the vicinity of minerals are freely accessible for surface binding. Such a molecular dynamics simulation gave a good correlation between the radial density of BPs and mineral binding for a small set of protein–BP conjugates. Whether such a correlation could be extended to a general population of BP conjugates, and especially tether lengths approaching “zero” where the strongest mineral affinities are expected, remains to be determined.
(3) It is possible that the interaction of the tether with a mineral surface might also influence conjugate binding. The commonly used poly(ethylene glycol) linker105,152 results in a highly hydrophilic tether that is expected to have little interaction with surfaces, and to actively impede binding of BP-conjugates to a surface. Hydrophobic tethers are sometimes known to contribute to the binding of a ligand to its receptor,162,163 especially when desolvation at the binding site is an impeding force. Considering that bone apatite primarily interacts via electrostatic means, tethers with polar or charged moieties are more likely to contribute directly to the surface binding.
 | ||
| Fig. 11 A. Effect of tether length on accessibility of a BP–protein conjugate to an apatite surface. Long tethers with a larger volume of distribution (dashed box) could facilitate binding to apatite surfaces that pose topographical barriers to conjugate binding. B. Shortest tethers utilized in the construction of BP–protein conjugates. Tethers between the ε-NH2 (protein) and α-carbon (BP) are shown. Conjugates in i and ii were described in ref. 148 and 152, respectively. A hypothetical ‘zero tether’ between these two groups is also shown as a reference. | ||
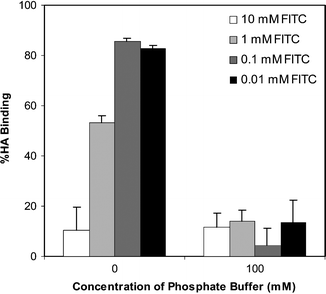 | ||
| Fig. 12 Effect of protein –NH2 modification on HA affinity. BSA was chemically modified at the –NH2 groups via different concentrations of FITC and HA binding of the modified BSA was determined in 0 (water) or 100 mM phosphate buffer (pH 7.0). As expected, protein binding to HA was minimal in 100 mM phosphate, but binding in 0 mM phosphate was significantly reduced as a function of FITC concentration used for BSA modification. Since native BSA binds strongly to HA (∼90%) in 0 mM phosphate, proteins whose –NH2 groups were extensively modified (>0.1 mM FITC) exhibited significant loss in HA affinity. | ||
On the other hand, the role of the R2 substituent on the α-carbon is more ambiguous. This substituent will be either the desired therapeutic agent (hence, no flexibility in its choice) or a tether that links the targeting moiety to a therapeutic agent of interest. The influence of tether hydrophobicity and length needs to be taken into account in that case. In constructing protein–BP conjugates, the BP side groups are usually modified for protein linkage and any differences in the mineral affinities of the original BPs do not necessarily translate into the imparted mineral affinity. For example, our studies indicated a significant difference in the in vitro HA affinity between 1-aminomethyl-1,1-bisphosphonatic acid and 2-(3-mercaptopropylsulfanyl)ethyl-1,1-bisphosphonic acid (Fig. 13A), but the conjugates prepared from these BPs did not lead to any observable difference in protein affinity in the same HA binding assay (Fig. 13B). Presumably the additional linkages introduced at the α-carbon dominated over the initial differences in the mineral affinities of these two BPs. Interestingly, proteins derivatized with the di-BPs and tetra-BPs did not exhibit a particularly superior HA affinity in vitro as compared to conjugates prepared with the conventional BPs.153,154 However, the bone targeting ability, especially for the tetra-BP conjugates, was superior after systemic administration in animals based on an equivalent number of BP molecules per protein substitution. We are therefore interpreting our in vitro HA binding results with caution.
 | ||
| Fig. 13 Effect of R2 side-group substituent on HA binding in vitro. A. Comparison of an amino-BP (1-aminomethylene-1,1-bisphosphonate, Scheme 2) and a thiol-BP (2-(3-mercaptopropylsulfanyl)ethyl-1,1-bisphosphonic acid, Scheme 3) binding to HA in different phosphate buffers. B. Correlation between the HA binding (in 100 mM phosphate) and the extent of amino-BP and thiol-BP substitution. Note that, despite differences in BP binding to HA, the conjugate binding did not depend on the nature of the substituent. Details of the experimental conditions were described in ref. 147. | ||
VII. Synopsis and future avenues of research
Given the scope of molecular species successfully designed for bone delivery, numerous musculoskeletal diseases are expected to benefit from bone-seeking medicinal agents. We are beginning an era where both therapeutic and diagnostic agents are being designed from first principles. Rather than screening efforts, research efforts are directed towards identifying molecules with a specific performance characteristic, and further engineering them for delivery specifically to bone. The current research in this field is at its infancy. Limited studies on chemical development or pharmacological assessment in preclinical animal models have been conducted. Protein targeting to bone is especially a new endeavor. The reported studies on this topic are limited, and do not fully probe the performance of reported BP–protein conjugates. Attention needs to be paid to several chemical aspects of BP-based agents for developing improved medicinal agents.Stability of chemical tether in bisphosphonate conjugates
The stability of the linkage between a BP and a therapeutic agent is expected to influence the in situ actions of the agent. A linkage with controlled degradation will enable release of an agent from the BP bound to the bone mineral surface, and allow binding of a drug to its receptor without steric hindrance. Although both of these premises are generally desirable, their validity remains to be demonstrated. With regards to free accessibility of agents to cell receptors, in vitro cell assays should be capable of exploring whether a drug in its BP-conjugate form is as potent as the free drug. With regards to release from bone mineral, BPs bound to bone mineral will be released into the local milieu during normal bone turnover (although natural turnover is likely to result in slower release than a rapidly degrading linkage). Studies with small molecules indicated cleavable conjugates could be based on several types of linkages, including ester, thioester, and amide linkages, and, in the case of esters, moieties adjacent to esters influenced the cleavage rate.18 The discouraging results with disulfide-linked protein–BP conjugates call for a better understanding of (i) rates of linkage cleavage in situ, and (ii) factors controlling protein desorption from minerals.Tailored bisphosphonates and other bone-seeking molecules
Protein–BP conjugates are composed of a small targeting moiety (<1 kDa) and a large bioactive molecule (10–200 kDa). Accordingly, multiple copies of BPs will need to be conjugated to each protein to impart a significant bone affinity. The accumulated data indicated the possibility of utilizing several sites on proteins for BP attachment, as well as distinct linkers for BP coupling. To identify the most effective strategy, BPs with different functional groups for diverse coupling reactions will need to be pursued, along with BPs with higher mineral affinities. If conventional BPs were to be used in bone targeting, the R1 substituent is likely to be –OH or –NH2 given their superior efficacy in bone targeting. The R2 substituents are likely to be short and hydrophilic, but their exact nature might be dictated by the chemical scheme for coupling to the payload. There might be opportunities to add functional moieties to the R2 chain that display a strong affinity to HA, such as –P(O)–(OH)2, –C(O)–OH, and –NH2 groups. These functional groups can act in concert with the substituted BPs in facilitating the conjugate retention on bone apatite.The alternative to conventional BPs is the molecules that bear multiple of copies of BPs; with a single attachment site, the extent of protein modification could be minimized by using such molecules. The dendritic BPs reported by the authors' group are exemplary for this effort, but the polymeric carriers in ref. 105 could also serve the same purpose. It is currently not known what other functional groups could substitute for BPs. One might imagine dendrimers with a high density of bone-binding moieties (see Section II) as a substitute for BP-bearing dendrimers. It is likely that the synthesis of the former molecules might be more convenient if the functional groups are simpler than the BPs.164 However, it must be stated that the studies that elucidated the effectiveness of these functional groups typically utilized simple binding conditions with no competing molecules, and relatively pure HA/bone apatite (i.e., without the components of the extracellular matrix). Will the effectiveness of these functional groups be retained under more-demanding physiological conditions? Will these functional groups be effective in seeking bone, which is a distinctly different process from bone binding? The answer for these questions is an unequivocal ‘yes’ in the case of BPs, but unknown in the case of molecules with a rich complement of bone-binding groups. Exploration of these issues might, nevertheless, lead to novel, dendritic bone-seeking molecules.
The current state of research does not provide any indication whether BPs are ideal agents for bone targeting. Although widely successful in delivering molecules to bones, the non-metabolizable nature of BPs is a concern especially if a medicinal agent needs to be administered on a regular basis. It might be possible to utilize mineral-binding protein motifs as a guide to develop more physiological molecules for bone targeting. Poly(aspartic acid) and poly(glutamic acids) are prototypical molecules for this reason, where the mineral affinity is afforded by totally metabolizable and endogenous moieties. In vivo targeting for small molecules, FITC98 and 17ß-estradiol,99 and recently a polymeric macromolecule,105 was successfully demonstrated using poly(amino acids). γ-Carboxyglutamic acid, phosphorylated serines/tyrosines, and lysine/arginine in an α-helix are other elements found in protein motifs that may be assembled into bone-seeking agents. Their potential to be physiologically metabolized with no trace of targeting moiety left behind makes them worthwhile to explore.
Generic bone carriers
An exciting area beginning to be explored is the development of bone-specific carriers that complex the medicinal agent without the need for its direct modification. The proof-of-principle for this approach has been already demonstrated with the delivery of radioisotopes and other imaging agents. In these cases, the bone-seeking BP moiety was separated from the carrier for the medicinal agent, so that the bone targeting efficiency of the BP was not compromised. Similar work with therapeutic agents will have a major impact. This goal will be more difficult to achieve since therapeutic molecules are larger in size, and designing a molecular host for them is not as straightforward as the simple metal-chelators. Nevertheless, endogenous molecules such as heparin and antibodies might be suitable to act as binders of larger therapeutic agents, and nanocarriers (e.g., polymeric or micellar) might be utilized for encapsulation of small molecular drugs. In these cases, it might be possible to isolate the payload from the bone-seeking agent, and independently engineer each component for an optimal performance. The developed systems are likely to be truly ‘generic’, in that a designed system should be able to deliver a range of therapeutic agents rather than molecule-specific that is often the case with current BP-conjugates.In conclusion, BPs have been unequivocally demonstrated to enhance delivery of numerous molecules to osseous tissue compared to administration of a therapeutic agent without any targeting mechanism. This is the first requirement for a ‘magic bullet’ for bone diseases. The fact that molecules with very different physicochemical properties were successfully utilized for bone delivery indicates the utility of BPs to serve as universal carriers of bone therapeutics. Given the spectrum of potential protein therapeutics, significant efforts from numerous academic investigators and, especially, industrial scientists will be required for full exploration of the potential of the BP-mediated bone targeting. It will be important to determine the relative bone targeting efficiency for a large number of proteins, so that further structure–function relationships could be pursued for targeting efficiency in terms of protein physicochemical features. In addition to skeletal activity, non-skeletal effects of therapeutic interventions (especially for proteins) need to be fully documented since it is the latter that will ultimately prove the benefit of BP-based targeting. Given the promise of the bone targeting approach, a more concerted effort in this area is expected in the near future, so as to overcome the current challenges and generate novel pharmaceutical agents specifically for musculoskeletal diseases.
Acknowledgements
The research in the authors' laboratory was supported by operating grants from the Canadian Institutes of Health Research (CIHR) and the Whitaker Foundation. Infrastructure support was provided by the Canadian Foundation for Innovation (CFI) and the Alberta Heritage Foundation for Medical Research (AHFMR). The authors thank our co-workers S. A. Gittens, J. E. I. Wright, and C. Kucharski for their contributions to our studies. We acknowledge the valuable input of numerous collaborators, including Dr Ronald F. Zernicke (Faculty of Kinesiology, University of Calgary), Dr Shelley Winn (Department of Surgery, Oregon Health and Sciences University), John R. Matyas (Department of Cell Biology, University of Calgary), and Dr Raimar Loebenberg (Faculty of Pharmacy & Pharmaceutical Sciences, University of Alberta).References
- Principles of Bone Biology, ed. J. P. Bilezikian, L. G. Raisz and G. A. Rodan, Academic Press, San Diego, USA, 2nd edn, 2002 Search PubMed.
- S. F. Siconolfi, R. J. Gretebeck, W. W. Wong, S. S. Moore and J. H. Gilbert, III., J. Appl. Physiol., 1998, 85, 1578 Search PubMed.
- D. Stepensky, L. Kleinberg and A. Hoffman, Clin. Pharmacokinet., 2003, 42, 863 Search PubMed.
- H. Fleisch and S. Bisaz, Am. J. Physiol., 1962, 203, 671 CAS.
- H. Fleisch, J. Maerki and R. G. G. Russell, Proc. Soc. Exp. Biol. Med., 1966, 122, 317 Search PubMed.
- H. Fleisch, R. G. G. Russell and F. Straumann, Nature, 1966, 212, 901 CrossRef CAS.
- H. Fleisch, D. Schibler, J. Maerki and I. Frossard, Nature, 1965, 207, 1300 CrossRef CAS.
- H. Fleisch, R. G. G. Russell, S. Bisaz, P.A. Casey and R. C. Muhlbauer, Calcif. Tissue Res., 1968, S2, 10 Search PubMed.
- H. Fleisch, R. G. G. Russell and M. D. Francis, Science, 1969, 165, 1262 CAS.
- H. Fleisch, R. G. G. Russell, B. Simpson and R. C. Muhlbauer, Nature, 1969, 223, 211 CrossRef CAS.
- M. D. Francis, R. G. G. Russell and H. Fleisch, Science, 1969, 165, 1264 CAS.
- C. A. Bassett, A. Donath, F. Macagno, R. Preisig, H. Fleisch and M. D. Francis, Lancet, 1969, 2, 845 CrossRef CAS.
- H. P. Pendergr, M. S. Potsaid and F. P. Castrono, Radiology (Oak Brook, IL, US), 1973, 107, 557 Search PubMed.
- E. B. Silberstein, E. L. Saenger, A. J. Tofe, G. W. Alexander, Jr. and H. M. Park, Radiology (Oak Brook, IL, US), 1973, 107, 551 Search PubMed.
- I. Fogelman, Eur. J. Nucl. Med., 1980, 5, 473 CAS.
- M. Eisenhut, J. Nucl. Med., 1984, 25, 1356 CAS.
- F. Wingen, H. Sterz, H. Blum, H. Moller, W. Pittermann, B. L. Pool, H. J. Sinn, H. Spring and D. Schmahl, J. Cancer Res. Clin. Oncol., 1986, 111, 209 CrossRef CAS.
- H. Uludag, Curr. Pharm. Des., 2002, 8, 1929 Search PubMed.
- S. A. Gittens, G. Bansal, R. F. Zernicke and H. Uludag, Adv. Drug Delivery Rev., 2005, 57, 1011 CrossRef CAS.
- H. Hirabayashi and J. Fujisaki, Clin. Pharmacokinet., 2003, 42, 1319 Search PubMed.
- Adv. Drug Delivery Rev., ed. D. Wang, S. C. Miller and J. Kopecek, 2005, 57(7) Search PubMed.
- L. Widler, K. A. Jaeggi, M. Glatt, K. Muller, R. Bachmann, M. Bisping, A. R. Born, R. Cortesi, G. Guiglia, H. Jeker, R. Klein, U. Ramseier, J. Schmid, G. Schreiber, Y. Seltenmeyer and J. R. Green, J. Med. Chem., 2002, 45, 3721 CrossRef.
- W. F. deJong, Recl. Trav. Chim. Pays-Bas, 1926, 45, 445 CAS.
- A. L. Boskey, in Bone tissue engineering, ed. J. O. Hollinger, T. A. Einhorn, B. A. Doll and C. Sfeir, CRC Press, Boca Raton, FL, 2005, ch. 4, pp. 111–114 Search PubMed.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130 CrossRef CAS.
- L. Pramatarova, E. Pecheva, R. Presker, M. T. Pham, M. F. Maitz and M. Stutzmann, Eur. Cells Mater., 2005, 9, 9 Search PubMed.
- P. W. Brown and R. I. Martin, J. Phys. Chem. B, 1999, 103, 1671 CrossRef CAS.
- K. Kandori, S. Sawai, Y. Yamamoto, H. Saitoand and T. Ishikawa, Colloids Surf., 1992, 68, 283 CrossRef CAS.
- I. S. Harding, N. Rashid and K. A. Hing, Biomaterials, 2005, 26, 6818 CrossRef CAS.
- M. S. Lorraine and W. J. Landis, Appl. Phys. Lett., 1992, 61, 2610 CrossRef CAS.
- J. Kirkham, J. Zhang, S. J. Brookes, R. C. Shore, S. R. Wood, D. A. Smith, M. L. Wallwork, O. H. Ryu and C. Robinson, J. Dent. Res., 2000, 79, 1943 CrossRef CAS.
- J. Vandiver, D. Dean, N. Patel, W. Bonfield and C. Ortiz, Biomaterials, 2005, 26, 271 CrossRef CAS.
- K. Kandori, M. Mukai, A. Yasukawa and T. Ishikawa, Langmuir, 2000, 16, 2301 CrossRef CAS.
- P. X. Zhu, Y. Masuda and K. Koumoto1, J. Colloid Interface Sci., 2001, 243, 31 CrossRef CAS.
- D. A. Smith, S. D. Connell, C. Robinson and J. Kirkham, Anal. Chim. Acta, 2003, 479, 39 CrossRef CAS.
- S. Mafe, J. A. Manzencres, H. Reiss, J. M. Thomann and P. Gramawn, J. Phys. Chem., 1992, 96, 861 CrossRef CAS.
- J. Zhang, J. Kirkham, M. L. Wallwork, D. A. Smith, S. J. Brookes, R. C. Shore, S. R. Wood and C. Robinson, Langmuir, 1999, 15, 8178 CrossRef CAS.
- C. Robinson, S. Connell, J. Kirkham, R. Shore and A. Smith, J. Mater. Chem., 2004, 14, 2242 RSC.
- W. J. Shaw, A. A. Campbell, M. L. Paine and M. L. Snead, J. Biol. Chem., 2004, 279, 40263 CrossRef CAS.
- N. Bouropoulos and J. Moradian-Oldak, Calcif. Tissue Int., 2003, 72, 599 CrossRef CAS.
- M. L. Wallwork, J. Kirkham, J. Zhang, S. J. Brookes, R. C. Shore, S. R. Wood, O. Ryu, C. Robinson and D. A. Smith, Langmuir, 2001, 17, 2508 CrossRef CAS.
- M. L. Wallwork, J. Kirkham, H. Chen, S.-X. Chang, C. Robinson, D. A. Smith and B. H. Clarkson, Calcif. Tissue Int., 2002, 71, 249 CrossRef CAS.
- H. Chen, M. B. Holl, B. G. Orr, I. Majoros and B. H. Clarkson, J. Dent. Res., 2003, 82, 443 CrossRef CAS.
- H. Chen, Y. Chen, B. G. Orr, M. B. Holl, I. Majoros and B. H. Clarkson, Langmuir, 2004, 20, 4168 CrossRef CAS.
- M. J. Dallemagne, in Bone and teeth, ed. H. J. J. Blackwood. Pergamon Press, Oxford, UK, Session III, 1964, pp. 171–175 Search PubMed.
- C. Robinson, S. Connell, J. Kirkham, R. Shore and A. Smith, J. Mater. Chem., 2004, 14, 2242 RSC.
- M. J. Rogers, D. J. Watts, R. G. G. Russell, X. Ji, X. Xiong, G. M. Blackburn, A. V. Bayless and F. H. Ebetino, J. Bone Miner. Res., 1994, 9, 1029 CrossRef CAS.
- B. Y. Klein, H. Ben-Bassat, E. Breuer, V. Solomon and G. Golomb, J. Cell. Biochem., 1998, 68, 186 CrossRef CAS.
- R. G. Russell, M. J. Rogers, J. C. Frith, S. P. Luckman, F. P. Coxon, H. L. Benford, P. I. Croucher, C. Shipman and H. A. Fleisch, J. Bone Miner. Res., 1999, 14, 53 CAS.
- D. Fernández, D. Vega and A. Goeta, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m543 CrossRef.
- D. Fernández, D. Vega and A. Goeta, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m494 CrossRef.
- E. Van Beek, M. Hoekstra, M. V. D. Ruit, C. Löwik and S. Papapoulos, J. Bone Miner. Res., 1994, 9, 1875 CrossRef CAS.
- R. G. G. Russell, R. C. Muhlbauer, S. Bisaz, D. A. Williams and H. Fleisch, Calcif. Tissue Res., 1970, 6, 183 Search PubMed.
- S. H. Szajnman, E. L. Ravaschino, R. Docampo and J. B. Rodriguez, Bioorg. Med. Chem. Lett., 2005, 15, 4685 CrossRef CAS.
- E. Van Beek, L. C. Wik, I. Que and S. Papapoulos, J. Bone Miner. Res., 1996, 11, 1492 CrossRef CAS.
- M. A. Lawson, J. T. Triffitt, F. H. Ebetino, R. J. Phipps, D. J. White and R. G. G. Russell, Bone, 2005, 36(S2), S308.
- G. H. Nancollas, R. Tang, R. J. Phipps, Z. Henneman, S. Gulde, W. Wu, A. Mangood, R. G. G. Russell and F. H. Ebetino, Bone, 2006 DOI:10.1016/j.bone.2005.05.003.
- F. H. Ebetino, G. H. Nicollas, Z. Hennerman, P. Emmerling, R. J. Phipps and B. L. Barnett, Bone, 2005, 36(S2), S187.
- E. C. Lisic, M. Phillips, D. Ensor, K. L. Nash, A. Beets and F. F. Knapp, Nucl. Med. Biol., 2001, 28, 419 CrossRef CAS.
- H. Hirabayashi, T. Sawamoto, J. Fujisaki, Y. Tokunaga, S. Kimura and T. Hata, Pharm. Res., 2001, 18, 646 CrossRef CAS.
- G. Subramanian and J. G. McAfee, Radiology (Oak Brook, IL, US), 1971, 98, 192 Search PubMed.
- K. Wang, Li Allen, E. Fung, C. C. Chan, J. C. S. Chan and J. F. Griffith, Clin. Nucl. Med., 2005, 30, 655 CrossRef CAS.
- A. Thakorlal, D. C. Wong and R. J. Anderson, Australas. Radiol., 2005, 49, 238 CrossRef CAS.
- E. Kawamura, J. Kawabe, T. Hayashi, A. Oe, J. Kotani, K. Torii, D. Habu and S. Shiomi, Clin. Nucl. Med., 2005, 30, 351 CrossRef.
- G. C. Mackie, Clin. Nucl. Med., 2003, 28, 851 CrossRef.
- W. B. G. Macdonald and R. G. Troedson, Clin. Nucl. Med., 2001, 26, 455 CrossRef CAS.
- H. Kaida, M. Ishibashi, K. Baba, H. Nishida, K. Matsuoka and N. Hayabuchi, Br. J. Radiol., 2004, 77, 869 Search PubMed.
- H. Stevens, J. W. G. Jacobs, P. P. Van Rijk and J. M. H. De Klerk, Clin. Nucl. Med., 2001, 26, 389 CrossRef CAS.
- Y. Imanishi, Y. Mitogawa, M. Takizawa, S. Konno, H. Samuta, A. Ohsawa, A. Kawaguchi, M. Fujikawa, H. Sakaida, T. Shinagawa and H. Yanashita, Clin. Nucl. Med., 1999, 24, 511 CrossRef CAS.
- Y. Y. Ho and Y. H. Chan, Australas. Radiol., 2005, 49, 21 CrossRef CAS.
- Y.-S. An, J.-K. Yoon, M.-H. Lee, C.-W. Joh and S.-N. Yoon, Clin. Nucl. Med., 2004, 29, 723.
- W.-J. Shih, A.-M. Wang, P. A. Domstad, S.-R. Cho and F. H. DeLand, Eur. J. Nucl. Med., 1985, 11, 43 CrossRef CAS.
- K. Libson, E. Deutsch and B. L. Barnett, J. Am. Chem. Soc., 1980, 102, 2476 CrossRef CAS.
- G. R. Storey, L. Ridley and H. Van Der Wall, Clin. Nucl. Med., 2001, 26, 538 CrossRef CAS.
- W. A. Volkert and S. Jurisson, Top. Curr. Chem., 1996, 176, 123 CAS.
- J. M. O'Sullivan, V. R. McCready, G. Flux, A. R. Norman, F. M. Buffa, S. Chittenden, M. Guy, K. Pomeroy, G. Cook, J. Gadd, J. Treleaven, A. Al-Deen, A. Horwich, R. A. Huddart and D. P. Dearnaley, Br. J. Cancer, 2002, 86, 1715 CrossRef CAS.
- P. Pripatnanont, P. Vittayakittipong, U. Markmanee, S. Thongmak and T. Yipintsoi, Int. J. Oral Maxillofac. Surg., 2005, 34, 364 CrossRef CAS.
- S. Saeed, S. Haq, M. Sohaib and A. N. Khan, Clin. Nucl. Med., 2001, 26, 930–932 CrossRef CAS.
- M. Oguchi, K. Higashi, M. Taniguchi, H. Tamamura, T. Okimura and I. Yamamoto, Clin. Nucl. Med., 1998, 23, 479 CrossRef CAS.
- G. R. Storey, L. Ridley and H. Van Der Wall, Clin. Nucl. Med., 2001, 26, 538 CrossRef CAS.
- K. Ogawa, T. Mukai, Y. Arano, M. Ono, H. Hanaoka, S. Ishino, K. Hashimoto, H. Nishimura and H. Saji, Bioconjugate Chem., 2005, 16, 751 CrossRef CAS.
- A. Zaheer, R. E. Lenkinski, A. Mahmood, A. G. Jones, L. C. Cantley and J. V. Frangioni, Nat. Biotechnol., 2001, 19, 1148 CrossRef CAS.
- R. E. Lenkinski, M. Ahmed, A. Zaheer, J. V. Frangioni and S. N. Goldberg, Acad. Radiol., 2003, 10, 1159 Search PubMed.
- V. Kubicek, J. Rudovsky, J. Kotek, P. Hermann, L. Vander Elst, R. N. Muller, Z. I. Kolar, H. T. Wolterbeek, J. A. Peters and I. Lukes, J. Am. Chem. Soc., 2005, 127, 16477 CrossRef CAS.
- A. El-Mabhouh, C. Angelov, A. McEwan, G. Jia and J. Mercer, Cancer Biother. Radiopharm., 2004, 19, 627 CAS.
- M. Malet-Martino and R. Martino, Oncologist, 2002, 7, 288 Search PubMed.
- A. El-Mabhouh and J. R. Mercer, Appl. Radiat. Isot., 2005, 62, 541 CrossRef CAS.
- E. Arstad, P. Hoff, L. Skattebol, A. Skretting and K. Breistol, J. Med. Chem., 2003, 46, 3021 CrossRef.
- J. K. Woodward, R. E. Coleman and I. Holen, Anti-Cancer Drugs, 2005, 16, 11 CrossRef CAS.
- P. Clezardin, F. H. Ebetino and P. G. Fournier, Cancer Res., 2005, 65, 4971 CrossRef CAS.
- Y. Hamma-Kourbali, M. Di Benedetto, D. Ledoux, O. Oudar, Y. Leroux, M. Lecouvey and M. Kraemer, Biochem. Biophys. Res. Commun., 2003, 310, 816 CrossRef CAS.
- M. Sebbah-Louriki, B. M. Colombo, D. El Manouni, A. Martin, J.-L. Salzmann, Y. Leroux, G. Y. Perret and M. Crepin, Anticancer Res., 2002, 22, 3925 CAS.
- P. Herczegh, T. Baxton, J. C. McPherson, A. Kovacs-Kulyassa, P. D. Brewe, F. Sztaricskai, G. S. Stroebel, K. M. Plowman, D. Farcasiu and J. F. Hartmann, J. Med. Chem., 2002, 45, 2338 CrossRef CAS.
- T. B. Buxton, D. S. Walsh, S. B. Harvey, J. C. McPherson, III., J. F. Hartmann and K. M. Plowman, Br. J. Surg., 2004, 91, 1192 Search PubMed.
- T. Miyazaki, A. Sanjay, L. Neff, S. Tanaka, W. C. Horne and R. Baron, J. Biol. Chem., 2004, 279, 17660 CrossRef CAS.
- E. G. Arias-Salgado, S. Lizano, S. Sarkar, J. S. Brugge, M. H. Ginsberg and S. J. Shattil, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13298 CrossRef CAS.
- C. A. Metcalf, III, M. R. Van Schravendijk, D. C. Dalgarno and T. K. Sawyer, Curr. Pharm. Des., 2002, 8, 2049 Search PubMed.
- M. Missbach, M. Jeschke, J. Feyen, K. Muller, M. Glatt, J. Green and M. Susa, Bone, 1999, 24, 437 CrossRef CAS.
- W. Shakespeare, M. Yang, R. Bohacek, F. Cerasoli, K. Stebbins, R. Sundaramoorthi, M. Azimioara, C. Vu, S. Pradeepan, C. Metcalf, III, C. Haraldson, T. Merry, D. Dalgarno, S. Narula, M. Hatada, X. Lu, M. R. Van Schravendijk, S. Adams, S. Violette, J. Smith, W. Guan, C. Bartlett, J. Herson, J. Iuliucci, M. Weigele and T. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9373 CrossRef CAS.
- C. B. Vu, G. P. Luke, N. Kawahata, W. C. Shakespeare, Y. Wang, R. Sundaramoorthi, C. A. Metcalf, III, T. P. Keenan, S. Pradeepan, E. Corpuz, T. Merry, R. S. Bohacek, D. C. Dalgarno, S. S. Narula, M. R. Van Schravendijk, M. K. Ram, S. Adams, S. Liou, J. A. Keats, S. M. Violette, W. Guan, M. Weigele and T. K. Sawyer, Bioorg. Med. Chem. Lett., 2003, 13, 3071 CrossRef CAS.
- Y. Wang, C. A. Metcalf, III, W. C. Shakespeare, R. Sundaramoorthi, T. P. Keenan, R. S. Bohacek, M. R. Van Schravendijk, S. M. Violette, S. S. Narula, D. C. Dalgarno, C. Haraldson, J. Keats, S. Liou, U. Mani, S. Pradeepan, M. Ram, S. Adams, M. Weigele and T. K. Sawyer, Bioorg. Med. Chem. Lett., 2003, 13, 3067 CrossRef CAS.
- R. Sundaramoorthi, W. C. Shakespeare, T. P. Keenan, C. A. Metcalf, III, Y. Wang, U. Mani, M. Taylor, S. Liu, R. S. Bohacek, S. S. Narula, D. C. Dalgarno, M. R. Van Schravendijk, S. M. Violette, S. Liou, S. Adams, M. K. Ram, J. A. Keats, M. Weigle, T. K. Sawyer and M. Weigele, Bioorg. Med. Chem. Lett., 2003, 13, 3063 CrossRef CAS; Erratum in: Bioorg. Med. Chem. Lett., 2003, 13, 4519 Search PubMed.
- J. B. Wang, C. H. Yang, X. M. Yan, X. H. Wu and Y. Y. Xie, Chin. Chem. Lett., 2005, 16, 859 CAS.
- L. Lazzarato, B. Rolando, M. L. Lolli, G. C. Tron, R. Fruttero, A. Gasco, G. Deleide and H. L. Guenther, J. Med. Chem., 2005, 48, 1322 CrossRef CAS.
- D. Wang, S. Miller, M. Sima, P. Kopeckova and J. Kopecek, Bioconjugate Chem., 2003, 14, 853 CrossRef CAS.
- S. A. Gittens, K. Bagnall, J. R. Matyas, R. Lobenberg and H. Uludag, J. Controlled Release, 2004, 98, 255 CrossRef CAS.
- K. A. Gonzalez, L. J. Wilson, W. Wu and G. H. Nancollas, Bioorg. Med. Chem., 2002, 10, 1991 CrossRef CAS.
- S. K. Bisland, C. Chien, B. C. Wilson and S. Burch, Photochem. Photobiol. Sci., 2006, 5, 31 RSC.
- S. Burch, A. Bogaards, J. Siewerdsen, D. Moseley, A. Yee, J. Finkelstein, R. Weersink, B. C. Wilson and S. K. Bisland, J. Biomed. Opt., 2005, 10, 034011 CrossRef CAS.
- N. Komerik, H. Nakanishi, A. J. MacRobert, B. Henderson, P. Speight and M. Wilson, Antimicrob. Agents Chemother., 2003, 47, 932 CrossRef CAS.
- K. Kusuzaki, H. Murata, T. Matsubara, S. Miyazaki, A. Okamura, M. Seto, A. Matsumine, H. Hosoi, T. Sugimoto and A. Uchida, Anticancer Res., 2005, 25, 1225 CAS.
- S. G. Rees, D. T. H. Wassell, R. J. Waddington and G. Embery, Biochim. Biophys. Acta, 2001, 1568, 118 CrossRef CAS.
- A. Tiselius, S. Hjertén and O. Levin, Arch. Biochem. Biophys., 1956, 65, 132 CrossRef.
- M. J. Gorbunoff, Anal. Biochem., 1984, 136, 425 CrossRef CAS.
- M. J. Gorbunoff, Anal. Biochem., 1984, 136, 433 CrossRef CAS.
- M. J. Gorbunoff and S. N. Timasheff, Anal. Biochem., 1984, 136, 440 CrossRef CAS.
- V. Hlady and H. Füredi-Milhofer, J. Colloid Interface Sci., 1979, 69, 460 CrossRef CAS.
- P. V. Hauschka and S. A. Carr, Biochemistry, 1982, 21, 2538 CrossRef CAS.
- R. Wallin, D. Cain, S. M. Hutson, D. C. Sane and R. Loeser, Thromb. Haemostasis, 2000, 84, 1039 Search PubMed.
- K. Ohta, H. Monma, J. Tanaka and H. Eda, J. Mater. Sci.: Mater. Med., 2002, 13, 633 CrossRef CAS.
- C. Robinson, S. Connell, S. J. Brookes, J. Kirkham, R. C. Shore and D. A. Smith, Arch. Oral Biol., 2005, 50, 267 CrossRef CAS.
- W. M. Rathman, M. J. Van Zeyl, P. A. M. Van Den Keybus and R. A. Bank, J. Biol. Buccale., 1989, 17, 199 Search PubMed.
- A. V. N. Amerongen, C. H. Oderkerk and E. C. I. Veerman, J. Biol. Buccale., 1989, 17, 85 Search PubMed.
- R. Fujisawa, Y. Wada, Y. Nodasaka and Y. Kuboki, Biochim. Biophys. Acta, 1996, 1292, 53 CrossRef CAS.
- D. A. Pampena, K. A. Robertson, O. Litvinova, G. Lajoie, H. A. Goldberg and G. K. Hunter, Biochem. J., 2004, 378, 1083 CrossRef CAS.
- C. E. Tye, K. R. Rattray, K. J. Warner, J. A. R. Gordon, J. Sodek, G. K. Hunter and H. A. Goldberg, J. Biol. Chem., 2003, 278, 7949 CrossRef CAS.
- H. A. Goldberg, K. J. Warner, M. C. Li and G. K. Hunter, Connect. Tissue Res., 2001, 42, 25 CrossRef CAS.
- M. Wuttke, S. Müller, D. P. Nitsche, M. Paulsson, F. G. Hanisch and P. Maurer, J. Biol. Chem., 2001, 276, 36839 CrossRef CAS.
- H. A. Goldberg, K. J. Warner, M. J. Stillman and G. K. Hunter, Connect. Tissue Res., 1996, 35, 385 CrossRef CAS.
- F. Poumier, P. Schaad, Y. Haikel, J. C. Voegel and P. Gramain, J. Biomed. Mater. Res., 1999, 45, 92 CrossRef CAS.
- A. A. Sawyer, D. M. Weeks, S. S. Kelpke, M. S. McCracken and S. L. Bellis, Biomaterials, 2005, 26, 7046 CrossRef CAS.
- D. Itoh, S. Yoneda, S. Kuroda, H. Kondo, A. Umezawa, K. Ohya, T. Ohyama and S. Kasugai, J. Biomed. Mater. Res., 2002, 62, 292 CrossRef CAS.
- S. Kasugai, R. Fujisawa, Y. Waki, K. Miyamoto and K. Ohya, J. Bone Miner. Res., 2000, 15, 936 CrossRef CAS.
- R. Fujisawa, Y. Kuboki and S. Sasaki, Calcif. Tissue Int., 1986, 39, 248 CAS.
- A. L. Boskey, M. Maresca, W. Ullrich, S. B. Doty, W. T. Butler and C. W. Prince, Bone Miner., 1993, 22, 147 CrossRef CAS.
- N. Ramasubbu, L. M. Thomas, K. K. Bhandary and M. J. Levine, Crit. Rev. Oral Biol. Med., 1992, 4, 363 Search PubMed.
- P. S. Stayton, G. P. Drobny, W. J. Shaw, J. R. Long and M. Gilbert, Crit. Rev. Oral Biol. Med., 2003, 14, 370–376 Search PubMed; P. A. Raj, M. Johnson, M. J. Levine and G. H. Nancollas, J. Biol. Chem., 1992, 267, 5968–5976 CAS.
- Q. Q. Hoang, F. Sicheri, A. J. Howard and D. S. C. Yang, Nature, 2003, 425, 977 CrossRef CAS.
- J. T. Stubbs, III, K. P. Mintz, E. D. Eanes, D. A. Torchia and L. W. Fisher, J. Bone Miner. Res., 1997, 12, 1210 CrossRef.
- A. Gericke, C. Qin, L. Spevak, Y. Fujimoto, W. T. Butler, E. S. Sorensen and A. L. Boskey, Calcif. Tissue Int., 2005, 77, 45 CrossRef CAS.
- M. T. Kaartinen, A. Pirhonen, A. Linnala-Kankkunen and P. H. Mäenpää, J. Biol. Chem., 1999, 274, 1729 CrossRef CAS.
- M. T. Kaartinen, A. Pirhonen, A. Linnala-Kankkunen and P. H. Mäenpää, J. Biol. Chem., 1997, 272, 22736 CrossRef CAS.
- H. Füredi-Milhofer, J. Moradian-Oldak, S. Weiner, A. Veis, K. P. Mintz and L. Addadi, Connect. Tissue Res., 1994, 30, 251 CrossRef CAS.
- I. Aizawa, N. Koganesawa, A. Kamakura, K. Masaki, A. Matsuura, H. Nagadome, Y. Terada, K. Kawano and K. Nitta, FEBS Lett., 1998, 422, 175 CrossRef.
- K. Kandori, N. Horigami, K. Kobayashi, A. Yasukawa and T. Ishikawa, J. Colloid Interface Sci., 1997, 191, 498 CrossRef CAS.
- H. Uludag, N. Kousinioris, T. Gao and D. Kantoci, Biotechnol. Prog., 2000, 16, 258 CrossRef CAS.
- G. Bansal, J. E. I. Wright, S. Zhang, R. F. Zernicke and H. Uludag, J. Biomed. Mater. Res., 2005, 74, 618.
- S. Gittens, J. R. Matyas, R. Zernicke and H. Uludag, Pharm. Res., 2003, 20, 978 CrossRef CAS.
- H. Uludag, B. Norrie, N. Kousinioris and T. Gao, Biotechnol. Bioeng., 2001, 73, 510 CrossRef CAS.
- C. L. Webb, F. J. Schoen and R. J. Levy, Exp. Mol. Pathol., 1989, 50, 291 CrossRef CAS.
- I. S. Alferiev, J. M. Connolly and R. J. Levy, J. Organomet. Chem., 2005, 690, 2543 CrossRef CAS.
- S. A. Gittens, P. I. Kitov, J. R. Matyas, R. Loebenberg and H. Uludag, Pharm. Res., 2004, 21, 608 CrossRef CAS.
- G. Bansal, S. Gittens and H. Uludag, J. Pharm. Sci., 2004, 93, 2788 CrossRef CAS.
- G. Bansal, J. E. I. Wright, C. Kucharski and H. Uludag, Angew. Chem., Int. Ed., 2005, 44, 3710 CrossRef CAS.
- H. Uludag and J. Yang, Biotechnol. Prog., 2002, 18, 604 CrossRef CAS.
- J. E. I. Wright, S. A. Gittens, G. Bansal, P. I. Kitov, D. Sindrey, C. Kucharski and H. Uludag, Biomaterials, 2006, 27, 769 CrossRef CAS.
- J. H. Lin, Bone, 1996, 18, 75 CrossRef CAS.
- S. A. Gittens, G. Bansal, C. Kucharski, M. Borden and H. Uludag, Mol. Pharmacol., 2005, 2, 392 CrossRef CAS.
- S. Zhang, J. E. I. Wright, G. Bansal, P. Cho and H. Uludag, Biomacromolecules, 2005, 6, 2800 CrossRef CAS.
- T. V. Ratto, K. C. Langry, R. E. Rudd, R. L. Balhorn, M. J. Allen and M. W. McElfresh, Biophys. J., 2004, 86, 2430 CrossRef CAS.
- P. I. Kitov, H. Shimuzu, S. W. Homans and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 3284 CrossRef CAS.
- A. A. Vaidya, B. S. Lele, M. G. Kulkarni and R. A. Mashelkar, Biotechnol. Bioeng., 1999, 64, 418 CrossRef CAS.
- Y. F. Han, C. P.-L. Li, E. Chow, H. Wang, Y. P. Pang and P. R. Carlier, Bioorg. Med. Chem., 1999, 7, 2569 CrossRef CAS.
- V. Maraval, R. Laurent, P. Marchand, A. M. Caminade and J.-P. Majoral, J. Organomet. Chem., 2005, 690, 2458 CrossRef CAS.
Footnotes |
| † Current address: Department of Biological Engineering, Massachusetts Institute of Technology, Boston, MA 02139, USA |
| ‡ Note that the release from a surface is also a consideration for conjugates constructed with non-labile linkages. Such conjugates need to be released from bone mineral surfaces in order to interact with their cell-surface receptors. |
| § Note that the zero tether length obtained with dendritic BPs was not considered for this analysis due to significant differences between the structures of these BPs and those of the conventional BPs. |
| This journal is © The Royal Society of Chemistry 2007 |