CO and NO in medicine
Brian E.
Mann
*a and
Roberto
Motterlini
b
aDepartment of Chemistry, University of Sheffield, Sheffield, UK S3 7HF. E-mail: b.mann@sheffield.ac.uk.; Fax: +44 (0)114 2738673; Tel: +44 (0)114 2229332
bVascular Biology Unit, Department of Surgical Research, Northwick Park Institute for Medical Research, Harrow, Middlesex, UK HA1 3UJ. E-mail: r.motterlini@imperial.ac.uk; Fax: +44 (0)208 8693538; Tel: +44 (0)208 8693181
First published on 14th June 2007
Abstract
The occurrence, role and consequences of CO and NO in biological systems are reviewed. This includes their syntheses by heme oxygenases and NO synthases, their biological targets and the physiological effects of their signals. The use of CO and NO gases in medicine are discussed and methods of delivery are illustrated with particular emphasis on the therapeutic properties of compounds that generate controlled amounts of NO and COin vivo.
 Brian E. Mann | Professor Brian E. Mann graduated from Oxford (BA in Chemistry, 1965; MA, DPhil in Chemistry, 1968; DSc in 1989) and was at the University of Leeds (1968–1973) before moving to the University of Sheffield in 1973. He has authored and co-authored over 200 publications and three books. He has been a visiting Professor at eight universities. He ran the EPSRC High Field NMR Service in Sheffield, 1980–1986. |
 Roberto Motterlini | Dr Roberto Motterlini obtained his doctoral degrees at the University of Milan and San Raffaele Scientific Institute (1986–1991) and was a post-doctoral fellow at the University of California San Diego (1992–1995). He is author of more than 100 publications and his major scientific interest focuses on studying the biological function of the heme oxygenase–carbon monoxide (CO) pathway. At Northwick Park Institute for Medical Research (Harrow, UK), where he is Head of the Vascular Biology Unit since 1996, Dr Motterlini pioneered and implemented the use of transition-metal carbonyls and boranocarbonate as CO-releasing molecules (CO-RMs) for therapeutic applications. |
Introduction
At first sight, the occurrence of carbon monoxide (CO) and nitric oxide (NO) as signalling molecules in our bodies is surprising. CO has a deservedly bad reputation as being ‘a silent killer’ and causes many deaths as a result of poorly maintained heaters. NO is the source of the polluting NOx found in the air of many cities. However, it is now known that, at very low concentrations, both CO and NO are essential molecules for life.NO in biology has been extensively reviewed. The book, ‘Life, Death and Nitric Oxide’, by Butler and Nicholson gives an excellent background to NO in biology.1 In addition there are a number of reviews on NO including ‘Nitric Oxide and Cyclic GMP in Cell Signaling and Drug Development’, by Murad,2 and ‘NO-Releasing Drugs’, by Napoli and Ignarro.3
For CO, recent reviews include ‘Metal carbonyls: a new class of pharmaceuticals?’, Mann, Motterlini et al.;4 ‘Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications’, by Ryter et al.;5 ‘CO–metal interaction: vital signalling from a lethal gas’, Motterlini et al.;6 and ‘Therapeutic applications of carbon-monoxide-releasing molecules’, Motterlini et al.7
A review of a wide range of gases in biology and medicine is entitled ‘Free radical biology and medicine: it's a gas man!’ by Pryor et al.8
The use of nitroprusside, [Fe(CN)5(NO)]2–, amyl nitrite and trinitroglycerine as pharmaceuticals to reduce blood pressure by dilating arteries has been known since the beginning of the nineteenth century but it was not until the 1970s that people began to realise that these pro-drugs work by the release of NO and that NO is the active species. In 1979, it was shown that NO relaxes precontracted bovine coronary artery.9 Following this discovery, numerous papers have appeared; there is now a good understanding of much of the biological role of NO and its mechanism of action.1,10
The formation of CO in humans was discovered in the 1940s11 and is one of the most commonly observed chemical processes in the human body, occurring whenever there is a bruise, see later. There were few investigations of the role of CO in mammals until the importance of NO was recognised and then, during the 1990s, research rapidly developed; now there are more than 1000 publications a year in the area of CO biology. As investigations into the biological effects of CO are approximately fifteen years behind that of NO, the subject is not as well developed and there are many more questions still to be answered.4,7,12 Studies on the molecular mechanisms by which CO exerts its biological function are at a very early stage and much remains to be discovered.
In many cases, the research is still developing and, when viewed with the eyes of a chemist, much of the mechanistic work at the molecular level is best viewed as plausible suggestions rather than proven facts.
Generation of NO in mammals
The principal natural source of NO is L-arginine. There are three isozymes of NO synthase that oxidise the L-arginine to L-citrulline and NO, see Scheme 1.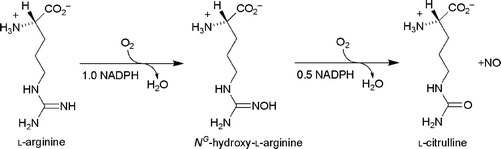 | ||
| Scheme 1 The formation of NO from L-arginine. | ||
The three isozymes of NO synthase are:
a. neuronal NOS (nNOS), which is found in neuronal cells and skeletal muscle;
b. endothelial NOS (eNOS), which is found in both endothelial and epithelial cells but also some neurons;
c. inducible NOS (iNOS), which is widespread throughout the body and can be highly expressed especially in macrophages, hepatocytes, astrocytes and smooth muscle cells.
In addition, NO can be generated in vivo from a wide range of organic nitrogen compounds such as nitrates, nitrites, S-nitrosothiols, NONOates such as Et2NN(O)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NONa, and oxadiazoles, e.g.3,4-dimethyl-1,2,5-oxadiazole 2-oxide, 1.
NONa, and oxadiazoles, e.g.3,4-dimethyl-1,2,5-oxadiazole 2-oxide, 1.

The wide range of compounds that generate NOin vivo has led to the development of compounds for pharmaceutical applications, vide infra.
Generation of CO in mammals
The major source of CO in mammals is from the oxidation of heme by heme oxygenase,13 see Scheme 2. | ||
| Scheme 2 The formation of biliverdin and CO from heme by heme oxygenase. | ||
In addition to CO, biliverdin, a green pigment, and FeII are produced during the degradation of heme by heme oxygenase. The biliverdin is subsequently reduced to bilirubin, 2, a yellow pigment.

This reaction can be observed by everyone and must be the most visible of all enzyme reactions as it takes place during the development of bruises, (Fig. 1).
 | ||
| Fig. 1 The changes in the appearance of a bruise with time. The pictures are of the thigh of an inline hockey player taken at various times after being hit by a puck. (a) 12 h, showing the red and purple of heme. (b) 2 days, showing green tinges due to the formation of biliverdin. (c) 5 days showing yellow due to the formation of bilirubin. (d) 10 days showing some residual bilirubin. The bright red colour in Fig 1d is due to carboxyhemoglobin. Reproduced from http://www.LondonSkaters.com with permission. | ||
The injury results in heme being liberated from hemoglobin and forming a dark red patch with O2 attached to each heme molecule. Heme oxygenase then catalyses the oxidation of the heme to biliverdin, FeII and CO. Examination of Scheme 2 shows that three molecules of O2 are consumed per heme, with the possibility for the tissues to become anaerobic. The O2 on the remaining unreacted heme is consumed, producing, after two days, a blue colour as is observed in deoxygenated venous blood (see Fig. 1b). Occasionally a green ring due to biliverdin is observed but this is converted to the yellow bilirubin, which is clearly observed after 5 days (Fig 1c). After 10 days (Fig. 1d) the colour of the remaining heme has changed to bright red, a colour markedly different from that observed in Fig 1a for the heme coordinated to O2. It is heme with CO coordinated, making the formation of CO easily visible. We are familiar with the formation of bilirubin also as the principal source of the yellow colour of urine.
Heme oxygenase accounts for about 86% of the CO produced in humans, with the remaining 14% of the CO generated coming from a mixture of sources, including photo-oxidation, lipid peroxidation, xenobiotics and bacteria.14
There are three isozymes of heme oxygenase: HO-1, which is inducible and is stimulated in response to stressful insults that cause a threat to cell homeostasis and survival;15,16HO-2, which is present in organs and tissues such as the brain, liver and endothelium and its function is associated with neurotransmission and regulation of vascular tone;15,17 and HO-3, which has been recently identified and found in the brain, heart, kidney, liver, testes and spleen of rats. The function of HO-3 is unknown as it does not possess any heme degrading activity.
The production of CO can be detected in exhaled air where an adult breathes out approximately 6 cm3 of CO per day. This can increase substantially in disease states18 such as asthma,19 bronchiectasis,20 cystic fibrosis,21 diabetes,22 and rhinitis.23
Sites of action of NO
Guanylyl cyclase
It is now well established that NO activates soluble guanylyl cyclase by binding to a heme and displacing a coordinated histidine.24 Soluble guanylyl cyclase catalyses the conversion of guanosine triphosphate to cyclic guanosine monophosphate, cGMP (see Scheme 3). | ||
| Scheme 3 The conversion of guanosine triphosphate to cyclic guanylyl monophosphate. | ||
The cyclic guanylyl monophosphate then enters a sequence of pathways. Selected ones that have been clearly related to medical conditions are given in Scheme 4, shown in green.25
 | ||
| Scheme 4 Mechanisms by which cGMP have been reported to ameliorate medical conditions (shown in green). cGKs, cGMP-dependent protein kinases, Iα and Iβ; IRAG, an IP3 receptor-associated cGKIβ substrate; VASP, vasodilator-stimulated phosphoprotein; PDEs, phosphodiesterases; cAMP, cyclic adenosine monophosphate.25 | ||
[NO]+, NO and [NO]–
NO can be converted to [NO]+ or [NO]– by a one-electron oxidation or reduction. Their roles in biological systems have been documented and circumstantial evidence suggests that the dynamic chemistry of the NO redox forms is important in determining specific physiological effects.26[NO]+ is iso-electronic with CO and is a very good ligand for heme iron, it can be transported effectively from S-nitrosothiols but has a very short life in aqueous solutions at physiological pH. [NO]– is also very unstable under physiological conditions although in vitro, in vivo and ex vivo analysis revealed that NO and [NO]– elicit distinct responses.27 Intriguingly, and this is perhaps the most striking link between NO and the heme oxygenase system, mammalian cells are highly susceptible to HO-1 induction by nitric oxide (NO) and its redox activated forms (NO+ and NO–).28,29 An excessive production of endogenous NO, which leads to “nitrosative stress”, also up-regulates HO-1 expression resulting in more CO being produced.30 Moreover, recent in vivo studies revealed that continuous administrations of organic nitrates and sodium nitroprusside are associated with both hepatic HO-1 expression and augmented carboxyhemoglobin levels.31 Notably, CO generated from enhanced HO-1 activity or exogenously applied has been demonstrated to provide cytoprotective effects against the toxicity caused by overproduction of NO.32,33 Thus, the increase in HO-1 expression by NO can act as a negative feedback on NO production to limit its cytotoxic effects.29NO is a highly reactive molecule and survives in vivo for only a few seconds. Much is known of the consequential chemistry, but the biological relevance is generally controversial. There are many known reactions of NO and consequential chemistry of the products1 and in this brief review only a few are described.
It readily reacts with superoxide, [O2]–, to give peroxynitrite, ONOO–, which is also an oxidising agent, but spontaneously decomposes to NO2 and [NO3]–, resulting in it being rapidly removed. Peroxynitrite is very reactive and is a much more powerful oxidant than NO. This explains why, in biological systems, peroxynitrite is considered a threat as it may oxidise many biomolecules when produced within the intracellular milieu. It can react directly with electron-rich groups, such as sulfhydryls, iron–sulfur centers, zinc-thiolates , and the active site sulfhydryl in tyrosine phosphatases. Moreover, the interaction with tyrosine to form nitrotyrosine is being regarded as a marker of peroxynitrite formation and nitrosative stress.34Peroxynitrite also reacts with carbon dioxide to form a transient intermediate, nitrosoperoxycarbonate, that rapidly decomposes homolytically to nitrogen dioxide and carbonate radical, which can initiate many damaging reactions within the cell.34
The reaction of NO with superoxide described above could be seen as a mechanism for protection against oxidation by [O2]–. Although conceptually the opposite is true, since superoxide inactivates NO and prevents its biological function as a signalling mediator, we cannot exclude that NO and superoxide can neutralise each other in conditions of uncontrolled oxidative and nitrosative stress.
Sites of action of CO
Guanylyl cyclase
Like NO, CO activates guanylyl cyclase to produce cGMP and similarly ameliorates medical conditions given in Scheme 4. It is only about 1/80th as effective as NO. However, there is a family of synthetic compounds of which YC-1, 3, is an example that, when combined with CO, are as effective as NO in activating guanylyl cyclase. This raises the possibility that there is a natural intracellular signalling molecule similar to YC-1.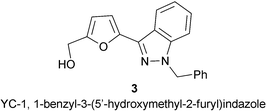
The enzyme, guanylyl cyclase has two domains, α and β. The heme is in the β-domain and is 5-coordinate. When it is activated by NO, the coordination of NO displaces the histidine-105 and the iron is still 5-coordinate, (Scheme 5). There is dispute as to whether the NO is on the same or the opposite side to His-105 in the activated form. It is probable that the NO binds to the heme in guanylyl cyclase to give what is in effect FeIII and [NO]–. Although there is no crystal structure, the structure of the five-coordinate NO adduct of cytochrome c′ indicates a bent Fe–N–O bond with an angle of 124° and 132° with a half-occupancy for each35 consistent with coordination of [NO]–. As it has been reported that the heme –NO complexes in both guanylyl cyclase and cytochrome c′ are very similar,36 then it can be anticipated that the Fe–N–O is also bent in the five-coordinate NO adduct of guanylyl cyclase. This is supported by a ν(NO) of 1677 cm–1.37 In contrast, when CO coordinates to guanylyl cyclase the iron becomes 6-coordinate and the iron almost certainly remains FeII. YC-1 interacts with the α-domain and the His-105 is displaced to produce a 5-coordinate iron in the heme-carbonyl and YC-1.38
 | ||
| Scheme 5 A pictorial representation of the activation of guanylyl cyclase by NO and CO/YC-1.38 | ||
Non-cGMP pathways
A number of additional intracellular pathways that could be targeted by CO have been identified but as yet have not received the same level of investigation as for the cGMP pathways. These are summarised in Scheme 6. | ||
| Scheme 6 Non-cGMP pathways for CO biological activity. KCa, calcium signalled potassium channels; JNK, c-Jun N-terminal kinase; p38 MAPK, p38 mitogen activated protein kinase; ERK1/2, extracellular signal-regulated kinases. | ||
The information available on the mechanism by which CO activates/inhibits these pathways at a molecular level is very limited. CO appears to modulate mitogen activated protein kinases (MAPK)-related pathways and this aspect has been reviewed elsewhere, showing specifically that CO activates p38 and down-regulates ERK1-2.39 Suppression of inflammatory cytokine production by CO could also involve the JNK pathway.40 It has also been shown that HO-2, and hence CO, is an essential part of the modulation of calcium-sensitive potassium channels by oxygen and is hence essential for respiratory control.41 It had been shown previously that heme is an essential part of this oxygen sensor,42 so it is probable that the heme acts as the CO receptor. From a chemical perspective, one wonders how CO can react directly with MAPK, ERK-1-2 or JNK as these proteins do not seem to contain a metal centre. As known, CO preferentially binds to and interacts with heme- or transition metal-containing proteins . Thus, until the crystal structures of these proteins have been revealed, we cannot exclude a priori that CO does not interact with MAPK or other proteins involved in signalling pathways. In contrast, it is important to emphasise that the ways CO interacts and controls the activity of some “classical” targets in mammalian cells (i.e. cytochrome oxidase) are being re-examined in the search for the precise mechanism(s) underlying its protective effects.6,43 These targets of CO include, but are not restricted to: (1) NADPH oxidase;44 (2) cytochrome oxidase and mitochondrial complexes;43–46 and (3) nitric oxide synthase.32,47,48 Circumstantial evidence also indicates that other hemoproteins, such as cytochrome P450 ,49 and proteins containing transition metals could be possible targets of CO.50
Hemes and cytochromes
Hemes and some cytochromes such as cytochrome P450 and cytochrome c oxidase bind CO and this provides a potential mechanism for CO to act by blocking enzymic activity. For example, it is well established that CO gives protection against reperfusion injury. In principle this protection can be provided by coordination of CO to a cytochrome such as cytochrome c oxidase to prevent the activation of oxygen.Other targets for CO
Although in mammals CO is only known to bind to hemes and cytochromes , there is chemical evidence of its binding to iron with other biologically relevant ligand systems. Indeed, it is known that CO can bind to iron, nickel and copper in bacteria, vide infra, and copper in cytochrome c oxidase.51 FeII reacts with cysteine to give [Fe(cysteinate)2] which readily absorbs CO to give [Fe(cysteinate)2(CO)2].52 Bleomycin, 4, reacts with FeII to give a complex and then CO can coordinate to the iron.53 The ligand set for the iron is formed from nitrogen and oxygen atoms. It is therefore probable that enzymes also contain a ligating set of atoms capable of binding FeII which then binds CO. This could be significant as heme oxygenase not only produces CO but also FeII. The production of FeII could be important as it seems strange that heme oxygenase catalyses the oxidation of heme to produce CO yet the iron is not oxidised to FeIII. Notably, FeII binds CO much more readily than FeIII.
CO as a reducing agent
Although bacteria use CO as a reducing agent via the water–gas shift reaction, vide infra, there is no evidence at present of this occurring in higher organisms. However, we know that CO is oxidised to CO2 by oxyferryl groups. This reaction could have a dual role: it removes CO from the system but at the same time it also destroys the highly oxidising oxyferryl groups. The relative importance of these two roles is not known.54CO in bacteria
Although the mechanism of CO action in mammals is poorly understood at a molecular level, three families of bacterial enzymes are based on metal carbonyls and the use of CO in these bacteria has been extensively investigated. These are hydrogenases, CO dehydrogenases and acetyl coenzyme A synthase. It is probable that these are very old enzymes, and originated at least 2.5 billion years ago when the Earth's atmosphere was anaerobic and probably contained significant quantities of CO.In hydrogenases, CO appears to act only as a ligand, but in CO dehydrogenases and acetyl coenzyme A synthase it acts as a reactive substrate.
Hydrogenases
The hydrogenase family of enzymes catalyses the reaction shown in Scheme 7.55 | ||
| Scheme 7 The reversible reduction of protons to dihydrogen. | ||
Hydrogenases are found in a wide range of bacteria and are involved in the production of dihydrogen, the use of dihydrogen as an energy source and as a dihydrogen sensor. Depending on the type(s) of metal at the active site, there are two classes of hydrogenases: [Fe]-only and [NiFe]-hydrogenases, (see Fig. 2).
![Active sites of [Fe]-only, (a), and [NiFe], (b), hydrogenases.](/image/article/2007/CC/b704873d/b704873d-f2.gif)
CO dehydrogenase/acetyl coenzyme A synthase
These are a pair of enzymes that probably originated in early organisms and are now found supporting anaerobic life over a wide range of environments including bogs, cow rumens and human intestines. This enzyme system enables micro-organisms such as Rhodospirillum rubrum, Moorella thermoacetica and Carboxydothermus hydrogenoformans to use CO as a carbon and energy source.The CO dehydrogenase enzymes catalyse the water–gas shift reaction (see Scheme 8).
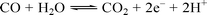 | ||
| Scheme 8 The water–gas shift reaction. | ||
There are two families of CO dehydrogenase enzymes. The molybdopterin Cu CO dehydrogenase contains a copper linked to a Mo-pterin, 5, 6.56 It is believed that the CO binds to copper.57

The second group is based on a nickel–iron cluster, 7, and further Fe4S4 clusters act as electron receivers/donors. There is FTIR evidence that the CO binds to nickel,58 and 17O ENDOR evidence that water binds to iron.59 The reaction mechanism in Scheme 9 has been proposed for the conversion of CO to CO2 at this cluster.

![The mechanism for the CO dehydrogenase reaction proposed by Ragsdale.56 [4Fe–4S] clusters are used for electron transfer.](/image/article/2007/CC/b704873d/b704873d-s9.gif) | ||
| Scheme 9 The mechanism for the CO dehydrogenase reaction proposed by Ragsdale.56 [4Fe–4S] clusters are used for electron transfer. | ||
This reaction can be used in either direction, either to produce electrons for reductions or to use CO2 as a source of CO. In the enzyme complex, CO2 is reduced to CO at CO dehydrogenase and passes through a channel to the acetyl coenzyme A synthase, where it inserts into a methyl–metal bond to generate MeC(O)–metal which is then transferred to the S– of coenzyme A, –SCoA, to generate MeC(O)SCoA. A possible mechanism for this reaction is shown in Scheme 10. The methyl group is transferred from a methylcobalamin complex. For further information, the reader is referred to a recent review.56
 | ||
| Scheme 10 A possible enzymic mechanism for the synthesis of MeC(O)SCoA.60 | ||
Comparison of roles of CO and NO
Lifetimes in vivo
CO is a relatively unreactive molecule in vivo. It is known to be oxidised by the oxyferryl group of hemes and cytochromes ,54 but it is not known if this reaction is biologically relevant in mammals. The amount of CO converted to CO2 in mammals is very unlikely to be significant. CO binds to hemes and cytochromes , but the reaction is reversible. For example, for myoglobin the half-life of bound CO before dissociation is around a minute.61 As, in the absence of external sources of CO, a healthy adult has ca. 0.6% of the heme of hemoglobin in the carbonoxy form, this provides a reservoir of CO in the body and is used as the transfer mechanism when CO gas is used as a pharmaceutical. It has been estimated that the half-life of CO in the body is in excess of an hour before it is breathed out.62NO is highly reactive, so that it only survives in solution for a very short time. It can be trapped by thiols as RSNO and this has been proposed as an important way to preserve NObioavailability and to enable the NOsignals to be transduced into specific biological functions.63 Nevertheless, NO is more reactive than CO and this may offer an advantage in stress conditions where NObioavailability will be compromised while CO can still exert its signalling properties.
A very significant paper has recently been published that shows a clear difference between CO and NO. It has been shown that NO synthase requires a normal oxygen concentration.64 As the system becomes anaerobic, the NO synthase enzymeceases to work, while heme oxygenase continues to produce CO at much lower oxygen levels. It is therefore possible that CO becomes a much more important signalling molecule than NO in anaerobic conditions
NO and CO in inflammation
As an example of the biological differences between NO and CO, one could look at the effects of these gaseous molecules in inflammation. Macrophages provide a major defence mechanism for the body against inflammation and invading microbes. They use inducible NOS synthase to generate NO, which reacts with superoxide, [O2]–, to give peroxynitrite, [ON–O–O]–, a powerful oxidising agent that destroys the unwanted cells.1Peroxynitrite has a very short life before it isomerises to nitrate. NO is also oxidised to N2O3, which is a nitrating agent and nitrates thiols to RSNO. As some microbial enzymes contain a thiol at the active centre, this could also be a mechanism to deactivate them. Once the concentration of NO starts to build up, enzymes in the macrophage are deactivated by nitration of tyrosine.
Inflammation that runs unchecked can lead to a host of diseases. An example of this is rheumatoid arthritis, where the cartilage is progressively destroyed by chronic inflammation. Chronic inflammation is a pathological condition typified by concurrent active inflammation, tissue destruction, and attempts at repair. Chronically inflamed tissue is characterised by the infiltration of mononuclearimmune cells, including monocytes and macrophages, which continuously generate inflammatory mediators such as NO. CO has been shown to inhibit NO production in macrophages and reduce inflammation. It is suggested that CO binds to the heme in NO synthase and prevents oxidation of arginine to citrulline and NO, see Scheme 1.
HO-1 mediated anti-inflammatory response is broader than inhibition of macrophages.65 It also reduces expression of the intercellular adhesion molecule-1, ICAM-1, and decreases pro-inflammatory cytokines such as TNF-α and increases crucial anti-inflammatory molecules such as IL-10. Both CO and biliverdin/bilirubin are involved in the anti-inflammatory response.
NO-releasing drugs
NO-releasing drugs utilised to treat angina pectoris are well established, with amyl nitrite being used since 1867,66 and nitroglycerine since 1879.67Sodium nitroprusside has also been used for many years for rapid reduction of hypertension, especially during surgical interventions. This compound has to be used with caution due to the associated risks of cyanide poisoning.More recently, a wide range of NO-releasing compounds have been introduced, including ones based on organic nitrates and nitrites and S-nitrosothiols, RSNO. Also, non-steroidal anti-inflammatory drugs have been modified so that they release NO. For example, two nitrato-aspirins, 8 and 9, have been shown to inhibit platelet aggregation.

It has been proposed that a number of other pharmaceuticals may, at least partly, work by modulating the NO-pathway. These include calcium channel blockers, ACE inhibitors and ANGII type 1 receptor antagonists, β-blockers and hydroxymethylglutaryl-CoA reductase inhibitors.
A more detailed review of NO-releasing drugs has been published recently.3
CO in medicine
The development of methods for delivering CO to treat human diseases is many years behind the NO-based pharmaceuticals. This reflects the fact that CO was discovered as a signalling molecule much later than NO. Nevertheless, with the beginning of the new century, scientists started to investigate experimentally the potential role of CO as therapeutic agent. The major strategic approaches that are being used to effectively deliver CO in mammals are summarised in the following sections.CO gas as therapeutic agent
The preclinical applications of CO gas have been reported for animal experiments. Most of the experiments have involved administering CO gas at 200 to 400 ppm in the air supply. Some highlights are reported here.In mice, CO suppresses the development of post-operative ileus in the murine small intestine of mice68 and is involved in the control of gastrointestinal contractile activity.69 In rats, CO suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury following angioplasty.70 Recently, in the same animals, CO has been shown to reverse established pulmonary hypertension.71
In pigs, CO improves cardiac energetics and safeguards the heart during reperfusion after cardiopulmonary bypass.72
Organ graft rejection is also suppressed by CO. A model of mouse-to-rat cardiac transplantation was performed. Rats were exposed to 400 ppm of CO gas for only 2 days following transplant and the graft survived for 50 days. In comparison, the control group, exposed to air, survived only 5 to 7 days.73
CO gas is entering phase 1 trials in the US for the prevention of lung inflammation.74 There is also some circumstantial evidence that CO gas can produce beneficial effects. Smokers self-administer CO along with many noxious chemicals. Statistical evidence shows that it gives partial protection against cell proliferation, which results in partial blocking of the artery following angioplasty.75 It also may partially protect women from pre-eclampsia.76
CO-releasing molecules (CORMs)
In contrast to CO gas, CO-releasing molecules (CORMs) have been designed to offer a safe and controlled method to administer CO to a specific part of the body. The challenge is to develop non-cytotoxic water-soluble compounds that release CO relatively fast with half-lives of less than two hours. Slower release is unlikely to result in a high enough CO concentration from an acceptable CO-releasing molecule concentration due to CO circulation within the body and the reduction in effective CO concentration by complexation with heme.The first CORMs to be identified were [Mn2(CO)10] and [Ru(CO)3Cl2]2.77 [Mn2(CO)10] is insoluble in water but releases CO on photolysis; it produces marked attenuation in coronary vasoconstriction in isolated rat hearts78 and increases blood carboxyhemoglobin levels, renal blood flow, glomerular filtration rate, and urinary cGMPexcretion.79 [Ru(CO)3Cl2]2 has been used as a solution in DMSO, which gives a mixture of [Ru(CO)3Cl2(DMSO)] and [Ru(CO)2Cl2(DMSO)2] (isomers).77 The [Ru(CO)3Cl2(DMSO)] rapidly releases CO to myoglobin and elicits profound vasodilatation in isolated rat aorta.77
The first water-soluble CORM to be identified was [Ru(CO)3Cl(glycinate)].80 It rapidly releases one CO group to myoglobin, t½ < 2 min even though it is relatively stable in distilled water with t½ > 24 h. It is believed that CO release in biological fluid involves coordination of a trans-labilising ligand such as a thiol . In vitro experiments have shown the rapid loss of CO from an aqueous solution of [Ru(CO)3Cl(glycinate)] on addition of cysteine or pyridine. Both the chloride and glycinate ligands are labile, being replaced by stronger ligands.81 This chemistry makes [Ru(CO)3Cl(glycinate)] convenient to administer as CO release is very slow until the compound meets suitable strong ligands in biological fluids.
The biological activity of [Ru(CO)3Cl(glycinate)] has received extensive investigation. For example, it produces vasodilatation in an isolated aortic ring precontracted with phenylephrine,82 protects an isolated rat heart from reperfusion damage following 30 min of ischemia,80 and markedly improves the survival time of mice following heart transplantation.80[Ru(CO)3Cl(glycinate)] also improves kidney function following cold ischemia occurring during organ preservation and protects against cisplatin-induced nephrotoxicity.46,83 More recently, in vitro studies revealed some important anti-inflammatory properties of this compound in the context of both vascular and neuro-inflammation,47,84 as well as inhibition of platelet aggregation.85
[Ru(CO)3Cl(glycinate)] has a complex solution chemistry, with hydroxide attacking one carbonyl at pH 3 to give [Ru(CO)2(CO2H)Cl(glycinate)]– (isomers), which then undergoes another pH-dependent reaction to give mainly [Ru(CO)2(CO2)Cl(glycinate)]2– (isomers) or [Ru(CO)2(CO2H)(OH)(glycinate)]– (isomers) at physiological pH, see Scheme 11.86
![The pH dependence of [Ru(CO)3Cl(glycinate)] in water. Due to the lability of the chloride it is not known if it remains coordinated in water.](/image/article/2007/CC/b704873d/b704873d-s11.gif) | ||
| Scheme 11 The pH dependence of [Ru(CO)3Cl(glycinate)] in water. Due to the lability of the chloride it is not known if it remains coordinated in water. | ||
CORMs are not restricted to transition metals. [H3BCO2]2– had been developed to convert [TcO4]– into [Tc(CO)3(OH2)3]+ for imaging.87[H3BCO2]2– releases CO with a t½ = 21 min at pH 7.4.88 The rate of reaction is pH dependent, see Scheme 12.
![The reaction between [H3BCO2]2– and acid.](/image/article/2007/CC/b704873d/b704873d-s12.gif) | ||
| Scheme 12 The reaction between [H3BCO2]2– and acid. | ||
[H3BCO2]2– produces vasodilatation in isolated vessels and promotes a reduction in mean arterial pressure in vivo at a rate that correlates with the CO release.88
Recently other CORMs have been identified, 10,89 [(η5-C5H4R)Fe(CO)3]+,90 and [Mn(CO)4X2]0/–.91 [(η5-C5H4R)Fe(CO)3]+, and [Mn(CO)4X2]0/– are particularly interesting, as the rate of CO loss can be controlled by choice of R or X and more than one CO is lost per metal, making it possible to use smaller concentrations of the compound.

[(η5-C5H5)Fe(CO)3]+ was identified as a CO-releasing molecule in 2001.92 It loses CO to myoglobin at 37 °C with t½ = 69 min. Unfortunately, it suffered from a problem. On CO release, the product precipitated. This immediately ruled out its use, as the precipitate could block micro-arteries and cause toxicity.93 Improved water-solubility has been introduced by substituting the cyclopentadienyl with relatively polar groups. At the same time, the rate of CO release is modulated, see Table 1. There is an approximate correlation between t½ and ν(CO).
The correlation between t½ and ν(CO) fails for [Mn(CO)4X2]+/0/–, see Table 2. Some of these molecules are excellent CO-releasing agents with t½ < 2 min, and many release more than one CO rapidly, enabling lower concentrations of compound to be used to generate CO. Cationic complexes such as [Mn(CO)4(bpy)]+ (bpy = 2,2′-bipyridine) are very slow CO releasers while anionic complexes such as [Mn(CO)4Br2]– are very fast CO releasers. This is despite [Mn(CO)4(bpy)]+ having the highest ν(CO) values of all the molecular ions in Table 2. The mechanism of CO release is unknown, but oxidation could possibly contribute to the liberation of CO from this type of compound.
Molecules catabolised to produce CO
CO is produced naturally by heme oxygenase oxidation of heme, (Scheme 2). This provides the possibility of enhancing CO production by providing more heme to be catabolised to give CO. For example, when spontaneously hypertensive rats are injected with heme or heme arginate, their blood pressure decreases.94Dichloromethane is catabolised in the liver to generate CO.95 It has been proposed that this provides a mechanism to supply CO as a pharmaceutical,96 but there are serious concerns about it causing liver and cardiac damage.97
Heme oxygenase-1 (HO-1) inducers
Heme oxygenase (HO-1) is a stress-inducible protein and its expression can be enhanced in cells and tissues by agents or conditions that cause an increase in oxidative stress.15 There is also a wide range of natural compounds present in fruits and vegetables that act as potent inducers of HO-1. Although it has not been proven experimentally, high intake of certain fruits or vegetables may eventually lead to increased production of CO in the body. This may explain why certain natural products appear to be beneficial for the cardiovascular system. Examples include curcumin98 found in turmeric, chalcones and L-sulforaphane99 present in broccoli, carnosol100 in rosemary, resveratrol101 in grape skin, as well as red wine and garlic-derived organosulfur compounds.102 It is also possible that HO-1 induction may contribute to the beneficial effects of drugs such as aspirin103 and some statins104 in the cardiovascular system, although direct experimental evidence for this link is still missing.Hydrogen sulfide
Recently, interest has been growing in H2S as a signalling molecule.105 As H2S has a pKa of 6.8, it is mainly present as H2S and [HS]– at physiological pH. It is more toxic than CO. In the brain, H2S is synthesised from cysteine, catalysed by cystathionine β-synthase, but it is probable that there are other pathways. H2S concentration is 50–160 µM in brain tissue and 10–100 µM in blood, which is much higher than is found naturally for either CO or NO. H2S is active in both the cardiovascular and central nervous systems. For example, it causes vasodilatation.106 As [HS]– readily coordinates to hemes and some cytochromes , it is not surprising that it is biologically active, but more research is necessary to establish whether or not it is an important signalling molecule.Conclusions
Both CO and NO are essential signalling molecules in the body. The importance of NO was discovered in the 1970s and has led to a much greater understanding of the biochemistry of NO and the development of NO-based pharmaceuticals. On account of the later discovery that CO is important, the understanding of the role of CO is lagging about 25 years behind NO. Most of the research on CO has been concentrated in the areas of the biological and medical effects of CO.The medical applications of CO are progressing rapidly and it is probable that CO will be used in human medicine in the near future, with an important role for CO-releasing molecules. The understanding of the mechanism of action of CO at a molecular level is now reasonably understood for guanylyl cyclase but the molecular mechanisms of action for the pathways in Scheme 6 leave much scope for further investigation.
References
- A. R. Butler and R. Nicholson, Life, Death and Nitric Oxide, The Royal Society of Chemistry, Cambridge, 2003 Search PubMed.
- F. Murad, N. Engl. J. Med., 2006, 355, 2003 CrossRef CAS.
- C. Napoli and L. J. Ignarro, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 97 CrossRef CAS.
- T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. Green and R. Motterlini, Angew. Chem., Int. Ed., 2003, 42, 3722 CrossRef CAS.
- S. W. Ryter, J. Alam and A. M. K. Choi, Physiol. Rev., 2006, 86, 583 CrossRef CAS.
- J. Boczkowski, J. J. Poderoso and R. Motterlini, Trends Biochem. Sci., 2006, 31, 614 CrossRef CAS.
- R. Motterlini, B. E. Mann and R. Foresti, Expert Opin. Invest. Drugs, 2005, 14, 1305 Search PubMed.
- W. A. Pryor, K. N. Houk, C. S. Foote, J. M. Fukuto, L. J. Ignarro, G. L. Squadrito and K. J. A. Davies, Am. J. Physiol., 2006, 291, 491 Search PubMed.
- C. A. Gruetter, B. K. Barry, D. B. McNamara, D. Y. Gruetter, P. J. Kadowitz and L. J. Ignarro, J. Cyclic Nucleotide Res., 1979, 5, 211 Search PubMed.
- S. Pfeiffer, B. Mayer and B. Hemmens, Angew. Chem., Int. Ed., 1999, 38, 1714 CrossRef.
- T. Sjostrand, Scand. J. Clin. Lab. Invest., 1949, 1, 201 CrossRef CAS.
- Heme Oxygenase: The Elegant Orchestration of Its Products in Medicine, ed. L. E. Otterbein and B. S. Zuckerbraun, Nova Medical, 2005 Search PubMed; Carbon Monoxide and Cardiovascular Functions, ed. R. Wang, CRC Press, Boca Raton, FL, 2002 Search PubMed.
- R. Tenhunen, H. S. Marver and R. Schmid, J. Biol. Chem., 1969, 244, 6388 CAS.
- H. J. Vreman, R. J. Wong and D. K. Stevenson, in Carbon Monoxide and Cardiovascular Functions, ed. R. Wang, CRC Press, Boca Raton, 2002, ch 15 Search PubMed.
- R. Foresti and R. Motterlini, Free Radical Res., 1999, 31, 459 CrossRef CAS.
- L. E. Otterbein and A. M. Choi, Am. J. Physiol., 2000, 279, L1029 Search PubMed; N. G. Abraham and A. Kappas, Free Radical Biol. Med., 2005, 39, 1 CrossRef CAS.
- M. D. Maine, Annu. Rev. Pharmacol. Toxicol., 1997, 37, 517 CrossRef CAS.
- M. Scharte, H. G. Bone, H. Van Aken and J. Meyer, Biochem. Biophys. Res. Commun., 2000, 267, 423 CrossRef CAS.
- K. Zayasu, K. Sekizawa, S. Okinaga, M. Yamaya and H. Sasaki, Am. J. Respir. Crit. Care Med., 1997, 156, 1140 Search PubMed; I. Horvath, L. E. Donelly, P. Paredi, S. A. Kharitonov and P. J. Barnes, Thorax, 1998, 53, 668 CrossRef CAS.
- I. Horvath, S. Loukides, T. Wodehouse, S. A. Kharitonov, P. J. Cole and P. J. Barnes, Thorax, 1998, 53, 867 CrossRef CAS.
- J. D. Antuni, S. A. Kharitonov, D. Hughes, M. E. Hodson and P. J. Barnes, Thorax, 2000, 55, 138 CrossRef CAS; P. Paredi, S. A. Kharatonov, D. Leak, P. L. Shah, D. Cremer, M. E. Hodson and P. J. Barnes, Am. J. Respir. Crit. Care Med., 2000, 161, 1247 Search PubMed.
- P. Paredi, W. Biernacki, G. Invernizzi, S. A. Kharitonov and P. J. Barnes, Chest, 1999, 116, 1007 Search PubMed.
- M. Monma, M. Yamaya, K. Sekizawa, K. Ikeda, N. Suzuki, T. Kikuchi, T. Takasaka and H. Sasaki, Clin. Exp. Allergy, 1999, 29, 1537 CrossRef CAS.
- L. J. Ignarro, G. Cirino, A. Alessandro and C. Napoli, J. Cardiovasc. Pharmacol., 1999, 34, 879 CrossRef CAS . See: A. Friebe and D. Koesling, Circ. Res., 2003, 93, 96 Search PubMed and references therein; M. Russwurm and D. Koesling, EMBO J., 2004, 23, 4443 Search PubMed.
- R. Feil and B. Kemp-Harper, EMBO Rep., 2006, 7, 149 Search PubMed.
- J. S. Stamler, D. J. Singel and J. Loscalzo, Science, 1992, 258, 1898 CrossRef CAS.
- D. A. Wink, K. M. Miranda, T. Katori, D. Mancardi, D. D. Thomas, L. Ridnour, M. G. Espey, M. Feelisch, F. A. Colton, J. M. Fukuto, P. Pagliaro, D. A. Kass and N. Paolocci, Am. J. Physiol., 2003, 285, H2264 Search PubMed.
- R. Motterlini, R. Foresti, M. Intaglietta and R. M. Winslow, Am. J. Physiol., 1996, 270, H107 Search PubMed; R. Foresti, J. E. Clark, C. J. Green and R. Motterlini, J. Biol. Chem., 1997, 272, 18411 CrossRef CAS; P. Naughton, R. Foresti, S. Bains, M. Hoque, C. J. Green and R. Motterlini, J. Biol. Chem., 2002, 277, 40666 CrossRef CAS.
- R. Motterlini, C. J. Green and R. Foresti, Antioxid. Redox Signaling, 2002, 4, 615 Search PubMed.
- I. A. Sammut, R. Foresti, J. E. Clark, D. J. Exon, M. J. J. Vesely, P. Sarathchandra, C. J. Green and R. Motterlini, Br. J. Pharmacol., 1998, 125, 1437 CrossRef CAS; R. Motterlini, R. Foresti, R. Bassi, V. Calabrese, J. E. Clark and C. J. Green, J. Biol. Chem., 2000, 275, 13613 CrossRef CAS.
- Y. Minamiyama, S. Takemura, K. Yamasaki, S. Hai, K. Hirohashi, Y. Funae and S. Okada, J. Pharmacol. Exp. Ther., 2004, 308, 729 CAS; J. Lopez-Herce, R. Borrego, A. Bustinza and A. Carrillo, Intensive Care Med., 2005, 31, 1235 CrossRef.
- K. Srisook, S. S. Han, H. S. Choi, M. H. Li, H. Ueda, C. Kim and Y. N. Cha, Biochem. Pharmacol., 2006, 71, 307 CrossRef CAS.
- B. M. Choi, H. O. Pae and H. T. Chung, Free Radical Biol. Med., 2003, 34, 1136 CrossRef CAS.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315 CrossRef CAS.
- D. M. Lawson, C. E. M. Stevenson, C. R. Andrew and R. E. Eady, EMBO J., 2000, 19, 5661 CrossRef CAS.
- J. R. Stone and M. A. Marletta, Biochemistry, 1994, 33, 5636 CrossRef CAS.
- G. Deinum, J. R. Stone, G. T. Babcock and M. A. Marletta, Biochemistry, 1996, 35, 1540 CrossRef CAS; S. Kim, G. Deinum, M. T. Gardner, M. A. Marletta and G. T. Babcock, J. Am. Chem. Soc., 1996, 118, 8769 CrossRef; B. L. Haymore and J. Ibers, Inorg. Chem., 1975, 14, 3060 CrossRef.
- E. Martin, K. Czarnecki, V. Jayaraman, F. Murad and J. Kincaid, J. Am. Chem. Soc., 2005, 127, 4625 CrossRef CAS; T. Uchida and T. Kitagawa, Acc. Chem. Res., 2005, 38, 662 CrossRef CAS; T. Kitagawa, J. Inorg. Biochem., 2004, 98, 824 CrossRef.
- S. P. Kim, S. W. Ryter and A. M. Choi, Annu. Rev. Pharmacol. Toxicol., 2006, 46, 411 CrossRef.
- D. Morse, S. E. Pischke, Z. Zhou, R. J. Davis, R. A. Flavell, T. Loop, S. L. Otterbein, L. E. Otterbein and A. M. Choi, J. Biol. Chem., 2003, 278, 36993 CrossRef CAS.
- S. E. J. Williams, P. Wooton, H. S. Mason, J. Bould, D. E. Iles, D. Riccardi, C. Peers and P. J. Kemp, Science, 2004, 306, 2093 CrossRef CAS.
- R. Wang and L. Wu, J. Biol. Chem., 1997, 272, 8222 CrossRef CAS.
- H. B. Suliman, M. S. Carraway, L. G. Tatro and C. A. Piantadosi, J. Cell Sci., 2006, 120, 299 Search PubMed.
- C. Taille, J. El-Benna, S. Lanone, J. Boczkowski and R. Motterlini, J. Biol. Chem., 2005, 280, 25350 CrossRef CAS.
- A. Sandouka, E. Balogun, R. Foresti, B. E. Mann, T. R. Johnson, Y. Tayem, C. J. Green, B. Fuller and R. Motterlini, Cell Mol. Biol. (Sarreguemines, Fr., Print), 2005, 51, 425 Search PubMed.
- A. Sandouka, B. J. Fuller, B. E. Mann, C. J. Green, R. Foresti and R. Motterlini, Kidney Int., 2006, 69, 239 CrossRef CAS.
- P. Sawle, R. Foresti, B. E. Mann, T. R. Johnson, C. J. Green and R. Motterlini, Br. J. Pharmacol., 2005, 145, 800 CrossRef CAS.
- P. Sawle, J. Hammad, I. J. Fairlamb, B. Moulton, C. T. O'Brien, J. M. Lynam, A. K. Duhme-Klair, R. Foresti and R. Motterlini, J. Pharmacol. Exp. Ther., 2006, 318, 403 CrossRef CAS.
- F. Coceani, N. C. Hamilton, J. Labuc and P. M. Olley, Am. J. Physiol., 1984, 246, H640 Search PubMed; F. Coceani, L. Kelsey and E. Seidlitz, Br. J. Pharmacol., 1996, 118, 1689 CAS.
- M. Desmard, M. Amara, S. Lanone, R. Motterlini and J. Boczkowski, Cell Mol. Biol. (Sarreguemines, Fr., Print), 2005, 51, 403 Search PubMed.
- S. Stavrakis, K. Koutsoupakis, E. Pinakoulaki, A. Urbani, M. Saraste and C. Varotsis, J. Am. Chem. Soc., 2002, 124, 3814 CrossRef CAS.
- M. P. Schubert, J. Am. Chem. Soc., 1933, 55, 4564.
- Y. Sugiura, T. Suzuki, H. Kawabe, H. Tanaka and K. Watanabe, Biochim. Biophys. Acta, 1982, 716, 38 CrossRef CAS; Y. Sugiura, J. Kuwahara and T. Suzuki, FEBS Lett., 1985, 182, 39 CrossRef CAS; N. J. Oppenheimer, L. O. Rodriguez and S. M. Hecht, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 5616 CAS; Y. Sugiura, T. Suzuki, Y. Muraoka, Y. Umezawa, T. Takita and H. Umezawa, J. Antibiot., 1981, 34, 1232 CAS; W. A. Herrmann, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, VCH, Weinheim, 1996, vol. 2, pp. 957–963 Search PubMed; R. M. Laine and R. B. Wilson, Aspects Homogeneous Catal., 1984, 5, 217 Search PubMed.
- Q. Ji, C. R. Lloyd, W. R. Ellis, Jr. and E. M. Eyring, J. Am. Chem. Soc., 1998, 120, 221 CrossRef CAS and references therein.
- See reviews in Coord. Chem. Rev., 2005, 249, issues 15–16 Search PubMed.
- S. W. Ragsdale, Chem. Rev., 2006, 106, 3317 CrossRef CAS and references therein.
- M. Gnida, R. Ferner, L. Gremer, O. Meyer and W. Meyer-Klaucke, Biochemistry, 2003, 42, 222 CrossRef CAS.
- C. L. Drennan, J. Heo, M. D. Sintchak, E. Schreiter and P. W. Ludden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11973 CrossRef CAS; H. Dobbek, V. Shetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2002, 298, 1281.
- V. J. DeRose, J. Tesler, M. E. Anderson, P. A. Lindahl and J. C. Fontecilla-Camps, Nat. Struct. Biol., 2003, 10, 271 CrossRef CAS.
- C. G. Riordan, JBIC, J. Biol. Inorg. Chem., 2004, 9, 509 CAS.
- R. J. Rohlfs, A. J. Mathews, T. E. Carver, J. S. Olson, B. A. Springer, K. D. Egeberg and S. G. Sligar, J. Biol. Chem., 1990, 265, 3168 CAS.
- L. K. Weaver, S. Howe, R. Hopkins and K. J. Chan, Chest, 2000, 117, 801 Search PubMed.
- L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221 CrossRef CAS; M. W. Foster, T. J. McMahon and J. S. Stamler, Trends Mol. Med., 2003, 9, 160 Search PubMed; J. S. Stamler, D. I. Simon, J. A. Osborne, M. E. Mullins, O. Jaraki, T. Michel, D. J. Singel and J. Loscalzo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 444 CAS.
- G. D'Amico, F. Lam, T. Hagen and S. Moncada, J. Cell Sci., 2006, 119, 2291 Search PubMed.
- F. A. D. T. G. Wagener, H. D. Volk, D. Willis, N. G. Abraham, M. P. Soares, G. J. Adema and C. G. Figdor, Pharmacol. Rev., 2003, 55, 551 CrossRef CAS.
- T. L. Brunton, Lancet, 1867, 90, 97 Search PubMed.
- W. Murrell, Lancet, 1879, 113, 80 Search PubMed; W. Murrell, Lancet, 1879, 113, 113 Search PubMed; W. Murrell, Lancet, 1879, 113, 151 Search PubMed; W. Murrell, Lancet, 1879, 113, 225 Search PubMed.
- B. A. Moore, L. E. Otterbein, A. Turler, A. M. K. Choi, A. J. Bauer and J. Anthony, Gastroenterology, 2003, 124, 377 CrossRef CAS.
- G. Farrugia, S. Lei, X. Lin, S. M. Miller, K. A. Nath, C. D. Ferris, M. Levitt and J. H. Szurszewski, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8567 CrossRef CAS.
- L. E. Otterbein, B. S. Zuckerbraun, M. Haga, F. Liu, R. Song, A. Usheva, C. Stachulak, N. Bodyak, R. N. Smith, E. Csizmadia, S. Tyagi, Y. Akamatsu, R. J. Flavell, T. R. Billiar, E. Tzeng, F. H. Bach, A. M. K. Choi and M. P. Soares, Nat. Med. (N. Y.), 2003, 9, 183 Search PubMed.
- B. S. Zuckerman, B. Y. Chin, B. Wegiel, T. R. Billiar, E. Czsimadia, J. Rao, L. Shimoda, E. Ifedigbo, S. Kanno and L. Otterbein, J. Exp. Med., 2006, 203, 2109 CrossRef CAS.
- M. Lavitrano, R. T. Smolenski, A. Musumeci, M. Maccherini, E. Slominska, E. di Florio, A. Bracco, A. Mancini, G. Stassi, M. Patti, R. Giovannoni, A. Froio, F. Simeone, M. Forni, M. L. Bacci, G. D'Alise, E. Cozzi, L. E. Otterbein, M. H. Yacoub, F. H. Bach and F. Calise, FASEB J., 2004, 18, 1093 CAS.
- K. Sato, J. Balla, L. Otterbein, R. N. Smith, S. Brouard, Y. Lin, E. Csizmadia, J. Sevigny, S. C. Robson, G. Vercellotti, A. M. Choi, F. H. Bach and M. P. Soares, J. Immunol., 2001, 166, 4185 CAS.
- http://clinicaltrials.gov/ct/show/NCT00094406 .
- M. Schillinger, M. Exner, W. Mlekusch, M. Haumer, S. Sabeti, R. Ahmadi, O. Wagner and E. Minar, Radiology (Oak Brook, IL, US), 2004, 231, 831 Search PubMed.
- S. A. Bainbridge, E. H. Sidle and G. N. Smith, Med. Hypotheses, 2005, 64, 17 CrossRef CAS.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, 1 Search PubMed.
- R. Motterlini, R. Foresti and C. Green, in Carbon Monoxide and Cardiovascular Functions, ed. R. Wang, CRC Press, Boca Raton, 2002, 249 Search PubMed.
- B. Arregui, B. Lopez, M. G. Salom, F. Valero, C. Navarro and F. J. Fenoy, Kidney Int., 2004, 65, 564 CrossRef CAS.
- J. E. Clark, P. Naughton, S. Shurey, C. J. Green, T. R. Johnson, B. E. Mann, R. Foresti and R. Motterlini, Circ. Res., 2003, 93, e2 Search PubMed.
- B. E. Mann and T. R. Johnson, unpublished results.
- R. Foresti, J. Hammad, J. E. Clark, T. R. Johnson, B. E. Mann, A. Friebe, C. J. Green and R. Motterlini, Br. J. Pharmacol., 2004, 142, 453 CrossRef CAS.
- Y. Tayem, T. R. Johnson, B. E. Mann, C. J. Green and R. Motterlini, Am. J. Physiol., 2006, 290, F789 Search PubMed.
- M. G. Bani-Hani, D. Greenstein, B. E. Mann, C. J. Green and R. Motterlini, J. Pharmacol. Exp. Ther., 2006, 318, 1315 CrossRef CAS.
- S. Chlopicki, R. Olszanecki, E. Marcinkiewicz, M. Lomnicka and R. Motterlini, Cardiovasc. Res., 2006, 71, 393 CrossRef CAS.
- T. R. Johnson, B. E. Mann, I. P. Teasdale, H. Adams, R. Foresti, C. J. Green and R. Motterlini, Dalton Trans., 2007, 1500 RSC.
- R. Alberto, R. Schibli, A. Egli, P. A. Schubiger, W. A. Herrmann, G. Artus, U. Abram and T. A. Kaden, J. Organomet. Chem., 1995, 493, 119 CrossRef CAS.
- R. Motterlini, P. Sawle, J. Hammad, S. Bains, R. Alberto, R. Foresti and C. J. Green, FASEB J., 2005, 19, 284 CAS.
- I. J. S. Fairlamb, A.-K. Duhme-Klair, J. M. Lynam, B. E. Moulton, C. T. O'Brien, P. Sawle, J. Hammad and R. Motterlini, Bioorg. Med. Chem. Lett., 2006, 16, 995 CrossRef CAS.
- D. Scapens, H. Adams, T. R. Johnson, B. E. Mann, P. Sawle, R. Aqil, T. Perrior and R. Motterlini, Dalton Trans., 2007 10.1039/b704832g.
- D. Scapens, T. R. Johnson, B. E. Mann, P. Sawle and R. Motterlini, unpublished work.
- B. E. Mann and R. Motterlini, Preparation of metal complexes for therapeutic delivery of carbon monoxide as vasodilator, PCT Int. Appl., WO 0292075 A2 20021121, 2002. Granted in US, US7045140, 2006 Search PubMed.
- B. E. Mann, T. R. Johnson and R. Motterlini, unpublished results.
- N. G. Abraham, S. Quan, S. Shenouda and A. Kappas, in Carbon Monoxide and Cardiovascular Functions, ed. R. Wang, CRC Press, Boca Raton, 2002, p. 233 Search PubMed.
- H. J. Vreman, R. J. Wong, M. L. Chan, B. W. Young and D. K. Stevenson, Pedriatr. Res., 2000, 47, 463 Search PubMed.
- C. Chauveau, D. Bouchet, J. C. Roussel, P. Mathieu, C. Braudeau, K. Renaudin, L. Tesson, J. P. Soulillou, S. Iyer, R. Buelow and I. Anegon, Am. J. Transplant., 2002, 2, 581 CrossRef CAS; P. N. Martins, A. Reuzel-Selke, A. Jurisch, K. Atrott, A. Pascher, J. Pratschke, R. Buelow, P. Neuhaus, H. D. Volk and S. G. Tullius, Transplant. Proc., 2005, 37, 379 CrossRef CAS.
- K. Mizutani, K. Shinomiya and T. Shinomiya, Forensic Sci. Int., 1988, 38, 113 CrossRef CAS; J. B. Leikin, D. Kaufman, J. W. Lipscomb, A. M. Burda and D. O. Hryhorczuk, Am. J. Emerg. Med., 1990, 8, 534 CrossRef CAS.
- R. Motterlini, R. Foresti, R. Bassi and C. J. Green, Free Radical Biol. Med., 2000, 28, 1303 CrossRef CAS; G. Scapagnini, R. Foresti, V. Calabrese, A. M. G. Stella, C. J. Green and R. Motterlini, Mol. Pharmacol., 2002, 61, 554 CrossRef CAS; E. Balogun, M. Hoque, P. Gong, E. Killeen, C. J. Green, R. Foresti, J. Alam and R. Motterlini, Biochem. J., 2003, 371, 887 CrossRef CAS.
- R. Foresti, M. Hoque, D. Monti, C. J. Green and R. Motterlini, J. Pharmacol. Exp. Ther., 2005, 312, 686 CAS; H. Abuarqoub, R. Foresti, C. J. Green and R. Motterlini, Am. J. Physiol., 2006, 290, C1092 Search PubMed; Y. S. Keum, S. Yu, P. P. Chang, X. Yuan, J. H. Kim, C. Xu, J. Han, A. Agarwal and A. N. Kong, Cancer Res., 2006, 66, 8804 CrossRef CAS.
- D. Martin, A. I. Rojo, M. Salinas, R. Diaz, G. Gallardo, J. Alam, C. M. De Galarreta and A. Cuadrado, J. Biol. Chem., 2004, 279, 8919 CrossRef CAS.
- S. H. Juan, T. H. Cheng, H. C. Lin, Y. L. Chu and W. S. Lee, Biochem. Pharmacol., 2005, 69, 41 CrossRef CAS.
- P. Gong, B. Hu and A. I. Cederbaum, Arch. Biochem. Biophys., 2004, 432, 252 CrossRef CAS.
- V. Nascimento-Silva, M. A. Arruda, C. Barja-Fidalgo, C. G. Villela and I. M. Fierro, Am. J. Physiol., 2005, 289, C557 Search PubMed.
- T. S. Lee, C. C. Chang, Y. Zhu and J. Y. Shyy, Circulation, 2004, 110, 1296 CrossRef CAS.
- R. Wang, FASEB J., 2002, 16, 1792 CrossRef CAS; R. Wang, Antioxid. Redox Signaling, 2003, 5, 493 Search PubMed.
- W. M. Zhoa, K. N. Houk and L. P. Olson, EMBO J., 2001, 20, 6008 CrossRef.
| This journal is © The Royal Society of Chemistry 2007 |