 Open Access Article
Open Access ArticleNovel octavalent cross-linker displays efficient trapping of protein –protein interactions†
Simon R.
Foster
a,
Alice
Pearce
b,
Alexander J.
Blake
a,
Melanie J.
Welham
b and
James
Dowden
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, UK NG7 2RD. E-mail: james.dowden@nottingham.ac.uk; Fax: 0115 95135654; Tel: 0115 9513566
bDepartment of Pharmacy and Pharmacology, University of Bath, Calverton Down, Bath, UK BA2 7AY
First published on 20th March 2007
Abstract
A novel octavalent, resorcin[4]arene derived, cross-linker designed to overcome some of the limitations of commercially available reagents is significantly more efficient for covalent stabilisation of protein –protein interactions.
Transient protein –protein interactions regulate a diversity of cellular responses.1,2 Currently, it is difficult to predict such interactions a priori from sequence information. Thus, methods to characterise protein –protein interactions are of significant interest for cell biology3 as well as the development of small molecule modulators of such interfaces.4 A comparison of wide-scope studies of the Saccharomyces cerevisiaeprotein interactions, including yeast two-hybrid5,6 and tandem affinity purification,7,8 has highlighted poor overlap of data-sets and the need for an intersection of data derived from diverse techniques.9
Chemical cross-linking10,11 provides covalent capture of transient protein –protein interactions and can facilitate topological analysis using mass spectrometry.12 A limitation of cross-linking reagents is that tether length and reactivity must be optimized for individual protein –protein complexes. Furthermore, it is difficult to discriminate between inter- vs. intra-molecular links or non-productive modifications arising from protein modification at one terminus of the cross-linker and, hydrolysis (for example) at the other. Recently an isotope coding strategy has been used to facilitate discrimination between some of these outcomes in the mass spectrum.13 A modular synthetic route that offers rapid access to a versatile arsenal of cross-linkers has also been reported, but the optimum reagent for each protein system must be selected by evaluation of each individual reagent.14
There remains an unmet need to augment the ratio of cross-linked compared to surface-labeled protein species.11 A reagent that does not require optimisation for individual protein systems would be of general utility. Increasing the efficiency of cross-linking reagents is therefore a key design criterion. This communication describes a novel cross-linker architecture that is a more efficient cross-linker than a representative panel of commercial reagents in preliminary comparative experiments.
We envisaged that a resorcinarene scaffold could be elaborated to display multiple functional groups over a large surface area. In turn, this should bias the reaction toward inter-protein links by offering a greater number and span of reactive functionality than contemporary cross-linkers, thus yielding greater efficiency. The well defined geometry available from the resorcinarene architecture offers potentially useful features for future developments, such as analysis of its ‘footprint’ at the protein surface by mass spectrometry. Furthermore, various aldehydes incorporated during resorcinarene synthesis could be easily varied to allow modular enhancement of the design, for example by attaching functionality that may assist in affinity purification.13–15
N-Hydroxysuccinimidylester octavalent cross-linker, SOXL 1, was rapidly synthesised over four linear steps in ∼40% overall yield on a gram scale. Reaction of 4-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)benzaldehyde16 with resorcinol in acidic absolute ethanol at 80 °C furnished the resorcinarene precursor in 78% yield after recrystallisation from hot ethanol (Scheme 1).17 Subsequent alkylation of all eight resorcin[4]arene phenols proceeded in good overall yield of the octaethyl ester 2 using a two molar excess of ethyl 2-bromoacetate for each phenolate . Saponification of the resulting ethyl esters provided the octa-acid 3, which was treated with oxalyl chloride, then N-hydroxysuccinimide (NHS) to yield the prototypical resorcin[4]arene derived succinimidyl octaester cross-linker SOXL 1. We found that employing the polymer-supported base piperidinomethyl polystyrene in this transformation provided high yields of essentially pure product 1.
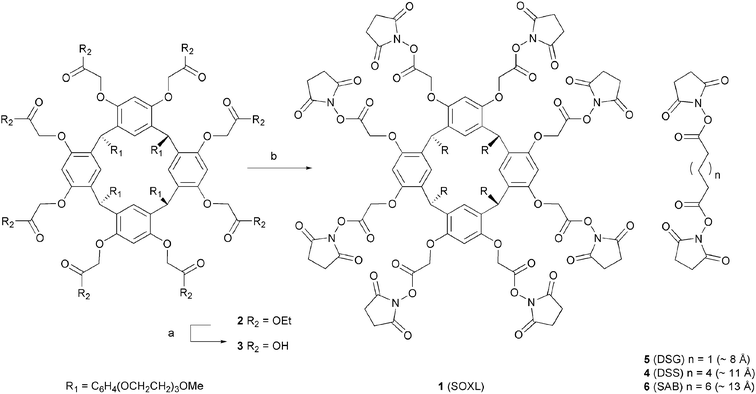 | ||
| Scheme 1 Reagents and conditions: (a) KOH, H2O/THF, rt; (b) (COCl)2, then N-hydroxysuccinimide, piperidinomethyl polystyrene, THF –10 °C. | ||
Comparison of NMR data with closely related polyether derivatives initially suggested a flattened cone structure,17 but we subsequently obtained crystals of the ethyl ester 2 in which the asymmetric unit contains two independent half molecules each lying about an inversion centre.18 This structure confirmed our assignment of the C2h isomer in which adjacent pairs of aldehyde derived groups sit on opposite faces of the central macrocycle . The terminal esters occupy a distorted rhomboid geometry, with C⋯C distances for the carbonyls ranging between 4.5 and 13.5 Å (Fig. 1).
 | ||
| Fig. 1 One centrosymmetric dimer from octaester 2 crystal structure.18 | ||
We then set out to compare the octaester 1 with a commercial homobivalent cross-linker disuccinimidyl suberate, DSS 4, for their efficiency to cross-link a known protein dimer. Glutathione S-transferases (GST) [E.C.2.5.1.18]19 exist as homodimers, although higher order oligomers have also been reported.20 Experiments were performed on the Schistosoma japonicum form of GST purified from E. coli transformed with the pGEX2T expression vector.21
Aliquots of between 1 and 8 molar equivalents of SOXL 1 were added to GST solution (2.7 µM), quenched after 1 h and separated through 10% poly-acrylamideSDS gel, with immunoblotting providing the principal means for visual inspection of the results. In the absence of cross-linker, only monomeric GST is apparent (Fig. 2(a), lane 13). At the same stoichiometry DSS was significantly less effective at achieving measurable cross-linking of GST. At 2, 4, 6 and 8 molar equivalents of cross-linker to GST, increased levels of dimer (∼56 kDa) are apparent in samples treated with SOXL 1 (Fig. 2(a), lanes 3, 5, 7 and 9) compared to DSS 4 (Fig. 2(a), lanes 4, 6, 8 and 10). For SOXL 1 treated lanes, an additional band consistent with addition of at least one copy of SOXL 1 (∼1.6 kDa) to GST monomer (∼27 kDa) is apparent. SOXL 1 treated GST-dimer has a slightly higher molecular weight than that treated with DSS 4, most likely due to the additional molecular weight contributed by SOXL 1. The nature of the higher molecular weight band in the DSS 4 treated lanes is currently not clear.
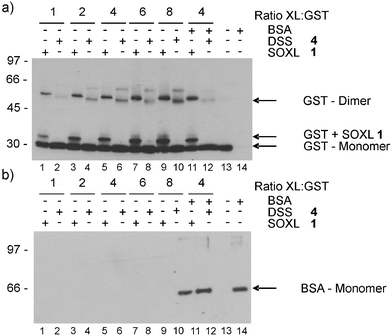 | ||
| Fig. 2 Cross-linking of GST was performed with molar equivalents of either SOXL 1 (lanes 1, 3, 5, 7, 9, 11) or DSS 5 (lanes 2, 4, 6, 8, 10, 12) as indicated. In lanes 11 and 12 an equimolar mixture of GST and BSA was used and a 4-fold excess of cross-linker added. Lanes 13 and 14 represent GST and BSA alone, respectively, in the absence of added cross-linkers. (a) Immunoblotting with anti-GST antibody. (b) Immunoblotting performed on the same gel using anti-BSA antibody. | ||
The improved efficiency is presumably due to presentation of multiple reactive groups that increase the effective molarity of the reactive NHS esters at the protein surface. Lanes 3 and 10 (Fig. 2(a)) offer comparative concentrations of NHS ester and apparently provide similar levels of GST dimer after 1 h. Kinetic measurements of protein cross linking at relevant concentrations of buffer are generally difficult,10,22 thus we were unable to compare the rate of reaction for DSS 4 and SOXL 1 with GST. Time-course experiments suggest that cross-linking with SOXL 1 but not DSS 4, is faintly detectable after 1 min of reaction, apparently reaching completion in 30–60 min (data not shown). The protein concentration and reagent stoichiometry are at the lower limit typically used for cross-linking experiments.10,22 Cross-linking with SOXL 1, however, can be observed in experiments run at lower protein concentrations (0.27 and 0.027 µM), although detection is rather faint, particularly at 0.027 µM GST (data not shown). Consistent observation of GST monomer modified with SOXL 1 suggests that much reagent is consumed by protein rather than hydrolysis, thus linking efficiency could be further improved.
Multiple reactive groups will increase the chance of reaction between any protein in addition to the target protein complex. Ideally, this outcome will be mitigated by the short lifetime of the reactive ester in water,10 which is the dominant reaction for NHS esters at low concentrations of protein and reagent.22 We therefore set out to explore whether non-neighboring proteins might be cross-linked by including bovine serum albumin (BSA), which does not form strong complexes with GST. Equimolar mixtures of BSA and GST exposed to either SOXL 1 or DSS 4 cross-linkers then examined by immunoblotting did not display altered GST-dimer formation (Fig. 2(a), lanes 11 and 12) and importantly, anti-BSA antibody only detected monomer (∼66 kDa), with no evidence of covalently linked GST-BSA (∼93 kDa, Fig. 2(b)) or higher complexes.
SOXL 1 was compared with commercially available cross-linkers of varied size in order to test whether the superior efficiency observed for SOXL 1 is due to the presentation of multiple reactive groups or simply because it spans a more appropriate distance for linking the GST homodimer than DSS 4. Neither five-carbon disuccinimidyl glutarate (DSG, 5 ∼8 Å) or ten-carbon sebacic acid bis(N-succinimidyl) ester (SAB, 6 ∼13 Å) performed any better than DSS 4 (∼11 Å, Fig. 3) and were significantly less efficient than SOXL 1, which discounts a simple distance effect and reinforces the benefit of the multivalent architecture.
 | ||
| Fig. 3 Cross-linking of GST was performed with 1, 2 and 4 molar equivalents of either SOXL 1 (lanes 2, 6 and 10), DSS 4 (lanes 3, 7 and 11), DSG 5 (lanes 4, 8 and 12), or SAB 6 (lanes 5, 9 and 13), as indicated. Lane 1 contains molecular weight markers only. | ||
Evaluation of SOXL 1 for cross-linking other protein –protein interactions is underway. Recently, collaborators have used higher concentrations of SOXL 1 in potentially reactive Tris buffer to define oligomerisation activity of N-terminal and C-terminal domains of the Bacillus subtilis DnaD protein .23 Meanwhile, we are actively exploiting the rapid, adaptable synthesis to achieve modular improvements to prototype 1, such as other protein reactive groups, modulation of solubility and incorporation of biotin for affinity purification. We anticipate that the defined geometry of SOXL 1 may be useful for topological analysis of protein complexes by mass spectrometry and are pursuing further improvement of its architecture to this end, the progress of which will be described in subsequent publications.
This work was supported by grants from BBSRC (E18957) and Wellcome Trust (069955MA). We acknowledge the use of the EPSRC Chemical Database Service at Daresbury.
Notes and references
- J. A. Papin, T. Hunter, B. O. Palsson and S. Subramaniam, Nat. Rev. Mol. Cell. Biol., 2005, 6, 99–111 CrossRef CAS.
- T. Pawson and P. Nash, Science, 2003, 300, 445–452 CrossRef CAS.
- R. B. Russell, F. Alber, P. Aloy, F. P. Davis, D. Korkin, M. Pichaud, M. Topf and A. Sali, Curr. Opin. Struct. Biol., 2004, 14, 313–324 CrossRef CAS.
- S. Fletcher and A. D. Hamilton, Curr. Opin. Chem. Biol., 2005, 9, 632–638 CrossRef CAS.
- T. Ito, T. Chiba, R. Ozawa, M. Yoshida, M. Hattori and Y. Sakaki, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4569–4574 CrossRef CAS.
- A. C. Gavin, M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J. M. Rick, A. M. Michon, C. M. Cruciat, M. Remor, C. Hofert, M. Schelder, M. Brajenovic, H. Ruffner, A. Merino, K. Klein, M. Hudak, D. Dickson, T. Rudi, V. Gnau, A. Bauch, S. Bastuck, B. Huhse, C. Leutwein, M. A. Heurtier, R. R. Copley, A. Edelmann, E. Querfurth, V. Rybin, G. Drewes, M. Raida, T. Bouwmeester, P. Bork, B. Seraphin, B. Kuster, G. Neubauer and G. Superti-Furga, Nature, 2002, 415, 141–147 CrossRef CAS.
- P. Uetz, L. Giot, G. Cagney, T. A. Mansfield, R. S. Judson, J. R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, A. Qureshi-Emili, Y. Li, B. Godwin, D. Conover, T. Kalbfleisch, G. Vijayadamodar, M. J. Yang, M. Johnston, S. Fields and J. M. Rothberg, Nature, 2000, 403, 623–627 CrossRef CAS.
- S. Maslov and K. Sneppen, Science, 2002, 296, 910–913 CrossRef CAS.
- C. von Mering, R. Krause, B. Snel, M. Cornell, S. G. Oliver, S. Fields and P. Bork, Nature, 2002, 417, 399–403 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, London, 1996, pp. 169–297 Search PubMed.
- J. W. Back, L. De Jong, A. O. Muijsers and C. G. De Koster, J. Mol. Biol., 2003, 331, 303–313 CrossRef CAS.
- (a) M. M. Young, N. Tang, J. C. Hempel, C. M. Oshiro, E. W. Taylor, I. D. Kuntz, B. W. Gibson and G. Dollinger, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5802–5806 CrossRef CAS; (b) F. X. Chu, S. O. Shan, D. T. Moustakas, F. Alber, P. F. Egea, R. M. Stroud, P. Walter and A. L. Burlingame, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16454–16459 CrossRef CAS.
- F. X. Chu, S. Mahrus, C. S. Craik and A. L. Burlingame, J. Am. Chem. Soc., 2006, 128, 10362–10363 CrossRef CAS.
- M. Trester-Zedlitz, K. Kamada, S. K. Burley, D. Fenyoe, B. T. Chait and T. W. Muir, J. Am. Chem. Soc., 2003, 125, 2416–2425 CrossRef CAS.
- S. M. Brittain, S. B. Ficarro, A. Brock and E. C. Peters, Nat. Biotechnol., 2005, 23, 463–468 CrossRef CAS.
- C. Lottner, K.-C. Bart, G. Bernhart and H. Brunner, J. Med. Chem., 2002, 45, 2079–2089 CrossRef CAS.
- A. J. Wright, S. E. Matthews, W. B. Fischer and P. D. Beer, Chem.–Eur. J., 2001, 7, 3474–3481 CrossRef CAS.
- Compound 2: (crystallizes with two independent half molecules in the asymmetric unit), colourless tablet, 1.12 × 1.12 × 0.58 mm, triclinic, P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.7830(8), b = 15.3349(11), c = 31.054(2) Å, α = 91.283(2), β = 91.879(2), γ = 102.979(2)°, V = 5462.3(6) Å3, Z = 2, Dc = 1.295 g cm–3, 2θmax = 55°, Mo-Kα, λ = 0.710
, a = 11.7830(8), b = 15.3349(11), c = 31.054(2) Å, α = 91.283(2), β = 91.879(2), γ = 102.979(2)°, V = 5462.3(6) Å3, Z = 2, Dc = 1.295 g cm–3, 2θmax = 55°, Mo-Kα, λ = 0.710![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 73 Å, ω scans, T = 150(2) K, 50321 reflections measured, all 24449 unique used in the refinement, no absorption or extinction corrections applied, structure solution by direct and difference Fourier methods using SHELXS97, structure refinement used SHELXL97, 1374 parameters, H atoms geometrically placed and refined using a riding model, R = 0.0758, wR = 0.235, full-matrix least squares on F2, final residual electron density 1.19 and –0.75 e Å–3. CCDC 297190. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b701542a.
73 Å, ω scans, T = 150(2) K, 50321 reflections measured, all 24449 unique used in the refinement, no absorption or extinction corrections applied, structure solution by direct and difference Fourier methods using SHELXS97, structure refinement used SHELXL97, 1374 parameters, H atoms geometrically placed and refined using a riding model, R = 0.0758, wR = 0.235, full-matrix least squares on F2, final residual electron density 1.19 and –0.75 e Å–3. CCDC 297190. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b701542a. - J. D. Hayes, J. U. Flanagan and I. R. Jowsey, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 51–88 CrossRef CAS.
- D. A. Fancy, K. Melcher, S. A. Johnston and T. Kodadek, Chem. Biol., 1996, 3, 551–559 CrossRef CAS.
- D. B. Smith, M. R. Rubira, R. J. Simpson, K. M. Davern, W. U. Tiu, P. G. Board and G. F. Mitchell, Mol. Biochem. Parasitol., 1988, 27, 249–256 CrossRef CAS.
- G. P. Smith, Bioconjugate Chem., 2006, 17, 501–506 CrossRef CAS.
- M. J. Carneiro, W. Zhang, C. Ioannou, D. J. Scott, S. Allen, C. J. Roberts and P. Soultanas, Mol. Microbiol., 2006, 60, 917–924 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details and characterisation data for compounds 1–3. See DOI: 10.1039/b701542a |
| This journal is © The Royal Society of Chemistry 2007 |