Synthesis of the most intensely fluorescent azobenzene by utilizing the B–N interaction†
Junro
Yoshino
,
Naokazu
Kano
and
Takayuki
Kawashima
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: takayuki@chem.s.u-tokyo.ac.jp; Fax: +81-3-5800-6899; Tel: +81-3-5800-6899
First published on 20th December 2006
Abstract
A boron-substituted azobenzene, (E)-[2-(4-methoxyphenylazo)phenyl]bis(pentafluorophenyl)borane, presented the most intense fluorescence among the azobenzene derivatives.
Azobenzene is one of the most common and frequently-used chromophores .1 Many azobenzene derivatives have been synthesized and used as dyes. Azobenzenes are also used as photoresponsive molecular switches by taking advantage of their photoisomerization .2 Their photoisomerization features inhibit another important property of chromophores , fluorescence. Since the photoisomerization process is very efficient in photoexcited azobenzenes, their radiative deactivation processes, which are slower, are not competitive.3 Hence, azobenzene itself virtually does not emit light.4 There have been only limited exceptional azobenzene derivatives that show fluorescence, however, most of them fluoresce only in a rigid matrix at low temperature and their fluorescence quantum yields, ΦF, are quite low.5Ortho-metalated azobenzene complexes6 and self-assembled aggregates of azobenzene derivatives7 have also been reported as emissive azobenzene derivatives and most of their fluorescence quantum yields at ambient temperature in the solution state are about 10–3, which are still low compared with organic fluorescent materials. In general, azobenzenes have remained as non-fluorescent chromophores while many other organic dye molecules such as rhodamines and fluoresceins have been developed as highly emissive chromophores and utilized for the labeling of biomolecules such as proteins to make them detectable.8 Azobenzenes would have promise to be useful as fluorescent materials, with applications in light-emitting devices, fluorescent probes, and molecular detection, if they emitted fluorescence because of the variety of easy methods for synthesizing azobenzenes and for tuning their properties.
Recently, Yamaguchi et al. reported that the fixation of π-conjugate systems of boryl-substituted thienylthiazoles into a planar conformation with the B–N interaction provided them with fluorescence.9 Meanwhile, during the course of our study on azobenzene derivatives bearing a main-group-element substituent,10 we have found the most intensely fluorescent azobenzene by utilizing the B–N interaction. We report here the synthesis, spectroscopic properties, and theoretical calculations of this azobenzene and related compounds.
Successive reaction of (E)-2-iodoazobenzene ((E)-1) with n-BuLi and fluoroborane 2a in Et2O at –112 °C gave a boron-substituted azobenzene (E)-3a (28%) as a brown solid, which can be handled in the air (Scheme 1). Compounds (E)-3b (31%) and (E)-3c (27%) were synthesized similarly from (E)-1 and 2b and from (E)-4 and 2a, respectively. Tetracoordination states of the boron atoms of (E)-3a–c were confirmed by the 11B NMR spectra in CDCl3 [(E)-3a: δB –0.8, (E)-3b: δB 7.0‡, (E)-3c: δB –0.6]. The X-ray crystallographic analysis of (E)-3a revealed that (E)-3a has a N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond length [1.2704(18) Å]11 which is almost the same as that of the previously reported (E)-azobenzenes (avg. 1.25 Å).12
N bond length [1.2704(18) Å]11 which is almost the same as that of the previously reported (E)-azobenzenes (avg. 1.25 Å).12
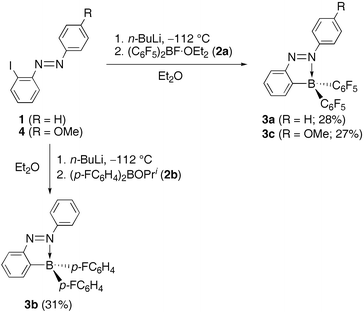 | ||
| Scheme 1 Synthesis of (E)-3a–c. | ||
In the UV–vis spectrum in hexane, (E)-3a showed an absorption maximum at 386 nm (ε = 1.9 × 104), assignable to the π–π* transition of the azo group, and no resolved band corresponding to the n–π* transition (Fig. 1(a)). This large bathochromic shift from the π–π* transition of unsubstituted (E)-azobenzene ((E)-5) (λ = 315 nm) suggests a considerable perturbation of the electronic structure of the azobenzene moiety of (E)-3a by the B–N interaction.
 | ||
| Fig. 1 (a) Absorption (blue line) and fluorescence (red line) spectra of (E)-3a and (E)-3c in hexane at r.t. (b) Photograph of hexane solution (ca. 5 µM) of (E)-3c under irradiation by 360 nm light. | ||
Irradiation (λ = 360 nm) of (E)-3a and (E)-3c in C6D6 at r.t. with a super-high-pressure mercury lamp (500 W) did not cause their photoisomerization to (Z)-3a and (Z)-3c, but caused strong green fluorescence, in contrast to the behavior of most azobenzene derivatives (Fig. 1(b)). In the fluorescence spectra in hexane, (E)-3a and (E)-3c showed emission maxima at 503 nm (excited at 386 nm) and 524 nm (excited at 439 nm), respectively (Fig. 1(a)). Matching between the peak of the excitation spectra corresponding to the emission at the wavelength of the emission maxima and the absorption maxima due to the π–π* transition for both (E)-3a and (E)-3c indicates that these emissions are assignable to the transition from the 1(π, π*) state to the ground state . The fluorescence quantum yields of (E)-3a and (E)-3c in hexane at r.t. were determined as ΦF = 0.23 and 0.76, respectively,13 which are extremely high compared with 2.53 × 10–5 for the unsubstituted azobenzene (E)-5;14 the latter value is higher than those of any azobenzene derivatives reported previously.5–7 In contrast, (E)-3b showed almost no fluorescent emission. The substituents on the boron atom should be responsible for the intensity of the fluorescence, considering the above results.
The electronic states and excitation energies of (E)-3a–c and unsubstituted azobenzene ((E)-5) were investigated by theoretical calculations using density-functional theory (DFT) and a time-dependent DFT (TD-DFT) method at the B3PW91/6-31+G(d)//B3PW91/6-31G(d) level of theory.15 The optimized structure of (E)-3a matches well with its crystal structure. In (E)-5, the π*, n and π orbitals are assignable to the LUMO , HOMO , and HOMO-1, respectively. However, the energy levels of the orbitals of (E)-3a–c that originate from the n orbital of the azo group (–8.9, –8.5, and –8.7 eV, respectively) are lower than those originating from the π orbital of the azo group (–7.1 to –7.3, –6.9 to –7.1, and –6.6 eV, respectively) (Fig. 2(a)). Lowering of the energy level of the n orbital would be caused by the B–N bonding interaction. As a reflection of this anomaly, the 1(π, π*) excited states of (E)-3a–c (2.8 to 3.2, 3.0 to 3.2, and 2.8 eV, respectively) are below the 1(n, π*) excited states (3.9, 4.1, and 4.0 eV, respectively) (Fig. 2(b)). This order of the energy levels is in contrast to that of (E)-5. Inversion of the order between the 1(n, π*) and 1(π, π*) states on the energy scale increases the transition probability from excited states to the ground state based on the orbital symmetries, and this inversion has been considered as one of the causes of fluorescence of protonated azobenzenes and 4-(dialkylamino)azobenzenes.5a,c–e In addition, the π orbitals of the azo group of (E)-3a and (E)-3c lie on their HOMO (Fig. 3(a) and (c)). As a result, the 1(π, π*) excited state of (E)-3a and (E)-3c occupies the lowest singlet excited state S1, and the transition between S1 and ground states is allowed. In contrast, the HOMO of (E)-3b is not on the azobenzene moiety but localized on the aryl groups attached to the boron atom (Fig. 3(b)). The relatively high energy level of the occupied orbitals of p-FC6H4 groups prevents the π orbital of the azo group of (E)-3b from occupying the HOMO and restricts its fluorescence quantum yield to quite low. Therefore, the characteristic emission behavior of (E)-3a and (E)-3c should be attributed to the inverted order of the 1(n, π*) and 1(π, π*) states and the low-lying occupied orbitals of the strong electron-withdrawing C6F5 group.
 | ||
| Fig. 2 Calculated (a) energy levels of frontier molecular orbitals and (b) singlet excitation energies of (E)-azobenzene ((E)-5) and (E)-3a–c at the B3PW91/6-31+G(d)//B3PW91/6-31G(d) level of theory. In (a), the orbitals indicated by gray–black and gray levels originate from the π (π*) and n orbitals of the azo group, respectively. In (b), gray–black and gray excited states have the character of 1(π, π*) and 1(n, π*) states, respectively. | ||
 | ||
| Fig. 3 Molecular orbital diagrams for the HOMO of (a) (E)-3a, (b) (E)-3b, and (c) (E)-3c (B3PW91/6-31+G(d)//B3PW91/6-31G(d)). | ||
In addition, the tight B–N coordination caused by fixation of the boron atom and the nitrogen atom at a suitable position to interact can provide more rigidity for the structure around the azo group than protonation of azo groups and substitution with amino groups.5a,c–e The enhanced rigidity of the structure of (E)-3a and (E)-3c suppresses the conformational change around the azo group like boryl-substituted thienylthiazoles.9 This suppression locks the photoisomerization process around the NN double bond, which is the mechanism responsible for the very efficient radiationless deactivation in the unsubstituted azobenzene,3 and helps provide (E)-3a and (E)-3c with the extremely high fluorescence quantum yield.
In conclusion, we have succeeded in synthesizing the most intensely fluorescent azobenzene. Both the intramolecular B–N dative bond and the strong electron-withdrawing C6F5 groups have been revealed to play an important role in providing the azobenzene with extremely high light-emitting ability, which could not be afforded by protonation, ortho-metalation, or self-assembled aggregation. This new design to provide azobenzene with fluorescence will expand the possibilities of utilizing azobenzenes for more versatile fluorescent probes of chemical sensors and light-emitting devices. Tuning of the emission maximum wavelengths and study on excited states and decay processes are underway.
We thank Tosoh Finechem Corp. and Central Glass Co., Ltd. for gifts of alkyllithiums and fluorine compounds, respectively. This work was partially supported by Grants-in-Aid for The 21st Century COE Program for Frontiers in Fundamental Chemistry (T.K.) and for Scientific Researches from the Ministry of Education, Culture, Sports, Science and Technology, Japan and the Japan Society for the Promotion of Science.
Notes and references
- (a) A. Stolz, Appl. Microbiol. Biotechnol., 2001, 56, 69 Search PubMed; (b) B. Armağan, O. Özdemir, M. Turan and M. S. Çelik, J. Chem. Technol. Biotechnol., 2003, 78, 725 CrossRef CAS.
- Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001, and references cited therein Search PubMed.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234 CrossRef CAS.
- H. Rau, Angew. Chem., Int. Ed. Engl., 1973, 12, 224 CrossRef.
- (a) H. Rau, Ber. Bunsen-Ges. Phys. Chem., 1967, 71, 48; (b) H. Rau, Ber. Bunsen-Ges. Phys. Chem., 1968, 72, 637 CAS; (c) H. Rau, Ber. Bunsen-Ges. Phys. Chem., 1971, 75, 1343 CAS; (d) H. Bisle, M. Römer and H. Rau, Ber. Bunsen-Ges. Phys. Chem., 1976, 80, 301 CAS; (e) M. Nepraš, S. Luňák, Jr., R. Hrdina and J. Fabian, Chem. Phys. Lett., 1989, 159, 366 CrossRef CAS.
- (a) Y. Wakatsuki, H. Yamazaki, P. A. Grutsch, M. Santhanam and C. Kutal, J. Am. Chem. Soc., 1985, 107, 8153 CrossRef CAS; (b) M. Ghedini, D. Pucci, G. Calogero and F. Barigelletti, Chem. Phys. Lett., 1997, 267, 341 CrossRef CAS; (c) M. Ghedini, D. Pucci, A. Crispini, I. Aiello, F. Barigelletti, A. Gessi and O. Francescangeli, Appl. Organomet. Chem., 1999, 13, 565 CrossRef CAS; (d) I. Aiello, M. Ghedini and M. La Deda, J. Lumin., 2002, 96, 249 CrossRef CAS.
- (a) M. Shimomura and T. Kunitake, J. Am. Chem. Soc., 1987, 109, 5175 CrossRef CAS; (b) M. Han and M. Hara, J. Am. Chem. Soc., 2005, 127, 10951 CrossRef CAS; (c) M. R. Han and M. Hara, New J. Chem., 2006, 30, 223 RSC.
- F. Wang, W. B. Tan, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, R1 CrossRef.
- A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170 CrossRef CAS.
- (a) N. Kano, F. Komatsu and T. Kawashima, J. Am. Chem. Soc., 2001, 123, 10778 CrossRef CAS; (b) N. Kano, M. Yamamura and T. Kawashima, J. Am. Chem. Soc., 2004, 126, 6250 CrossRef CAS; (c) N. Kano, J. Yoshino and T. Kawashima, Org. Lett., 2005, 7, 3909 CrossRef CAS; (d) M. Yamamura, N. Kano and T. Kawashima, J. Am. Chem. Soc., 2005, 127, 11954 CrossRef CAS; (e) N. Kano, F. Komatsu, M. Yamamura and T. Kawashima, J. Am. Chem. Soc., 2006, 128, 7097 CrossRef CAS.
- Crystallographic data for (E)-3a: C24H9BF10N2, M = 526.14, triclinic, a = 9.861(7), b = 10.732(6), c = 10.822(7) Å, α = 99.976(7), β = 113.714(8), γ = 91.109(7)°, U = 1027.7(11) Å3, T = 120 K, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, µ(Mo-Kα) = 0.164 mm–1, 6375 reflections measured, 3488 unique (Rint = 0.0191) which were used in all calculations. The final R1 (I > 2σ(I)) and wR2 (all data) were 0.0371 and 0.1068, respectively. GOF = 1.089. CCDC 607217. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b615966d.
, Z = 2, µ(Mo-Kα) = 0.164 mm–1, 6375 reflections measured, 3488 unique (Rint = 0.0191) which were used in all calculations. The final R1 (I > 2σ(I)) and wR2 (all data) were 0.0371 and 0.1068, respectively. GOF = 1.089. CCDC 607217. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b615966d. - R. Allmann, in The Chemistry of the Hydrazo, Azo and Azoxy Groups, ed. S. Patai, John Wiley & Sons, London, 1975, ch. 2, p. 43 Search PubMed.
- B. Valeur, in Molecular Fluorescence, Wiley-VCH, Weinheim, 2002, ch. 6, p. 161 Search PubMed.
- T. Fujino, S. Yu. Arzhantsev and T. Tahara, J. Phys. Chem. A, 2001, 105, 8123 CrossRef CAS.
- Gaussian 03, Revision B.01, M. J. Frisch, et al., Gaussian, Inc., Pittsburgh PA, 2003. See Supporting Information.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of (E)-3a–c, the X-ray crystallographic analysis for (E)-3a, Cartesian coordinates and molecular orbital diagrams for (E)-3a–c. See DOI: 10.1039/b615966d |
| ‡ The VT-NMR revealed that the equilibrium in the solution between the tricoordinate and tetracoordinate boron state of (E)-3b is negligible because its chemical shift was almost unchanged in the temperature range from –60 °C to +60 °C. Our preliminary theoretical calculations on the 11B NMR chemical shifts of (E)-3a and (E)-3b support the proposal that the difference between their observed chemical shifts should be attributed not to the equilibrium but to the difference in the substituents on the boron atom. |
| This journal is © The Royal Society of Chemistry 2007 |