
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric propargyl- and allylboration of hydrazonoesters: a metal-free approach to sterically encumbered chiral α-amino acid derivatives†
Sybrand J. T.
Jonker‡
 a,
Colin
Diner‡
a,
Göran
Schulz
a,
Hiroaki
Iwamoto
a,
Lars
Eriksson
b and
Kálmán J.
Szabó
*a
a,
Colin
Diner‡
a,
Göran
Schulz
a,
Hiroaki
Iwamoto
a,
Lars
Eriksson
b and
Kálmán J.
Szabó
*a
aDepartment of Organic Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: kalman.j.szabo@su.se
bDepartment of Materials and Environmental Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden
First published on 24th October 2018
Abstract
A new asymmetric catalytic propargyl- and allylboration of hydrazonoesters is reported. The reactions utilize allenyl- and allylboronic acids in the presence of the inexpensive parent BINOL catalyst. The reactions can be performed under mild conditions (0 °C) without any metal catalyst or other additives affording sterically encumbered chiral α-amino acids. This is the first metal-free method for the asymmetric propargyl- and allylboration of hydrazonoesters.
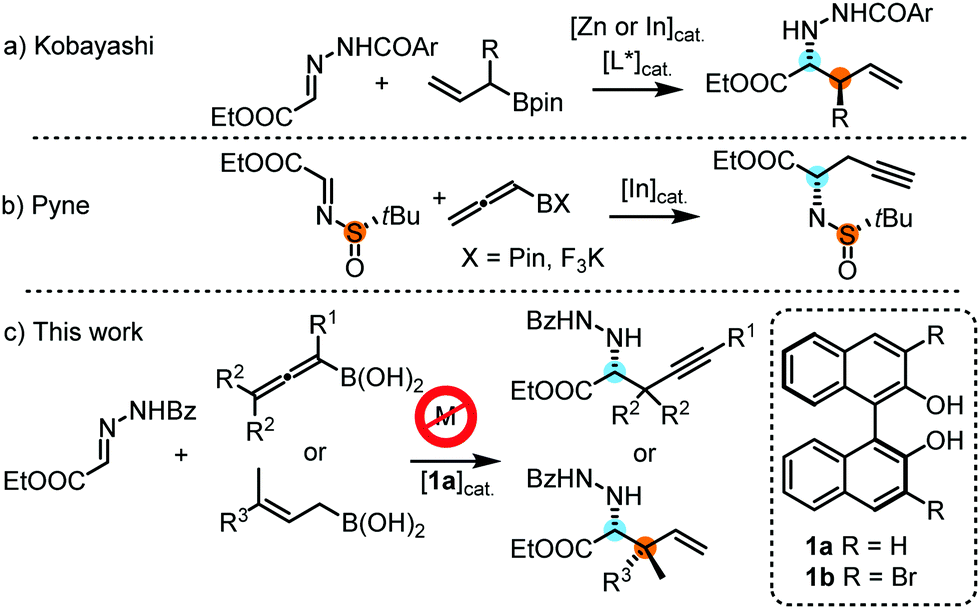
Unnatural α-amino acids represent a very important class of bioactive compounds in life sciences. As a consequence, there has been great interest in developing new methodologies for their synthesis, in particular for the synthesis of chiral α-amino acids.1 However, finding efficient methods for the enantio- and diastereoselective de novo formation of stereocenters in amino acid products is still a challenging task.2 One strategy towards this goal is the enantioselective addition of organoboron species3 to α-iminoesters (Fig. 1). The highly selective allyl-4 and propargylboration5 of ketones and imines has been demonstrated in many recent studies.6 However, the allyl- and propargylboration of iminoesters for the direct synthesis of α-amino acids is less developed. Kobayashi and co-workers7 have shown that Zn-catalyzed allylboration of hydrazonoesters can be used for the asymmetric synthesis of allylated α-amino acids (Fig. 1a). Subsequently, an In-catalyzed variant of this reaction was reported.8 Recently, Pyne and co-workers9 demonstrated that N-tert-butylsulfinyl imines undergo In-catalyzed couplings with allenylboron species (Fig. 1b). This reaction proceeds with high stereoselectivity affording propargylated α-amino acids. Related methods can also be used for the synthesis of racemic α-amino acids.10
 | ||
| Fig. 1 Asymmetric propargyl- and allylboration of iminoesters for the synthesis of α-amino acid derivatives. | ||
Recently, we have published several studies on the application of allyl-11 and allenylboronic12 acids for asymmetric allyl-6a,b and propargylations12 under metal-free conditions using BINOL-based organocatalysts. These methods take advantage of the unusually compact cyclic transition states provided by allylboron reagents that enable high levels of enantio- and diastereoselectivity. In addition, the high reactivity of allyl- and allenylboronic acids allows coupling of densely substituted organoboron species opening a new route for the synthesis of sterically encumbered chiral α-amino acids. Here we present the first report on asymmetric synthesis and catalysis using metal-free allyl- and propargylboration of hydrazone substrates.
Beyond its obvious environmental impact, developing metal-free allyl- and propargylborations has important implications regarding the stereochemical model of this class of reactions. One of the main reasons for using Zn or In catalysis (Fig. 1a and b) in allyl- and propargylborations is that replacing the boron atom with a different metal can make the reagent more reactive or enable a stronger interaction with a chiral element (ligand or coordinating moiety on electrophiles) to achieve efficient stereoinduction. The flip side of this strategy is that the interaction distance between the metal atom and the Lewis-base (nitrogen or oxygen center) will be longer13 than that with the original boron reagent. DFT calculations on the allylboration of imines and ketones indicate short (1.5–1.7 Å) C–O and C–N bonds in the TS.14 A compact TS is beneficial for stereoselectivity because it capitalizes steric repulsions between the interacting substituents in the competing TS structures. Here, we report our new method for a highly selective metal-free propargyl- and allylboration reaction affording α-amino acids with one or two new stereocenters (Fig. 1c).
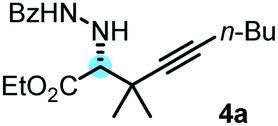
After optimization of the reaction conditions, we observed that benzoyl hydrazone 2 reacted with allenylboronic acid 3a in the presence of BINOL 1a (10 mol%) to afford the propargylated α-amino acid 4a with high enantioselectivity (e.r. 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) and in good yield (79%) when the reaction was conducted in toluene at 0 °C (Table 1, entry 1). The reaction proceeds smoothly with a sterically encumbered (tetrasubstituted) allenylboronic acid derivative (3a) without any metal catalyst or additive. Deviation from the above optimal conditions led to either lower selectivity or yield (entries 2–8) for the formation of 4b. When the reaction time of 48 hours was decreased to 24 hours, product 4a was obtained in a lower yield without any significant change in the enantioselectivity (entry 2). Application of a larger amount (20 mol%) of the BINOL (1a) catalyst did not alter the yield or the selectivity (entry 3). At room temperature, the enantioselectivity (e.r. 88:12) was significantly lower than that at 0 °C (e.r. 96:4, entry 4). In our previous studies on asymmetric allyl- and propargylborations, we observed that the addition of aliphatic alcohols, such as EtOH, led to an increase in enantioselectivity.6a,b,12 However, the addition of only 2 equiv. of EtOH to the reaction mixture led to a major decrease in enantioselectivity from an e.r. of 96:4 (entry 1) to 50:50 (entry 5). This drastic change in selectivity is a clear indication of the mechanistic differences for the allylboration of hydrazones (such as 2) and other species, such as ketones and imines. In the allylation- and propargylation of imines and ketones with allyl- and propargylboronic acids, substituted BINOLs, in particular bromo-BINOL 1b, always outperformed the parent BINOL 1a in regards to enantioselectivity. However, the enantioselectivity with bromo-BINOL 1b (e.r. 64:36, entry 6) was much lower than that with BINOL 1a (e.r. 96:4, entry 1). In addition, the application of 1b instead of 1a led to a major decrease in yield (25%, entry 6). We did not observe product formation (4a) in the absence of BINOL catalysts (entry 7). Under these conditions (0 °C), 2 was practically insoluble in toluene. At room temperature, racemic 4a was formed with 68% yield without BINOL catalysts (entry 8). In previous studies, we have noticed15 that hydrazones have a poor solubility in toluene, and therefore the racemic allylboration reactions were conducted in DMSO as a solvent.15 These studies also indicated that only benzoyl hydrazones (such as 2) reacted with allylboronic acids, while other hydrazones did not give any coupling product.15
:4) and in good yield (79%) when the reaction was conducted in toluene at 0 °C (Table 1, entry 1). The reaction proceeds smoothly with a sterically encumbered (tetrasubstituted) allenylboronic acid derivative (3a) without any metal catalyst or additive. Deviation from the above optimal conditions led to either lower selectivity or yield (entries 2–8) for the formation of 4b. When the reaction time of 48 hours was decreased to 24 hours, product 4a was obtained in a lower yield without any significant change in the enantioselectivity (entry 2). Application of a larger amount (20 mol%) of the BINOL (1a) catalyst did not alter the yield or the selectivity (entry 3). At room temperature, the enantioselectivity (e.r. 88:12) was significantly lower than that at 0 °C (e.r. 96:4, entry 4). In our previous studies on asymmetric allyl- and propargylborations, we observed that the addition of aliphatic alcohols, such as EtOH, led to an increase in enantioselectivity.6a,b,12 However, the addition of only 2 equiv. of EtOH to the reaction mixture led to a major decrease in enantioselectivity from an e.r. of 96:4 (entry 1) to 50:50 (entry 5). This drastic change in selectivity is a clear indication of the mechanistic differences for the allylboration of hydrazones (such as 2) and other species, such as ketones and imines. In the allylation- and propargylation of imines and ketones with allyl- and propargylboronic acids, substituted BINOLs, in particular bromo-BINOL 1b, always outperformed the parent BINOL 1a in regards to enantioselectivity. However, the enantioselectivity with bromo-BINOL 1b (e.r. 64:36, entry 6) was much lower than that with BINOL 1a (e.r. 96:4, entry 1). In addition, the application of 1b instead of 1a led to a major decrease in yield (25%, entry 6). We did not observe product formation (4a) in the absence of BINOL catalysts (entry 7). Under these conditions (0 °C), 2 was practically insoluble in toluene. At room temperature, racemic 4a was formed with 68% yield without BINOL catalysts (entry 8). In previous studies, we have noticed15 that hydrazones have a poor solubility in toluene, and therefore the racemic allylboration reactions were conducted in DMSO as a solvent.15 These studies also indicated that only benzoyl hydrazones (such as 2) reacted with allylboronic acids, while other hydrazones did not give any coupling product.15
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | t (h) | T (°C) | Yield (%) | e.r. |
| 1 | 10 mol% 1a | None | 48 | 0 | 79 | 96:4 |
| 2 | 10 mol% 1a | None | 24 | 0 | 67 | 95:5 |
| 3 | 20 mol% 1a | None | 48 | 0 | 78 | 97:3 |
| 4 | 10 mol% 1a | None | 24 | r.t. | 83 | 88:12 |
| 5 | 10 mol% 1a | 2 equiv. EtOH | 48 | 0 | 52 | 50:50 |
| 6 | 10 mol% 1b | None | 48 | 0 | 25 | 64:36 |
| 7 | No catalyst | None | 48 | 0 | 0 | N/A |
| 8 | No catalyst | None | 24 | r.t. | 68 | 50:50 |
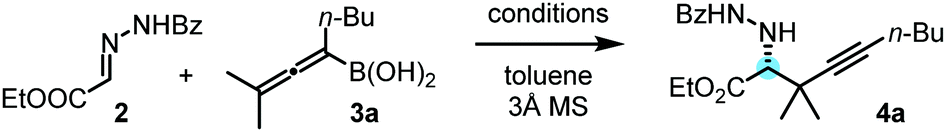
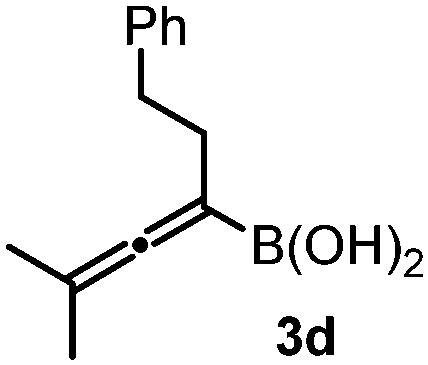
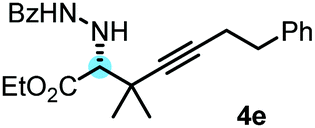
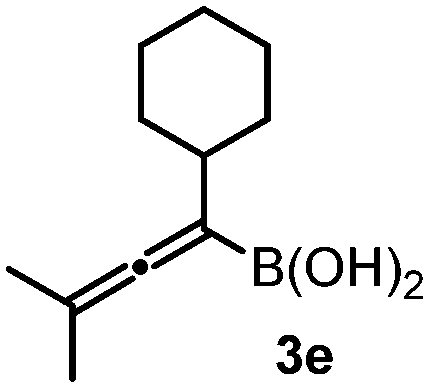
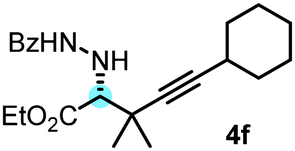
With the above optimization results in hand, we studied the synthetic scope of the reaction (Table 2). First, we studied the effects of replacement of 3a with other tetrasubstituted allenylboronic acids (entries 1–5) and replacement of 1a with its enantiomer. As expected, when (R)-BINOL (entry 2) was employed instead of (S)-BINOL 1a (entry 1), 4b was formed, which is the enantiomeric form of 4a. The yield and selectivity were nearly identical in the two reactions, confirming that our method is suitable for the efficient synthesis of both enantiomers of the propargylated α-amino acids. Trimethyl substituted allenylboronic acid 3b reacted with a bit lower selectivity (e.r. 92:8) than 3a (entry 3). The replacement of the terminal methyl groups with a cyclohexyl ring (3c) did not alter the enantioselectivity (c.f. entries 4 and 1). Likewise, the replacement of the butyl substituent on the borylated carbon of 3a, such as in 3d–e, allowed for highly selective reactions to the corresponding α-amino acid derivatives 4d–e (entries 5 and 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Boronic acid | Product | t (h) | Yieldb (%) | e.r. |
| a Unless otherwise stated, 2 (0.1 mmol), 3 (0.12 mmol) and 1a (0.01 mmol, 10 mol%) were reacted in toluene at 0 °C in the presence of molecular sieves (3 Å) MS. b Isolated yields. c 1 mmol of 2 was applied. d (R)-BINOL (10 mol%) was used as a catalyst instead of (S)-BINOL 1a. e The reaction temperature was −10 °C. | |||||
| 1 |
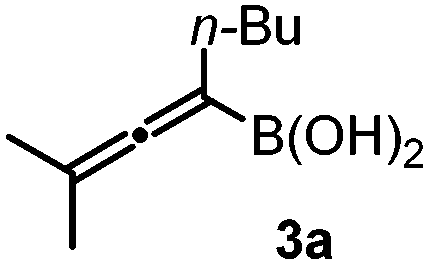
|
|
48 | 79 | 96:4 |
| 75c | |||||
| 2d | 3a |

|
48 | 81 | 96:4 |
| 3e |

|

|
48 | 66 | 92:8 |
| 4 |

|

|
24 | 63 | 96:4 |
| 5 |
|
|
72 | 66 | 95:5 |
| 6 |
|
|
48 | 56 | 96:4 |
| 7 |
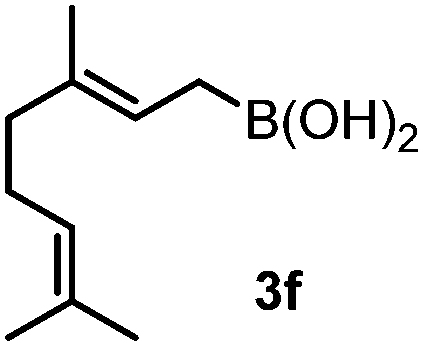
|

|
48 | 71 | 92:8 (97:3 dr) |
| 8d | 3f |

|
48 | 78 | 92:8 (97:3 dr) |
| 9 |

|

|
48 | 81 | 92:8 (98:2 dr) |
| 10d | 3g |

|
48 | 75 | 92:8 (98:2 dr) |

To our delight, even allylboronic acids, such as 3f–g, can be employed in this asymmetric functionalization of hydrazonoester 2. Under the same conditions, geranylboronic acid113f gave α-amino acid 4g in good selectivity (e.r. 92:8) and yield (71%). As 3f is a γ-disubstituted allylboronic acid, two adjacent stereocenters were formed, one of which being quaternary (entry 7). The selective formation of quaternary all carbon stereocenters in acyclic molecules is still considered a challenge in organic synthesis.16 An important requirement in modern drug development is finding synthetic routes to all stereoisomeric forms of the potentially bioactive compounds with two or multiple stereocenters.17 We have found that all four stereoisomeric α-amino acids 4g–j can be synthesized in good selectivity/yield using this method. Application of (R)-BINOL leads to the selective formation of 4h, which is the enantiomer of 4g. An epimer of 4g, compound 4i, can be prepared from nerylboronic acid113g (which is the stereoisomer of 3f). α-Amino acid 4j (enantiomer of 4i and epimer of 4g and 4h) can be prepared by allylboration of hydrazonoester 2 with nerylboronic acid 3g in the presence of catalytic amounts of (R)-BINOL (entry 10). The reactions can be easily scaled up. A 10-fold scale-up of the reaction of 2 with 3a afforded 4a with practically the same yield and selectivity, like the reaction on a 0.1 mmol scale (entry 1).
We were not able to obtain crystals of products 4a–j, which would be suitable for X-ray structure determination, and therefore the absolute configurations were determined by analysis of the corresponding Mosher amides (see the ESI†).18 A similar method was used for determining the absolute configuration of the amino substituted stereocenter in 4g. The relative configuration of the stereocenters was determined by derivatization of 4h (see the ESI†), which gave a crystalline product suitable for X-ray analysis.
A stereoinduction model for the propargylborylation reaction is given in Fig. 2. For simplicity, we selected the reaction of 2 with 3a in the presence of 1a in this model. Similarly to our previous models on the propargylboration of ketones, we suggest that esterification of allenylboronic acid 3a leads to the formation of boronic ester 5. Previously, we have performed NMR studies, which indicated that the B(OH)2 functionality in allylboronic acids undergoes diesterification with BINOL derivatives.6a In addition, our DFT modeling studies on the asymmetric allylboration of imines with allylboronic acids have shown14b that the formation of BINOL esters (such as 5) leads to a considerable increase in the Lewis acidity of the boron atom, and thus an acceleration of the reactions. The steric interactions at the TS are dependent on which face of prochiral 2 is attacked by chiral species 5. In the case of the Si-face attack (TS1), the C3′–H bond of (S)-BINOL 1a and the COOEt moiety are in close proximity. This (or any other) clash is absent in the Re-face arrangement (TS2), which leads to the favored product 4a. Indeed, 4a was formed in high selectivity (e.r. 96:4) in the above reaction (Table 1, entry 1). As mentioned above, 2 has a very poor solubility in toluene at 0 °C, which may explain why racemic propargylboration in the absence of BINOL did not occur (Table 1, entry 7). The poor performance of bromo-BINOL 1b (Table 1, entry 6) is probably due to possible clashes even in a TS2-like transition state, for example by the (long) C3–Br bond and the NHCOPh group of 2. Accordingly, when bromo-BINOL 1b is applied as a catalyst, the C3′–Br bond is supposed to clash with the COOEt moiety in a TS1-type of transition state, while the C3–Br bond and the NHCOPh group would clash in TS2 rendering both transition states sterically hindered. The relatively low yield (25%) in the case of application of bromo-BINOL 1b (Table 1, entry 6) indicates a slow reaction, which is probably the consequence of the relatively high activation barrier raised by both the TS1- and TS2-types of transition states.
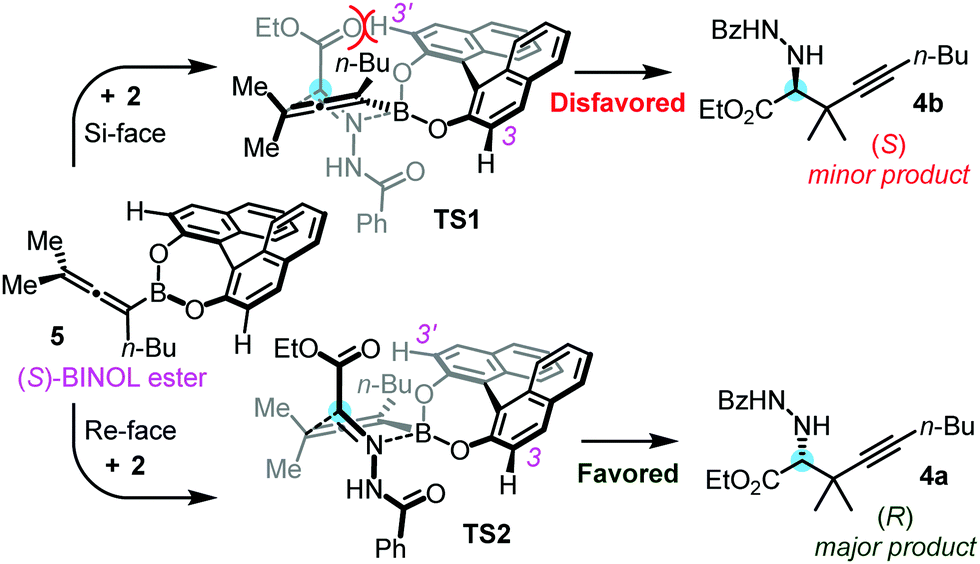 | ||
| Fig. 2 Proposed stereoinduction model for the asymmetric propargylation of hydrazonoester 2 with BINOL ester 5. | ||
A simplified stereoinduction model is presented for the allylboration reaction of 2 and 3f in the presence of (S)-BINOL 1a in Fig. 3. In the case of the Re-face approach (TS3), there is no major clash between the COOEt moiety of 2 and the C3′–H bond of (S)-BINOL ester 6. The relative stereochemistry of the newly formed stereocenters is controlled by the E-configuration of the COOEt and NHCOPh functionalities along the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of 2. A proposed mechanism for the catalytic cycle for propargylboration of 2 with 3a in the presence of BINOL 1a is presented in the ESI.†
N bond of 2. A proposed mechanism for the catalytic cycle for propargylboration of 2 with 3a in the presence of BINOL 1a is presented in the ESI.†
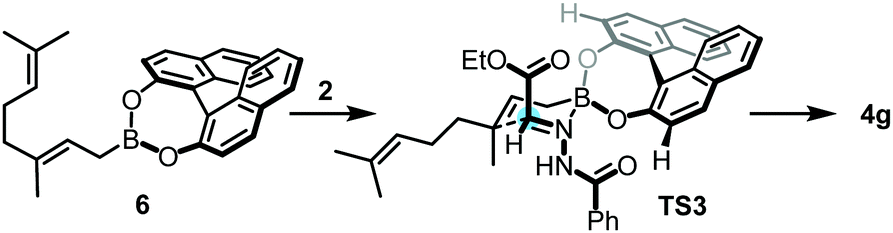 | ||
| Fig. 3 Proposed stereoinduction model for the asymmetric allylation of hydrazonoester 2 with BINOL ester 6. | ||
In conclusion, we have shown that the catalytic asymmetric propargyl- and allylboration of hydrazonoester 2 can be performed in very high selectivity in the presence of inexpensive BINOL. The reaction results in α-amino acids with high enantioselectivity and in generally good yields. The reactions can be performed without the application of metal catalysts, which is probably beneficial for the very high selectivity. In fact, this study presents the first metal-free asymmetric propargyl- and allylboration of hydrazones. In this reaction densely substituted allenyl- and allylboronic acids can be employed opening a new route to asymmetric synthesis of sterically encumbered α-amino acids. Thus, the present study further expands the synthetic scope of propargyl- and allylboronic acids, which are useful species in natural product synthesis19 and in the asymmetric synthesis of complex molecules.6a,b
We are grateful to Mr Erik Ulriksson Ardehed for performing some of the experiments. Support from the Swedish Research Council is greatly appreciated. An ERASMUS stipend for G. S. is gratefully acknowledged. H. I. thanks the Japan Society for the Promotion of Science (JSPS) for financial support. A generous gift of B2(OH)4 from Allychem is highly appreciated.
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. A. Soloshonok and K. Izawa, Current Frontiers in Asymmetric Synthesis and Application of alpha-Amino Acids, ACS Symposium Series, Oxford University Press, Oxford, 2009 Search PubMed.
- (a) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS PubMed; (b) H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS PubMed; (c) J. L. Aceña, A. E. Sorochinsky and V. A. Soloshonok, Synthesis, 2012, 1591 CrossRef; (d) U. Kazmaier, S. Maier and F. L. Zumpe, Synlett, 2000, 1523 CAS; (e) Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 5127 CrossRef CAS; (f) T. Kanayama, K. Yoshida, H. Miyabe and Y. Takemoto, Angew. Chem., Int. Ed., 2003, 42, 2054 CrossRef CAS PubMed; (g) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080 CrossRef CAS PubMed; (h) R. Berger, K. Duff and J. L. Leighton, J. Am. Chem. Soc., 2004, 126, 5686 CrossRef CAS PubMed.
- (a) D. G. Hall, Boronic Acids, Wiley, Weinheim, 2011 CrossRef; (b) D. G. Hall and H. Lachance, Allylboration of Carbonyl Compounds, Wiley, Hoboken, New Jersey, 2012 Search PubMed.
- (a) C. Diner and K. J. Szabó, J. Am. Chem. Soc., 2017, 139, 2 CrossRef CAS PubMed; (b) H.-X. Huo, J. R. Duvall, M.-Y. Huang and R. Hong, Org. Chem. Front., 2014, 1, 303 RSC; (c) T. R. Ramadhar and R. A. Batey, Synthesis, 2011, 1321 CAS.
- T. Thaima, F. Zamani, C. J. T. Hyland and S. G. Pyne, Synthesis, 2018, 1461 Search PubMed.
- (a) R. Alam, T. Vollgraff, L. Eriksson and K. J. Szabó, J. Am. Chem. Soc., 2015, 137, 11262 CrossRef CAS PubMed; (b) R. Alam, C. Diner, S. Jonker, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 14417 CrossRef CAS PubMed; (c) R. J. Morrison and A. H. Hoveyda, Angew. Chem., Int. Ed., 2018, 57, 11654 CrossRef CAS PubMed; (d) F. W. van der Mei, H. Miyamoto, D. L. Silverio and A. H. Hoveyda, Angew. Chem., Int. Ed., 2016, 55, 4701 CrossRef CAS PubMed; (e) D. L. Silverio, S. Torker, T. Pilyugina, E. M. Vieira, M. L. Snapper, F. Haeffner and A. H. Hoveyda, Nature, 2013, 494, 216 CrossRef CAS PubMed; (f) J. L. Y. Chen and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 10992 CrossRef CAS PubMed; (g) K. Yeung, R. E. Ruscoe, J. Rae, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 11912 CrossRef CAS PubMed; (h) Y. Jiang and S. E. Schaus, Angew. Chem., Int. Ed., 2017, 56, 1544 CrossRef CAS PubMed; (i) S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2006, 128, 12660 CrossRef CAS PubMed; (j) S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2007, 129, 15398 CrossRef CAS PubMed; (k) H. Wu, F. Haeffner and A. H. Hoveyda, J. Am. Chem. Soc., 2014, 136, 3780 CrossRef CAS PubMed; (l) N. Balasubramanian, T. Mandal and G. R. Cook, Org. Lett., 2015, 17, 314 CrossRef CAS PubMed; (m) T. R. Wu and J. M. Chong, J. Am. Chem. Soc., 2005, 127, 3244 CrossRef CAS PubMed; (n) J. M. Chong, L. Shen and N. J. Taylor, J. Am. Chem. Soc., 2000, 122, 1822 CrossRef CAS.
- M. Fujita, T. Nagano, U. Schneider, T. Hamada, C. Ogawa and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 2914 CrossRef CAS PubMed.
- A. Chakrabarti, H. Konishi, M. Yamaguchi, U. Schneider and S. Kobayashi, Angew. Chem., Int. Ed., 2010, 49, 1838 CrossRef CAS PubMed.
- R. K. Chambers, N. Chaipukdee, T. Thaima, K. Kanokmedhakul and S. G. Pyne, Eur. J. Org. Chem., 2016, 3765 CrossRef CAS.
- (a) F. Liepouri, G. Bernasconi and N. A. Petasis, Org. Lett., 2015, 17, 1628 CrossRef CAS PubMed; (b) S. Kobayashi, T. Kitanosono and M. Ueno, Synlett, 2010, 2033 CrossRef CAS; (c) N. Selander, A. Kipke, S. Sebelius and K. J. Szabó, J. Am. Chem. Soc., 2007, 129, 13723 CrossRef CAS PubMed.
- M. Raducan, R. Alam and K. J. Szabó, Angew. Chem., Int. Ed., 2012, 51, 13050 CrossRef CAS PubMed.
- J. Zhao, S. J. T. Jonker, D. N. Meyer, G. Schulz, C. D. Tran, L. Eriksson and K. J. Szabo, Chem. Sci., 2018, 9, 3305 RSC.
- (a) R. W. Hoffmann, Angew. Chem., Int. Ed., 1982, 21, 555 CrossRef; (b) D. A. Evans, J. V. Nelson and T. R. Taber, Top. Stereochem., 1982, 13, 1 CAS.
- (a) R. Alam, A. Das, G. Huang, L. Eriksson, F. Himo and K. J. Szabó, Chem. Sci., 2014, 5, 2732 RSC; (b) G. Huang, C. Diner, K. J. Szabó and F. Himo, Org. Lett., 2017, 19, 5904 CrossRef CAS PubMed.
- A. Das, R. Alam, L. Eriksson and K. J. Szabó, Org. Lett., 2014, 16, 3808 CrossRef CAS PubMed.
- (a) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef CAS PubMed; (b) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed.
- (a) D. A. Allen, A. E. Tomaso, O. P. Priest, D. F. Hindson and J. L. Hurlburt, J. Chem. Educ., 2008, 85, 698 CrossRef CAS; (b) T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451 CrossRef CAS PubMed.
- (a) Z. Lu, M. Yang, P. Chen, X. Xiong and A. Li, Angew. Chem., Int. Ed., 2014, 53, 13840 CrossRef CAS PubMed; (b) E. P. Farney, S. S. Feng, F. Schäfers and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 1267 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and compound characterization data are given. CCDC 1850578. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc07908k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |