NMR-based screening: a powerful tool in fragment-based drug discovery†
Jochen
Klages
a,
Murray
Coles
b and
Horst
Kessler
a
aDepartment Chemie, TU München, Lichtenbergstr. 4, 85747 Garching, Germany
bDepartment of Protein Evolution, Max-Planck-Institute for Developmental Biology, Spemannstrasse 35, 72076 Tübingen, Germany
First published on 5th May 2006
1 Introduction
NMR has become a powerful tool in the pharmaceutical industry for a variety of applications. Instrumental improvements in recent years have contributed significantly to this development. Digital recording, cryogenic probes, autosamplers, and higher magnetic fields shorten the time for data acquisition and improve the spectral quality. In addition, new experiments and pulse sequences make a vast amount of information available for the drug discovery process. As a consequence, new screening technologies involving NMR have become indispensable for modern drug discovery. These developments have been summarized in several recent reviews.1–12The drug development process is usually divided into the following familiar phases. Initially, a target is identified that may be involved in initiating or blocking a certain physiological effect. Most drug targets are proteins, but RNA and DNA also can serve as targets for drug discovery. When a suitable target is identified, it is screened for compounds that may cause the desired biological effect. Assays involving binding to the target, either directly or in competition with labelled ligands, are often used in place of functional (cellular) tests. The molecules that are identified as hits from binding screens are validated for the desired biological effect, e.g. inhibition of enzymatic activity. The remaining leads are then optimized by structural refinement to increase the affinity and selectivity for the receptor and for bioavailability and other in vivo pharmacological and physiological properties, which may eventually result in a drug candidate.
NMR can assist the above process at different stages, namely, in hit finding, hit validation, and lead optimization.13 These three areas of application will be discussed in detail in the following sections. The drug discovery process is summarized in Figure 1, in which shaded boxes indicate where incorporation of NMR techniques is possible.
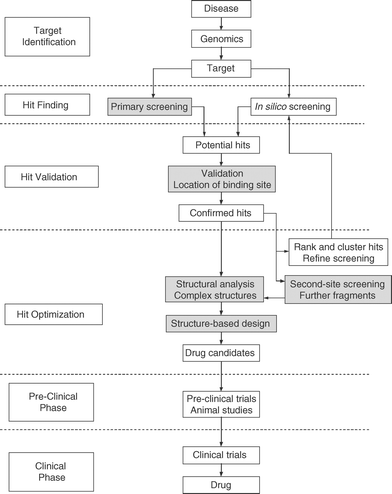 | ||
| Fig. 1 A flowchart outlining the drug discovery process; steps allowing the application of NMR techniques are shaded. | ||
NMR offers some unique features that make it an attractive alternative for applications in drug research. The primary, intrinsic advantage, however, is the ability to detect weak intermolecular interactions, e.g. between a ligand and a target, with unmatched sensitivity. This ability makes NMR ideal for fragment-based screening.1,14–18 In a fragment-based approach, comparably small and simple molecules are screened for binding to a target. These compounds often reveal only a weak affinity. However, when several fragments for different binding sites of the target are identified, they can be linked to form higher affinity ligands. Although the linkage of fragments is an additional difficulty, this approach accesses a larger structural space because each fragment can be optimized separately.
In contrast, structural space is significantly restricted in conventional high throughput screening (HTS), in which the identified hits often are hydrophobic and possess relatively high molecular masses already, and it is difficult to improve their activity without further increasing either their hydrophobicity or molecular weight, or both. Lipinski's “rule of five”19 (Table 1) requires a certain window of lipophilicity, a maximum of five hydrogen bond donors and <10 hydrogen bond acceptors, and in addition a molecular weight below 500 Da. Clearly, the fragment-based approach carries a higher potential for designing efficient drug candidates within the limits set by the “rule of five”. The advantage of the fragment-based approach over other techniques like HTS is illustrated in Figure 2.
 | ||
| Fig. 2 Schematic representation of the advantage of the fragment-based approach over conventional HTS screening.14 The range of affinities and molecular weights of average HTS hits and fragments are illustrated. The thickly dashed line indicates the size limit set by Lipinski's “rule of five”. Whereas HTS hits tend to have high molecular masses, fragments show much lower molecular weights but only slightly lower affinities, which conveys higher lead-like character. | ||
| “Rule of five” criteria19 |
| • Molecular weight ≤500 Da |
| • Log P ≤ 5 |
| • Hydrogen bond donors (OH and NH) ≤ 5 |
| • Hydrogen bond acceptors (lone-pairs of hetero-atoms, like O and N) ≤10 |
| Other criteria(Verber-rules77) |
| • Number of heavy atoms 10–70 |
| • Rotatable bonds 2–8 |
| • Number of rings 1–6, aromatic ≤3 |
| • Molar refractivity 40–130 |
| • Size of hydrophobic area |
Additionally, high throughput screens usually work as functional or competitive binding assays with labelled ligands that are often derived from the natural binding molecules. Establishing a functional assay can be tedious, whereas binding can be measured easily. An advantage of NMR is that it is applicable even when no functional assay or known binders are available; moreover, NMR can be used for validation of hits from functional HTS screening.
A further key advantage of NMR is its ability to provide additional structural information, which is welcome even at the earliest stages of the drug development process. Structural information supports rational lead design or the design of biased libraries based on known structure activity relationships (SARs).
The number of compounds that can be screened by NMR is limited to about 103–104depending on the technology used – much lower than for HTS and in silico screening methods (105–106compounds). Hence, it is not the number of compounds that makes NMR an attractive method, but rather the possibility to use fragment-based screening. In combination with the structural information that can be extracted simultaneously, NMR experiments can open up new, unexplored regions of chemical diversity in the search for drugs.
2 NMR screening: general aspects
It is clear that to carry out ligand screening by NMR, the library compounds (ligands) must be distinguished from the target. One of the most important methods of achieving this is through their vastly different molecular masses. Several NMR parameters are dependent on the rotational and translational diffusion rates of the subject molecule, and therefore on molecular mass. Notably, the transverse (entropic) relaxation of the NMR signal is dependent on the rotational diffusion rate. Large, slowly tumbling target molecules relax much faster than rapidly tumbling small molecules. As the transverse relaxation rate is directly related to linewidth, small molecules can be distinguished by their narrow lines. Also, filters suppressing broad signals can be used to minimize signals from the target. Other relaxation-related parameters, such as the nuclear Overhauser effect (NOE), are also related to rotational diffusion rates; small molecules show a positive enhancement and large molecules a negative enhancement. In contrast to rotational diffusion, translational diffusion can be measured directly by NMR. This technique allows rapidly diffusing small molecules to be distinguished from the slowly diffusing target by suppressing the signal from the former.These differences in NMR properties between small and large molecules form the basis of many NMR screening experiments. When a small ligand binds to a large target, it adopts the properties of the target to an extent dependent on the residence time of the binding event (Figure 3). NMR screening experiments can therefore be designed to detect molecules with intermediate properties. It is clear that distinction on the basis of molecular mass would fail if the ligands were not considerably smaller than the target. It should also become clear in the following sections that the use of relaxation or translational diffusion filters to suppress ligand and/or target signals places considerable affinity limits on the techniques involved.
 | ||
| Fig. 3 Alteration of the physicochemical properties during the process of binding. On binding, the ligand adopts the properties of the large target molecule due to drastic increase in the effective molecular mass. | ||
A second major method of distinguishing ligand and target signals does not rely on molecular mass, but on the much greater dispersion of chemical shifts usually observed for the target. This property can be exploited to create non-equilibrium magnetization specifically on the target, which can be transferred to binding ligands and subsequently detected. By far, the most robust method of distinguishing ligands and target, however, takes this a step further, using specific isotope labelling of one or the other component to create a unique chemical shift range. This strategy allows some of the simplest NMR screening techniques to be used, relying on the change in chemical environment induced by the binding ligand. This change in environment affects the chemical shifts of both the target and a binding ligand, predominantly at the binding site. Thus, chemical shift changes observed in a (15N- or 13C-labelled) target can localize and partially characterize the binding site. Distinction via isotope labelling avoids the use of relaxation- and/or diffusion-based filters to suppress the unwanted component, giving these techniques a much wider affinity range. These techniques are also ideally suited to competitive screening applications, which represent some of the most promising new developments in the field.
3 Ligand-vs. target-detected methods
NMR screening can be divided into ligand- and target-detected methods. In the first class, changes induced in the ligand's NMR signals by binding to a large target are observed, whereas in the second class, the influences of ligands on the spectra of the target are detected. Both techniques have their intrinsic advantages for different applications, making them largely complementary.Ligand-based methods predominantly make use of one-dimensional (1D) NMR spectra and therefore are comparably fast, allowing higher throughput in screening. The detection of the ligand signals offers the opportunity to screen mixtures of ligands without the need for deconvolution, as long as the signals of the ligands do not overlap. Moreover, the amount of target required for the screening process is smaller and there are fewer restrictions on the properties of the target. Commonly, the target is not isotopically labelled and its molecular mass is practically unlimited; in some techniques (e.g. saturation transfer difference (STD), see below), the target can even be immobilized in membranes. Finally, some crude information about the binding epitope and the binding mode can be extracted by ligand-based methods.20
Most ligand-detected methods are limited to low and medium affinities because of the need to suppress the signals of the target molecule. Ligands that bind too tightly are indistinguishable from the target and thus are suppressed as well, resulting in false negatives. In contrast, non-specific binding can result in the appearance of false positives.
Target-based methods can easily distinguish between non-specific and specific binding. In addition, effects caused by aggregation and pH-changes can be excluded. The detectable affinity range is much higher than in ligand-detected techniques, particularly at the high-affinity end. However, the most valuable advantage is the possibility of extracting detailed structural information about the ligand–target complex from the spectra. For target-detected experiments comparably large amounts of the protein target are needed, because the observation of its NMR signals requires concentrations in the range of 0.1–1 mM. These targets usually have to be labelled with NMR active isotopes (15N and/or 13C) making these techniques quite costly. In contrast to ligand-detected screening methods, it is not possible to screen mixtures of compounds without deconvolution. The standard implementation of these experiments includes two-dimensional (2D) spectra, which require longer acquisition times. As the targets usually have high molecular masses (>10 kDa), the signals are subject to fast relaxation. This limitation restricts the size of targets that can be observed to molecular weights <100 kDa, even if techniques like transverse relaxation-optimized spectroscopy (TROSY)21 or cross-relaxation-induced polarization transfer (CRIPT)22 are included. Although this limit may be overcome with specific methyl group labelling in the future, interesting targets might still remain intractable with these techniques.
3.1 Sample requirements
Because the interaction between the ligand and the target usually takes place in aqueous solution, the ligands have to be soluble in water to an appropriate level; otherwise, aggregation effects lead to false positives during the screening process. For standard ligand-based techniques, a concentration of 100 μM is found to be satisfactory for a reasonable signal-to-noise ratio. The target, which is usually unlabelled, is sufficiently concentrated when it is within a range of 1–50 μM; for a 0.5 mL solution, this corresponds to 0.1–5 mg of protein with a molecular weight of 20 kDa. In target-detected experiments, however, labelling is essential to avoid signal overlap. Moreover, higher concentrations are necessary, raising the amount of target needed to 100–300 μM. Depending on the size and the relaxation properties of the target, different labelling schemes are necessary. For relatively small proteins, uniform 15N-labelling is sufficient. For larger proteins, 13C-labelling and/or deuteration may be required. Site- or amino acid-selective labelling23 can extend the size of the proteins, which can be studied as overlap is reduced. Selective 13C-labelling of the methyl groups of valine, leucine, and isoleucine24–27 has proved to be especially advantageous because these residues are at the surface of hydrophobic areas, which are often involved in binding events. In addition, methyl group signals are usually narrower and therefore better resolved.4 Incorporation of NMR into the drug discovery process
In the following detailed discussion of the application of NMR techniques to drug discovery, a short description for hit finding, hit validation, and hit (lead) optimization is given. In addition, the corresponding requirements for the NMR experiments and the resulting implementations are described. Only typical experiments are given explicitly, but other techniques are summarized in Table 2.| Method | Application | Limits and requirements | Identification of | ||||
|---|---|---|---|---|---|---|---|
| Target MW limit | Affinity limit | Labelled target req. | Binding site on target | Binding epitope on ligand | Binding comp. in mixtures | ||
| Diffusion filtering | Hit finding | Lower | U/L | No | No | No | Yes |
| Relaxation filtering | Hit finding | Lower | U/L | No | No | No | Yes |
| TrNOE | Hit finding | Lower | U/L | No | No | No | Yes |
| NOE pumping | Hit finding | Lower | U/L | No | No | No | Yes |
| Rev. NOE pumping | Hit finding | Lower | U/L | No | No | Yes | Yes |
| WaterLOGSY | Hit finding | Lower | U/L | No | No | Yes | Yes |
| STD | Hit finding | Lower | U/L | No | No | Yes | Yes |
| 19F-screening | Hit finding | None | None | 19F ligand | No | No | Yes |
| CSM | Hit validation | Upper | None | 15N or 13C | Yes | No | No |
| Comp. screening | Hit optimization | None | None | No | Yes | No | No |
| Comp. 19F-screening | Hit optimization | None | None | 19F ligand | Yes | No | No |
The classification of drug discovery into different phases is to a certain extent artificial – the different phases overlap or can be applied simultaneously – but the general outline indicated here shows that more complex NMR techniques are needed when the drug discovery process is more advanced.
4.1 Hit finding
In hit finding, a large number of compounds, the so-called library, is screened vs. a target to identify components that bind. Libraries can meet different types of requirements, and efforts are made to optimize them with respect to diversity, solubility, drug-like character (see Table 1), and the synthetic accessibility of the compounds they contain.28,29 Substance libraries can contain small organic fragments, synthetic compounds of diverse chemical structures, or natural products.30Experiments for hit finding by NMR spectroscopy should allow high throughput, since substance libraries can easily contain up to 104 compounds. Hence, time-optimized techniques are required that not only reduce experimental time, but also the time for sample preparation and data analysis. Consequently, lengthy 2D or three-dimensional (3D) techniques are unfavourable, as are complex experiments that require fine tuning for each sample. For an industrial application, the ability to identify binding components from large mixtures without deconvolution is nearly indispensable. Moreover, labelled samples and target-consuming techniques should be avoided for cost reduction. These requirements make 1D ligand-based experiments the most favourable alternative.
STD is one of the most useful experiments for NMR screening. Here, non-equilibrium magnetization is achieved by selective excitation (on-resonance) of target resonances. The magnetization is transferred within the target via spin diffusion and eventually to a bound ligand (Figure 4). When this ligand dissociates from the target into solution, the magnetization change transferred in the bound state is retained in the free ligand. The difference from a reference spectrum taken without on-resonant irradiation thus yields a spectrum containing only those ligands that have been perturbed by binding to the target. The reference spectrum is obtained by off-resonance irradiation in a spectral window where no target signals appear (Figure 5).
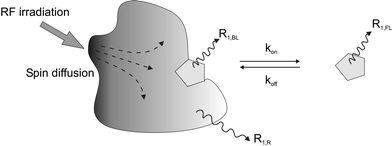 | ||
| Fig. 4 Illustration of the effect of on-resonant irradiation of protein signals. Due to spin diffusion, the resulting non-equilibrium magnetization spreads out across the target molecule and is transferred to the ligand. The magnetization of the bound ligand decays rapidly with a rate of R1,BL, which is of the same order as the rate for the target molecule R1,R. On dissociation into solution, the relaxation properties of the ligand change and the acquired non-equilibrium magnetization now decays with a rate of R1,FL for the free ligand.12 | ||
 | ||
| Fig. 5 Schematic representation of the procedure for the measurement of STD experiments.12 Polygons and stars represent binding and non-binding components, respectively. During the off-resonant experiment (top panel), no magnetization is transferred from the binding ligand to the target molecule. If the irradiation changed to “on-resonant frequencies” (middle panel), magnetization transfer from the bound ligand to the target occurs, which lowers the signal intensity of the ligand. The difference between the two spectra only contains the signals of binding components. | ||
STD spectroscopy offers several advantages. First, there is almost no upper limit for the size of the target, even membrane proteins or immobilized targets are applicable.32 On the contrary, since the correlation time increases with increasing molecular mass, spin diffusion becomes more and more effective, which in turn leads to stronger STD effects. For this technique, the amount of target and ligand needed to give reasonable results is relatively small. In larger ligands, the part of the molecule that is in direct contact with the target is most strongly affected. Hence, structural information of the complex from the ligand side is provided. Estimations of the binding constants can also be drawn.20 STD spectra are usually recorded as 1D spectra, but they can be easily extended to 2D or 3D versions if necessary.37 This option offers a diverse tool for a variety of different tasks in screening. STD fundamentally depends on an effective spin-diffusion mechanism and therefore on proton density. Hence, the observed STD effect might not be large enough to be detected, especially when small targets are used. As it is the free fraction of the ligand that is observed, one normally sees a dramatic amplification effect if the ligand concentration is higher than that of the target. However, similar to other ligand-based techniques, STD is not applicable to high-affinity binders.
If the target does not provide a large enough proton density, as is the case with nucleic acids,12 the WaterLOGSY (water-ligand observed via gradient spectroscopy) technique might be preferred. The large bulk water magnetization is used for an effective transfer via the ligand–target complex to the free ligand in solution. By selectively inverting or saturating the water resonance, the magnetization is transferred to the target and then finally to the ligand. The exact details of the transfer are more complicated and will not be discussed in this review. The important point is the difference in the cross-relaxation properties of the water with non-binding ligands and binding ligands. In the first case, the relaxation includes small correlation times leading to positive cross-relaxation rates, in contrast to the second case in which positive cross-relaxation rates are observed. As a consequence, non-binding and binding ligands show different signs in the spectrum. This combination can lead to an erroneous interpretation if positive and negative peaks cancel each other out.
Of course, the need for 19F-labels seems to be a profound drawback. However, about 10% of all drugs on the market already contain fluorine10 mainly for modification of metabolic stability. Additionally, the small fluorine atom can be often replaced by hydrogen without loss of binding affinity.
4.2 Hit validation
For all compounds found in the primary screen, the binding has to be validated to remove false positives, which is especially important for hits of functional assays. False positives might originate from changes in the pH, aggregation, or non-specific binding. By monitoring binding on a molecular level, interactions with the wrong binding site and chemical reactions with the drug target can also be excluded. The number of compounds investigated during the validation process is much lower than for the hit finding process. Therefore, the problems of deconvolution, concentration, and experimental time can be less rigorous, whereas a large window of affinities is still essential. Even more important is the lack of ambiguity of the experiment with respect to the binding of the ligands. To extract true binders from the hits, it is advisable to apply several experiments to confirm the mode of interaction.Chemical shift perturbations are most commonly tracked in 13C- and 15N-HSQC spectra, although HNCO spectra have also been used.1515N-labelling is relatively cheap if an appropriate overexpression system has been established, while in contrast, uniformly 13C-labelling is expensive. Nowadays, new labelling schemes enable the selective incorporation of 13C-labels in the methyl groups of valine, leucine, and isoleucine.27 Isotopic enrichment with 13C at specific locations forms a convenient alternative to 15N-labelling, especially considering that the apparent signal intensity of methyl groups is three times that of amide protons. The chemical shift dispersion of the methyl moieties is merely somewhat smaller than that of the amide protons. The amide protons visible in the 15N-HSQC spectrum are hydrophilic, whereas the methyl groups of the above-mentioned residues are hydrophobic. Therefore, both techniques are partially complementary.
4.3 Hit optimization
For hit optimization, the confirmed hits from the validation process are first ranked and clustered. Specific properties, e.g. Lipinski's “rule of five”, have to be considered to develop a promising lead structure (Table 1).13,41 This assessment includes the solubility, molecular weight, chemical accessibility, and affinities (binding constants). For a further improvement of the binders, structural information is indispensable.15 The exact binding site as well as the precise binding mode has to be extracted from the experiments. For the design of new ligands, analogs of the lead structure are explored, often assisted by combinatorial chemistry. The resulting compounds are subjected to a screening comparable to the hit finding process, but with more thorough evaluation. As targets often have more than one binding site, these have to be identified and screened too. If ligands for adjacent binding sites can be linked, large increases in their affinities can be expected.Depending on the stage of the hit optimization process, the experiments should provide structural information of different qualities. Whereas at the beginning, even crude SARs are sufficient, more detailed information is required at an advanced stage of drug development. Therefore, the techniques change from ligand- to target-based methods and more complex pulse sequences are applied. Target-based techniques have the additional advantage of offering the possibility of monitoring high-affinity ligands, as low-affinity binders are of minor interest at this stage. Of course, the target needs to be NMR accessible, which sets some limitations in size.
 | ||
| Fig. 6 Illustration of the SAR-by-NMR technique. High-affinity binders are identified on the basis of two medium-affinity binders of different but adjacent binding sites. | ||
Another interesting approach is the observation of binding within a living cell.44–4615N-HSQC spectra are recorded directly of the bacterial slurry, resulting in spectra of practical resolution. This result is of particular interest because many compounds have high affinity in vitro but show much weaker effects in vivo.
2D spectra sometimes are not sufficient to visualize the binding process because of remaining signal overlaps. If the binding site is already known, site-selective labelling might help to avoid this problem. Single types of amino acids are selectively labelled to simplify the spectra and focus it on the binding pocket.
The labelling schemes described are of an immense value for protein targets; however, there are still no comparable labelling techniques that make target-based experiments appropriate for nucleic acid targets.
An alternative approach is competition-based screening.47–49 A known medium-affinity binder (reporter ligand) is added to the solution of the target. If other ligands (screening ligands) are present in this solution as well, the reporter ligand will be displaced according to the affinity of the screening ligands (Figure 7). The prerequisite of fast exchange is only valid for the reporter ligand and therefore the range of affinities for competing ligands is not limited. For the detection of weaker binders, the concentration of the reporter ligand has to be decreased. During the experiment the focus is on the reporter ligand, since only signals of this species are detected. This limitation raises the problem of deconvoluting mixtures of ligands if binders are discovered. With competition-based experiments, only ligands that bind to the same site as the reporter ligand are detectable, also making this application relevant to the hit validation process. All competition experiments can be used to estimate the dissociation constant of the screened ligands if the KD of the reporter ligand is known. An NMR titration has to be performed with the ligand of interest while the concentration of the target and the reporter ligand are kept fixed. The change of the intensity of the reporter ligands signal and subsequent fitting to corresponding equations12 gives the required information.
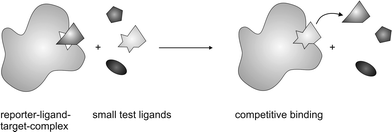 | ||
| Fig. 7 Depiction of competition-based screening. Low- to medium-affinity binders are displaced from their binding site by higher affinity binders. In this experiment, only the signals of these low- to medium-affinity binders (reporter ligands) are monitored. | ||
In principle, all ligand-based techniques can be extended to a competition type of experiment. However, experiments using simple 1D spectra are especially worth considering, e.g. WaterLOGSY,47 STD, and 19F-screening,40 which rank among the most powerful ligand-based screening techniques. Their advantage that they only require a small amount of target and their high sensitivity is adopted by the competition experiment. Competition-based fluorine screening has to be highlighted in this context as it combines the advantages of most techniques. Fluorine chemical shift anisotropy and exchange for screening (FAXS) is one implementation of this concept. The relaxation parameters of the reporter ligand change dramatically on the displacement by other ligands. Clean spectra are easily obtained without suppression of target or solvent signals making it a highly effective tool for screening, even in hit finding.
 | ||
| Fig. 8 Introduction of paramagnetic spin labels for second site screening. Paramagnetic spin labels drastically alter the relaxation properties of binding components. The large gyromagnetic ratio of unpaired electrons amplifies the relaxation via dipole–dipole interaction, which is represented by the arrows. Stars at the edges of the binding ligand illustrate the increased transverse relaxation. | ||
5 Representative case studies
In the following section, examples of NMR-based screening are presented. The first two examples include the screening of libraries containing 400 and 10,000 compounds, respectively. One involves fluorine techniques as a method for primary screening, while the other uses target-based methods for detection. In the second example, the fragment-based approach is described where NMR methods for hit finding, hit validation, and hit optimization are included. The last example deals with a new technique (saturation transfer double difference (STDD)) that might have a considerable impact on drug development in future. The examples reflect the techniques we regard as the most promising alternatives for the drug discovery process.5.1 Fluorine screening
Recently, we screened riboflavin synthase (RiSy) against a small library of 19F-containing ligands. The library encompasses about 400 ligands containing either an aromatic fluorine or a trifluoromethyl moiety.RiSy catalyzes the final step of riboflavin (vitamin B2) biosynthesis. During the last step, two lumazine units (1, DMRL) are fused to form riboflavin (2) and 5-amino-6-ribitylamino-2,4-(1H,3H)-pyrimidinedione (3, ARP) (see Figure 9). ARP is recycled afterwards in the biosynthetic pathway via lumazine synthase. The disproportionation reaction that results in the formation of vitamin B2 involves the transfer of a C4-fragment between the two DMRL molecules. The xylene ring system of riboflavin is obtained by a head-to-tail assembly of the two substrate molecules.
 | ||
| Fig. 9 Biosynthesis of riboflavine (2) by catalysis of RiSy. Two molecules of lumazine (1) form one molecule of riboflavine and one molecule of 5-amino-6-ribitylamino-2,4-(1H, 3H)-pyrimidinedione (3) by disproportionation. | ||
RiSy consists of three identical subunits denoted α.51,52 They form a complex of pseudo-D3 symmetry with a total molecular mass of about 75 kDa. The a-subunit (23 kDa) shows an internal sequence similarity resulting in a 26% identity of the C- and N-terminus. The N-terminal domain can be expressed separately and forms a homo-dimer that binds the substrate with a similar affinity to the complete trimeric protein.53–55
This enzyme forms an attractive target for medical chemistry to create an anti-infective drug. Selective suppression of this enzyme will result in a deficiency of vitamin B2. While mammals have the ability to absorb riboflavin from their nutrition, gram-negative bacteria and various yeasts lack an equivalent mechanism56–59 and hence depend completely on the endogenous biosynthesis of this vitamin. This requirement facilitates the development of a specific drug, as the target is present in bacteria and fungi but not in the host.
Several inhibitors mimicking the scaffold of lumazine (1) have been identified,60 but none of them consisted of new structural features. To overcome this drawback, a screening of fluorinated ligands was performed.
The 19F-chemical shift was measured in advance for each member of the library. These reference experiments were carried out in D6-DMSO solutions with trifluoroethanol (TFE) as an internal standard. The 400 ligands were divided into groups of about 10–30 compounds, resulting in 15 mixtures. The combinations were chosen such as to avoid reactions and overlap of 19F-chemical shifts. For the composition of these mixtures, stock solutions in D6-DMSO were set up and added to buffer solutions (50 mM KH2PO4, 10 mM EDTA, 10 mM Na2SO3, pH 7.1). To test for binding, a portion of the stock solution was added to a solution of RiSy (0.1 mM) in the same buffer. For both samples 1D 19F-spectra were acquired at 14.09 T (564 MHz 19F-frequency).
Compounds binding to RiSy experienced a significant line broadening (Figure 10); this effect was observed for 11 out of the 400 ligands (Table 3). The six best (approximated from the amount of broadening) were combined in a new mixture, which was added in excess to a small amount of enzyme. In the case of competitive binding it is assumed that the binder with the highest affinity displaces all the other ligands. From this experiment it was possible to identify the two strongest binders, compounds 4 and 5 (Figure 11).
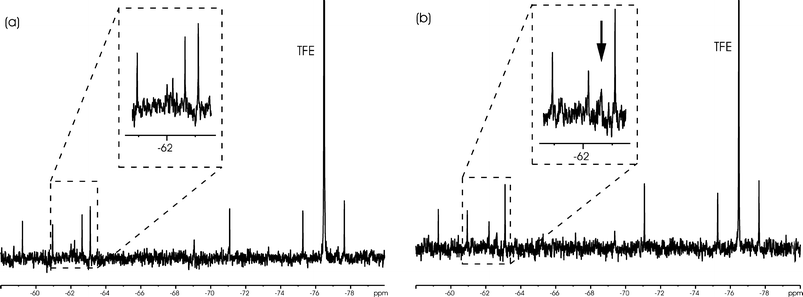 | ||
| Fig. 10 Typical 19F spectrum of a screening mixture with (a) and without (b) protein. In the expansion, the change of the chemical shift as well as the line broadening of the centre signal is visible. | ||
 | ||
| Fig. 11 19F spectrum of all compounds showing an interaction with the protein during the first screening with (a) and without (b) protein. It is apparent that compounds 4 and 5 are the highest affinity binders among the ligands investigated. | ||

|
An enzymatic assay of RiSy revealed a Ki value of about 20–30 μM for compound 4, which could serve as a starting point for further optimizations to generate an appropriate lead structure.
This example shows the ease of performing fluorine-based screenings. Once the 19F-library is characterized (knowledge about all 19F-chemical shifts) and standard mixtures can be used for screening, it becomes a rather efficient technique. The experimental time to acquire a spectrum of one mixture took about 75 min in this case, but with higher concentrations, experimental times can be reduced to 30 min or even less. This estimate includes the sample set up, data acquisition, and the change of the sample by the autosampler. Therefore, a throughput of 400–1000 compounds per day can easily be achieved if 20 components are assumed per mixture.
5.2 SAR-by-NMR
SAR-by-NMR1 represents a very intuitive way of rational, fragment-based drug design. Two or more independently optimized ligands of different binding sites are combined to give a high-affinity binder. The example presented here deals with the development of a selective inhibitor for the protein tyrosine phosphatase 1B (PTP1B).61PTP1B operates in the insulin-signalling pathway. When insulin binds to its receptor, there is a conformational change of intracellular region of the protein, which results in the O-phosphorylation of three tyrosine residues as the first step in the cascade of the insulin signalling.62 PTP1B is reckoned to be responsible for the dephosphorylation of the insulin receptor.63–67 Dephosphorylation results in the down regulation of the insulin receptor and therefore selective inhibition of PTP1B may enhance insulin activity.
The exogenous regulation of this enzyme is of general interest as disturbed insulin signalling may lead to type II diabetes. Currently, about 130 million people suffer from this disease and it is expected that this number will rise during the next years.68 Nowadays, the treatment encompasses the application of insulin and hypoglycaemic drugs, but none of them is sufficient to assuage the disease completely.69
Many processes in eukaryotic cells are regulated by reversible phosphorylation. Hence, compounds interacting with this class of proteins are regarded as promising candidates for the therapy of many diseases. PTP1B belongs to one subclass of these proteins, the tyrosine phosphatases. Unfortunately, the catalytic domain is generally conserved among these enzymes, complicating selective inhibition.
First, a primary screen of 10,000 compounds was performed to find new scaffolds that bind to the active site of PTP1B. This screen involved a shortened form of the protein (292 residues) that was either uniformly 15N-labelled or selectively labelled with δ-13CH3 isoleucine. Chemical shift perturbations in the 15N-HSQC and 13C-HSQC spectra (Figure 12) were used for the detection of binding ligands. From the screen, compound 15 (Figure 13) could be identified as a ligand. Comparison with the natural ligand phosphotyrosine (pTyr) revealed that both ligands have equivalent binding modes. For further improvement of the affinity, a derivative of the new ligand with a bulkier group was synthesized (naphthyloxamic acid, 16). This ligand showed an increase of the Ki value from 293 to 39 μM in a para-nitrophenyl phosphate (pNPP) assay. Furthermore, covalent binding of 16 to PTP1B could be excluded by enzyme kinetics. An X-ray analysis confirmed that naphthyloxamic acid also binds in the same fashion as the natural ligand.
 | ||
| Fig. 12 13C-HSQC of PTP1B with ligand (red) and without (black) ligand. The chemical shift change for the isoleucine residue 219 indicates binding of the ligand to the corresponding amino acid. | ||
 | ||
| Fig. 13 Structure evolution of a high-affinity binder (18) for PTP1B via SAR-by-NMR screening. | ||
Apparent from the X-ray structure (Figure 14) is a groove in the protein surface connecting the active site with a second, inactive site. Further derivatization of the lead with the goal of finding a linker for the two sites resulted in compound 17. A diamido chain at 4-position gave a 40-fold boost in affinity (Ki = 1.1 μM).
 | ||
| Fig. 14 X-ray structure of compounds 15–18 with PTP1B (a–c) revealing the same binding mode. Also visible is the groove in the protein surface that connects the catalytic and the non-catalytic binding sites. | ||
The second, non-active site is also a binding site for pTyr. This non-catalytic site offers great potential to increase the selectivity of the new ligand for PTP1B. Another NMR screen, also involving 10,000 compounds, provided several hits, mostly small fused-ring aromatics. This time, the screening was performed with the truncated protein consisting of selectively 13C-labelled methionine. Residue Met258 is present in the second binding site and is therefore an excellent probe for a binding event. The fusion of 3-hydroxy-2-naphtoic acid and 17 resulted in a nanomolar binder (18) with a 50-fold higher affinity (Ki = 22 nM).
Finally, this inhibitor was cross-checked against five other phosphatases to determine the selectivity. Among those analyzed, TCPTP is closely related to PTP1B on the basis of sequence homology. Therefore, a difference in the affinity of the evaluated compounds for these two enzymes would be a remarkable achievement. The relative selectivities are summarized in Table 4, indicating that compound 18 is indeed able to “distinguish” between PTP1B and TCPTP.
This example underscores the efficiency of SAR-by-NMR and moreover illustrates the fragment-based approach.
| Compound | ||||
|---|---|---|---|---|
| Phosphatase | 15 | 16 | 17 | 18 |
| PTP1B | 1 | 1 | 1 | 1 |
| TCPTP | 1 | 1 | 1 | 2 |
| LAR | >3 | 1 | 6 | 36 |
| SHP-2 | >3 | >7 | 27 | 104 |
| CD45 | >1 | >4 | 380 | 2700 |
| Calicineurin | >7 | >270 | >13000 | |
5.3 Saturation transfer double difference
STD spectroscopy9,31 is a frequently used tool to test ligands for binding. The reason is the large variability and the high sensitivity of this experiment and because it can also be applied to the investigation of integral membrane proteins.32 These proteins are especially difficult to study as they usually lose their biological activity if removed from their natural environment. Therefore, this class of proteins is reintegrated into liposomes for NMR analysis to simulate the physiological conditions. Nevertheless, most screening techniques are not able to monitor the interaction between these targets and a ligand.Membrane-bound proteins are of highest interest for the pharmaceutical industry, as about 30% of proteins in the mammalian cells belong to this class. A subclass of these proteins are the G-protein coupled receptors (GPCRs),70 which play an important role in signal transfer pathways. One example is the αIibβ3 integrin, which occurs on the surface of platelets where it is the most prevalent component of the exposed glycoproteins. The integrin is composed of two non-covalently linked α and β subunits and is able to bind proteins and peptides consisting of an RGD motif.71,72 Platelets are of crucial importance in the blood clotting process by forming platelet plugs. This process can be inhibited by RGD-containing peptides and non-peptidic peptidomimetics. A cyclic peptide that shows high activity (low nanomolar range) for the αvβ3 integrin is cyclo(-RGDfV-).73,74 However, this peptide shows the desired lower affinity (micromolar range) for the αIibβ3 integrin, which is required for the STD experiment to achieve a sufficiently high STD amplification by rapid exchange of bound and free ligand molecules.
For a proof of concept, Meinecke et al.75 reinvestigated this interaction by STD spectroscopy on liposome, embedded αIibβ3 integrin. Recently, a further example that probably represents a key step towards in-cell screening was published by Claasen et al.76 They investigated the binding of the above-mentioned system in vivo. Intact human platelets were mixed with cyclo(-RGDfV-) and the resulting solution was analyzed with a new technique called STDD spectroscopy (see below).
Suspensions of human platelets were prepared from concentrated blood donations. The work up yielded two samples, both containing the same composition of platelets in a D2O-TBS (deuterated TRIS saline) buffer. The amount of platelets was estimated to 7 × 109, representing an effective concentration of 100–600 pM if a number of 1–5 × 104 receptors is assumed per platelet. One sample (A) was treated with the cyclic peptide at a concentration of 150 nM, while the other one (B) remained unaltered. The normal STD analysis of sample A containing a 250–1500-fold excess of the ligand did not result in an interpretable spectrum. Even a T1ρ-filter element integrated into the pulse sequence to suppress resonances of large-size macromolecules did not lead to significant improvement. The problem was primarily the result of the fact that the suspension consisted of many small and large molecules showing a lot of binding events. The resulting STD spectrum obviously contained signals of these compounds. To filter these contributions out of the spectrum, a reference STD spectrum that included exactly these ligands was recorded and subtracted afterwards. For this reference, an STD spectrum of sample B was acquired. Subtraction of the two resulted in the spectrum shown in Figure 15D. This double difference method also has the advantage that residual signals of macromolecules are eliminated without the need for a relaxation filter element, thus increasing the signal-to-noise ratio. The quality of the resulting spectra is apparent by the comparison of the STDD spectrum and the simple 1D spectrum of the pure cyclic peptide (Figure 15E).
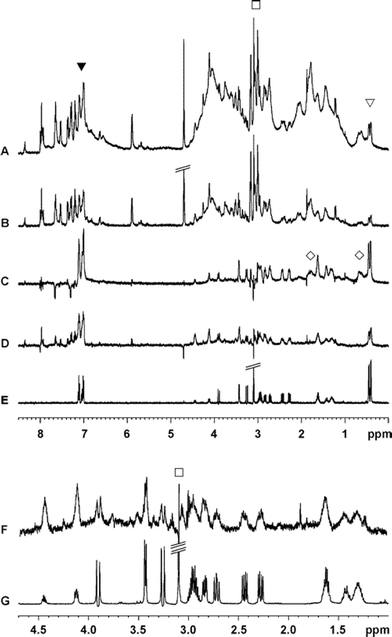 | ||
| Fig. 15 Filter effect of the STDD technique. In panel A, the crowded 1H STD NMR spectrum of a platelet suspension with cyclo(-RGDfv-) is shown, where ▼and ▽ denote the only quantifiable signals. The unsatisfactory filter effect of a T1ρ-element to suppress signals of the platelets resulted in spectrum B. Spectrum C (700 MHz) and D (500 MHz) show the filter effect of the saturation double difference, when compared to the spectrum of the ligand only (E). The platelet signals are marked with ◇ Spectrum F and G show expansions of spectra D and E to highlight the strong filter effect of the STDD method. Signals of TRIS included in deuterated TBS buffer are indicated by □. | ||
Compared to the STD spectrum of cyclo(-RGDfV-) in the presence of integrin αIibβ3 integrated into liposomes, the STDD spectrum of the native integrin showed a fivefold higher STD effect. Moreover, a difference in the STD response can be observed, reflecting a slightly different binding mode. These findings emphasize the value of this experiment, particularly if all of the capabilities are considered.
Of course, this technique is not useful for a primary screen, as the ligands may interact with many targets and in addition to the desired one. However, if ligands are found with a high enough affinity and an accordingly high selectivity in vitro, this experiment may serve as a control to distinguish scaffolds of high activity in vivo from those with a low activity at an early stage of the drug discovery process.
6 Conclusion
NMR spectroscopy has evolved into an important method for screening ligand mixtures for binders to medicinal relevant protein or nucleic acid targets. A number of new technologies have been established for this purpose in the last decade; the most efficient ones are STD, waterLOGSY, and the screening of fluorine-containing libraries. Moreover, NMR spectroscopy can assist the hit validation process and provide structural information on the ligand–target complex.To a large extent, NMR is, complementary to other techniques. The ability to detect weak binders under quasi-natural conditions is a particular advantage, making it ideally suited for the fragment-based approach for developing lead structures.
References
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Science, 1996, 274, 1531 CrossRef CAS.
- P. A. Keifer, Curr. Opin. Biotechnol., 1999, 10, 34 CrossRef CAS.
- G. Roberts, Drug Discovery Today, 2000, 5, 230 CrossRef CAS.
- A. Ross, G. Schlotterbeck, W. Klaus and H. Senn, J. Biomol. NMR, 2000, 16, 139 CrossRef CAS.
- T. Diercks, M. Coles and H. Kessler, Curr. Opin. Chem. Biol., 2001, 5, 285 CrossRef CAS.
- M. Heller and H. Kessler, Pure Appl. Chem., 2001, 73, 1429 CrossRef CAS.
- B. J. Stockman and C. Dalvit, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 41, 187 CrossRef CAS.
- M. Pellecchia, D. S. Sem and K. Wüthrich, Nat. Rev. Drug Discovery, 2002, 1, 211 CrossRef CAS.
- B. Meyer and T. Peters, Angew. Chem., Int. Ed., 2003, 42, 864 CrossRef CAS.
- M. Coles, M. Heller and H. Kessler, Drug Discovery Today, 2003, 8, 803 CrossRef CAS.
- W. Jahnke and H. Widmer, Cell. Mol. Life Sci., 2004, 61, 580 CAS.
- J. W. Peng, J. Moore and N. Abdul-Manan, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 44, 225 CrossRef CAS.
- J. R. Huth and C. H. Sun, Comb. Chem. High Throughput Screening, 2002, 5, 631 CAS.
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nat. Rev. Drug Discovery, 2004, 3, 660 CrossRef CAS.
- M. van Dongen, J. Weigelt, J. Uppenberg, J. Schultz and M. Wikstrom, Drug Discovery Today, 2002, 7, 471 CrossRef.
- J. Fejzo, C. A. Lepre, J. W. Peng, G. W. Bemis, Ajay, M. A. Murcko and J. M. Moore, Chem. Biol., 1999, 6, 755 CrossRef CAS.
- J. W. Peng, C. A. Lepre, J. Fejzo, N. Abdul-Manan and J. M. Moore, Nucl. Magn. Reson. Biol. Macromol., Pt A, 2001, 338, 202 Search PubMed.
- P. J. Hajduk, A. Gomtsyan, S. Didomenico, M. Cowart, E. K. Bayburt, L. Solomon, J. Severin, R. Smith, K. Walter, T. F. Holzman, A. Stewart, S. McGaraughty, M. F. Jarvis, E. A. Kowaluk and S. W. Fesik, J. Med. Chem., 2000, 43, 4781 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef.
- M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108 CrossRef CAS.
- K. Pervushin, R. Riek, G. Wider and K. Wuthrich, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12366 CrossRef CAS.
- R. Riek, G. Wider, K. Pervushin and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4918 CrossRef CAS.
- J. Weigelt, M. Wilkstrom, J. Schultz and M. J. P. van Dongen, Comb. Chem. High Throughput Screening, 2002, 5, 623 CAS.
- K. H. Gardner and L. E. Kay, J. Am. Chem. Soc., 1997, 119, 7599 CrossRef CAS.
- N. K. Goto, K. H. Gardner, G. A. Mueller, R. C. Willis and L. E. Kay, J. Biomol. NMR, 1999, 13, 369 CrossRef CAS.
- V. Tugarinov and L. E. Kay, J. Am. Chem. Soc., 2003, 125, 13868 CrossRef CAS.
- P. J. Hajduk, D. J. Augeri, J. Mack, R. Mendoza, J. G. Yang, S. F. Betz and S. W. Fesik, J. Am. Chem. Soc., 2000, 122, 7898 CrossRef CAS.
- C. A. Lepre, Drug Discovery Today, 2001, 6, 133 CrossRef CAS.
- C. A. Lepre, J. Peng, J. Fejzo, N. Abdul-Manan, J. Pocas, M. Jacobs, X. L. Xie and J. M. Moore, Comb. Chem. High Throughput Screening, 2002, 5, 583 CAS.
- F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206 CrossRef CAS.
- M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784 CrossRef CAS.
- J. Klein, R. Meinecke, M. Mayer and B. Meyer, J. Am. Chem. Soc., 1999, 121, 5336 CrossRef CAS.
- C. Dalvit, P. Pevarello, M. Tato, M. Veronesi, A. Vulpetti and M. Sundstrom, J. Biomol. NMR, 2000, 18, 65 CrossRef CAS.
- C. Dalvit, G. Fogliatto, A. Stewart, M. Veronesi and B. Stockman, J. Biomol. NMR, 2001, 21, 349 CrossRef CAS.
- A. Chen and M. J. Shapiro, J. Am. Chem. Soc., 1998, 120, 10258 CrossRef CAS.
- A. D. Chen and M. J. Shapiro, J. Am. Chem. Soc., 2000, 122, 414 CrossRef CAS.
- M. Vogtherr and T. Peters, J. Am. Chem. Soc., 2000, 122, 6093 CrossRef CAS.
- J. W. Peng, J. Magn. Reson., 2001, 153, 32 CrossRef CAS.
- C. Dalvit, P. E. Fagerness, D. T. A. Hadden, R. W. Sarver and B. J. Stockman, J. Am. Chem. Soc., 2003, 125, 7696 CrossRef CAS.
- C. Dalvit, M. Flocco, M. Veronesi and B. J. Stockman, Comb. Chem. High Throughput Screening, 2002, 5, 605 CAS.
- G. M. Rishton, Drug Discovery Today, 2003, 8, 86 CrossRef CAS.
- B. Wang, K. Raha and K. M. Merz, J. Am. Chem. Soc., 2004, 126, 11430 CrossRef CAS.
- M. A. McCoy and D. F. Wyss, J. Am. Chem. Soc., 2002, 124, 11758 CrossRef CAS.
- Z. Serber, A. T. Keatinge-Clay, R. Ledwidge, A. E. Kelly, S. M. Miller and V. Dotsch, J. Am. Chem. Soc., 2001, 123, 2446 CrossRef CAS.
- Z. Serber and V. Dötsch, Biochemistry, 2001, 40, 14317 CrossRef CAS.
- Z. Serber, R. Ledwidge, S. M. Miller and V. Dotsch, J. Am. Chem. Soc., 2001, 123, 8895 CrossRef CAS.
- C. Dalvit, M. Fasolini, M. Flocco, S. Knapp, P. Pevarello and M. Veronesi, J. Med. Chem., 2002, 45, 2610 CrossRef CAS.
- C. Dalvit, M. Flocco, S. Knapp, M. Mostardini, R. Perego, B. J. Stockman, M. Veronesi and M. Varasi, J. Am. Chem. Soc., 2002, 124, 7702 CrossRef CAS.
- W. Jahnke, P. Floersheim, C. Ostermeier, X. L. Zhang, R. Hemmig, K. Hurth and D. P. Uzunov, Angew. Chem., Int. Ed., 2002, 41, 3420 CrossRef CAS.
- W. Jahnke, L. B. Perez, C. G. Paris, A. Strauss, G. Fendrich and C. M. Nalin, J. Am. Chem. Soc., 2000, 122, 7394 CrossRef CAS.
- A. Bacher, R. Baur, U. Eggers, H. D. Harders, M. K. Otto and H. Schnepple, J. Biol. Chem., 1980, 255, 632 CAS.
- S. Gerhardt, A. K. Schott, N. Kairies, M. Cushman, B. Illarionov, W. Eisenreich, A. Bacher, R. Huber, S. Steinbacher and M. Fischer, Structure, 2002, 10, 1371 CrossRef CAS.
- D. I. Liao, Z. Wawrzak, J. C. Calabrese, P. V. Viitanen and D. B. Jordan, Structure, 2001, 9, 399 CrossRef CAS.
- W. Meining, G. Tibbelin, R. Ladenstein, S. Eberhardt, M. Fischer and A. Bacher, J. Struct. Biol., 1998, 121, 53 CrossRef CAS.
- V. Truffault, M. Coles, T. Diercks, K. Abelmann, S. Eberhardt, H. Lüttgen, A. Bacher and H. Kessler, J. Mol. Biol., 2001, 309, 949 CrossRef CAS.
- S. V. Bandrin, M. Y. Beburov, P. M. Rabinovich and A. I. Stepanov, Genetika, 1979, 15, 2063 Search PubMed.
- J. C. Sircar, T. Capiris, S. J. Kesten and D. J. Herzig, J. Med. Chem., 1981, 24, 735 CrossRef CAS.
- O. Oltmanns and F. Lingens, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1967, 22, 751 CAS.
- J. C. Roberts, H. Gao, A. Gopalsamy, A. Kongsjahju and R. J. Patch, Tetrahedron Lett., 1997, 38, 355 CrossRef CAS.
- M. K. Otto and A. Bacher, Eur. J. Biochem., 1981, 115, 511 CAS.
- B. G. Szczepankiewicz, G. Liu, P. J. Hajduk, C. Abad-Zapatero, Z. H. Pei, Z. L. Xin, T. H. Lubben, J. M. Trevillyan, M. A. Stashko, S. J. Ballaron, H. Liang, F. Huang, C. W. Hutchins, S. W. Fesik and M. R. Jirousek, J. Am. Chem. Soc., 2003, 125, 4087 CrossRef CAS.
- M. P. Czech and S. Corvera, J. Biol. Chem., 1999, 274, 1865 CrossRef CAS.
- K. A. Kenner, E. Anyanwu, J. M. Olefsky and J. Kusari, J. Biol. Chem., 1996, 271, 19810 CrossRef CAS.
- H. Chen, S. J. Wertheimer, C. H. Lin, S. L. Katz, K. E. Amrein, P. Burn and M. J. Quon, J. Biol. Chem., 1997, 272, 8026 CrossRef CAS.
- F. Ahmad, P. M. Li, J. Meyerovitch and B. J. Goldstein, J. Biol. Chem., 1995, 270, 20503 CrossRef CAS.
- H. Chen, L. N. Cong, Y. H. Li, Z. J. Yao, L. Wu, Z. Y. Zhang, T. R. Burke and M. J. Quon, Biochemistry, 1999, 38, 384 CrossRef CAS.
- S. Walchli, M. L. Curchod, R. P. Gobert, S. Arkinstall and R. H. van Huijsduijnen, J. Biol. Chem., 2000, 275, 9792 CrossRef CAS.
- P. Zimmet, K. Alberti and J. Shaw, Nature, 2001, 414, 782 CrossRef CAS.
- J. M. Nuss and A. S. Wagman, Annu. Rep. Med. Chem., 2000, 35, 211 Search PubMed.
- A. Wise, K. Gearing and S. Rees, Drug Discovery Today, 2002, 7, 235 CrossRef CAS.
- R. O. Hynes, Cell, 1987, 48, 549 CrossRef CAS.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491 CrossRef CAS.
- M. Pfaff, K. Tangemann, B. Muller, M. Gurrath, G. Muller, H. Kessler, R. Timpl and J. Engel, J. Biol. Chem., 1994, 269, 20233 CAS.
- L. Marinelli, A. Lavecchia, K. E. Gottschalk, E. Novellino and H. Kessler, J. Med. Chem., 2003, 46, 4393 CrossRef CAS.
- R. Meinecke and B. Meyer, J. Med. Chem., 2001, 44, 3059 CrossRef CAS.
- B. Claasen, M. Axmann, R. Meinecke and B. Meyer, J. Am. Chem. Soc., 2005, 127, 916 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615 CrossRef CAS.
Footnote |
| † This is Chapter 12 taken from the book Exploiting Chemical Diversity for Drug Discovery (Edited by M. Entzeroth and P. A. Bartlett) which is part of the RSC Biomolecular Sciences series. |
| This journal is © The Royal Society of Chemistry 2007 |