Capillary electrophoresis –mass spectrometry of proteins at medium pH using bilayer -coated capillaries
Jonatan R.
Catai
,
Javier Sastre
Toraño
,
Gerhardus J.
de Jong
and
Govert W.
Somsen
*
Department of Biomedical Analysis, Utrecht University, P.O. Box 80082, 3508 TB Utrecht, The Netherlands. E-mail: g.w.somsen@pharm.uu.nl; Fax: +31-30-2535180; Tel: +31-30-2536951
First published on 18th October 2006
Abstract
The feasibility of using noncovalently bilayer -coated capillaries for capillary electrophoresis –mass spectrometry (CE –MS ) of acidic proteins was investigated using background electrolytes (BGEs) of medium pH. The capillary was coated by successively rinsing the capillary with solutions of the oppositely charged polymers polybrene (PB) and poly(vinyl sulfonic acid) (PVS). Volatile BGEs containing ammonium formate and/or N-methyl morpholine were tested at pH 7.5 and 8.5. Overall, these BGEs provided relatively fast protein separations (analysis times of ca. 12 min) and showed high efficiencies (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300000 plates) when the ionic strength was sufficiently high. Migration-time reproducibilities were very favorable with RSDs of less than 1.0%. Infusion experiments showed satisfactory MS responses for studied proteins dissolved in ammonium formate (pH 8.5), however, high concentrations of N-methyl morpholine appeared to seriously suppress the MS protein signals. Evaluation of the CE –MS system was performed by analyzing a mixture of intact proteins yielding efficient separations and good-quality mass spectra . CE –MS analysis of a reconstituted formulation of the biopharmaceutical recombinant human growth hormone (rhGH) which was stored for a prolonged time, revealed one degradation product which was provisionally identified as desamido rhGH. Based on the MS responses the amount of degradation was estimated to be ca. 25%.
000–300000 plates) when the ionic strength was sufficiently high. Migration-time reproducibilities were very favorable with RSDs of less than 1.0%. Infusion experiments showed satisfactory MS responses for studied proteins dissolved in ammonium formate (pH 8.5), however, high concentrations of N-methyl morpholine appeared to seriously suppress the MS protein signals. Evaluation of the CE –MS system was performed by analyzing a mixture of intact proteins yielding efficient separations and good-quality mass spectra . CE –MS analysis of a reconstituted formulation of the biopharmaceutical recombinant human growth hormone (rhGH) which was stored for a prolonged time, revealed one degradation product which was provisionally identified as desamido rhGH. Based on the MS responses the amount of degradation was estimated to be ca. 25%.
Introduction
Capillary electrophoresis (CE ) is a powerful separation technique which shows attractive characteristics for the analysis of biopolymers such as proteins.1–4 In CE , separations are based on differences in electrophoretic mobility , which is a function of charge and size of the studied ionic species. When a protein undergoes degradation or binds to other molecules (e.g. drugs), this might induce changes in the protein's net charge and/or shape, which may lead to altered electrophoretic migration. Separations in CE are performed in open tubes and analysis of proteins in principle can be carried out under mild pH circumstances using conditions that do not significantly affect their conformation. Such ‘nondenaturing’ conditions may be difficult to achieve with other separation methods like liquid chromatography (LC ) and slab-gel electrophoresis . Although size-exclusion or ion-exchange LC can be carried out using mild buffers, (unknown) interactions with the stationary phase may alter the protein conformation. Furthermore, CE can provide high separation efficiencies, relatively short analysis times and requires only minute amounts of sample. Due to these characteristics, CE gradually has become an essential separation technique for proteins,5 with applications in, for instance, proteomics,6,7 food analysis8 and analysis of biopharmaceuticals.9–11 Its significance is further illustrated by the recent introduction of CE methods in the European Pharmacopoeia monographs of the pharmaceutical proteins erythropoietin (EPO)12 and human growth hormone (somatropin).13 Especially in the biopharmaceutical field, there is a growing need for separation techniques that use relatively mild conditions (i.e. no extreme pH, no stationary phase ) in order to avoid potential protein changes induced by the separation medium.CE analyses are normally performed using fused-silica capillaries. However, particularly during protein analysis, interactions between the analytes and the inner capillary surface can occur, leading to band broadening, unstable electroosmotic flow (EOF) and poor migration-time reproducibilities . Therefore, in protein CE the use capillary coatings is often advocated.1–5 An elegant means of coating a capillary is to physically adsorb one or more layers of charged polymers on the internal capillary wall.14–22 This type of coating are relatively easy to prepare by successively rinsing the capillary with solutions of polymers of opposite charge, thereby creating monolayers ,14–17bilayers ,18 and multiple layers .19,20Bilayer coatings have been used successfully for small molecular-weight compounds and proteins, yielding strong and constant EOFs over a wide pH range (2–11).18,21,22 We have demonstrated the usefulness of capillaries coated with a bilayer of polybrene (PB) and poly(vinyl sulfonic acid) (PVS) for the analysis of peptides at pH 2.5 and proteins at pH 7–8.5.23–25 The PB–PVS coating showed very stable migration-time (RSDs < 1.0%) for these compounds. This constancy in migration-time is difficult to obtain with bare-fused silica capillaries even with extensive capillary rinsing. Moreover, better separation efficiencies (higher plate numbers) could be achieved with bilayer -coated capillaries.
Mass spectrometry (MS ) has become an important technique for the analysis and characterization of intact proteins and protein complexes.26–30 Matrix-assisted laser desorption ionization (MALDI) is frequently used for molecular weight determination of proteins,31 but it is less suitable for coupling with flow systems. Electrospray ionization (ESI) of proteins provides multiple charged ions with m/z values that often fall within the range of conventional mass analyzers as quadrupoles and ion traps. Furthermore, ESI is a soft ionization method which does not induce protein fragmentation and even may leave the conformation of the analyzed protein unchanged,27,32 which can be important when structural integrity has to be probed. The combination of CE with ESI-MS clearly provides a tool that can be very useful for purity and stability studies of proteins under mild separation conditions. Until now, however, CE –MS of proteinaceous samples has mainly been performed using acidic BGEs (pH < 4.5) in combination with bare fused-silica capillaries or with capillaries coated with a positively-charged or neutral polymer .6,33,34 So far, the use of medium-pH BGEs with coated capillaries for protein analysis by CE –MS has been quite limited. Simó et al.35 have used an ammonium acetate BGE (pH 5.5) in combination with EPyM-DMA-coated (positively-charged polymer ) capillaries for the analysis of basic proteins in food products. He et al.36 used Tris acetate (pH 6.0) with capillaries coated with a neutral polymer for the analysis of degraded cytochrome c. Quite a number of proteins have isoelectric points lower than 7 and, thus, carry a net negative charge in medium pH buffers. Such acidic proteins would preferably be analyzed by CE using a negatively charged coating providing both electrostatic repulsion and a significant electroosmotic flow (EOF). Hitherto, such CE –MS systems have not been described in literature.
In this work, the potential of capillaries coated with PB–PVS for the CE –MS analysis of proteins using volatile BGEs of medium pH is investigated. Recently, we have demonstrated that PB–PVS coated capillaries can be used for CE –MS of peptides with a BGE of low pH, yielding highly efficient and reproducible separations.23 As indicated above, we are interested to analyze proteins under much milder conditions (i.e. medium pH). Besides, at low pH, most proteins are heavily positively charged and will strongly interact with the negative PB–PVS coating, yielding broad and tailing peaks. Lately, we showed the feasibility of the PB–PVS coating for protein analysis using a Tris phosphate BGE of medium pH.25 As this nonvolatile BGE is not compatible with MS detection, a fully new method optimization, including CE -MS interfacing, had to be carried out. Therefore, the influence of the type, concentration and pH of the BGE on plate number, migration-time reproducibility and MS signal intensity of proteins was examined. The performance of the CE –MS system was evaluated using several test proteins and the biopharmaceutical recombinant human growth hormone (rhGH). Finally, the usefulness of the system is illustrated by the analysis of a solution of a commercial rhGH formulation which had been stored for a prolonged time.
Experimental
Chemicals
Polybrene (hexadimethrine bromide) and a 25% (m/v) aqueous solution of poly(vinyl sulfonate) sodium salt (PVS) (Mr = 4000–6000) were from Sigma-Aldrich (Steinheim, Germany). A 1% (v/v) PVS solution was prepared by diluting the purchased PVS solution with Milli-Q water (18.2 MΩ). Polybrene was dissolved to a final concentration of 1% (m/v) in Milli-Q water. Formic acid was from Riedel-de Haën (Seelze, Germany). Ammonium hydroxide (25%) was from Merck (Darmstadt, Germany) and N-methyl morpholine (99.5%) from Sigma-Aldrich. The test proteins α-lactalbumin (bovine milk), insulin (porcine), and carbonic anhydrase were from Sigma-Aldrich. Recombinant human growth hormone (rhGH) protein (somatropin CRS) was from the European Directorate for the Quality of Medicines (Strasbourg) and contained unspecified amounts of glycine, mannitol, lactose, and sodium bicarbonate. Formulated rhGH (Humatrope, Eli Lilly, Houten, The Netherlands) was reconstituted in water 18 months before the start of this study, and stored at 4 °C. The resulting solution contained rhGH (1.5 mg mL–1), glycine (1.5 mg mL–1), mannitol (4.5 mg mL–1), disodium hydrogen phosphate (1 mg mL–1), 0.3% metacresol, 0.29% glycerin and glycerol. Stock solutions of test proteins (3 mg mL–1) were weekly prepared in Milli-Q water and stored at 4 °C. The insulin stock solution contained 0.2% acetic acid. A protein test sample of insulin, carbonic anhydrase and α-lactalbumin was prepared by diluting aliquots of stock solutions to the desired concentrations . rhGH CRS was diluted to 1.5 mg mL–1 with Milli-Q water. BGEs were prepared by diluting ammonium hydroxide or N-methyl morpholine to the desired concentration and the pH was adjusted to 7.5 or 8.5 with formic acid. All BGEs were passed through a 0.22 µm hydrophilic filter from Sartorius (Göttingen, Germany) before use.CE systems
Capillaries with an internal diameter (ID) of 50 µm were from Composite Metal Services (the Chase, Hallow, UK). CE experiments with UV detection were performed using a Beckman-Coulter (Fullerton, CA, USA) P/ACE MDQ capillary electrophoresis instrument equipped with a diode array detector. Capillaries had a total and effective length of 70 and 60 cm, respectively. Samples were injected for 7 s at 34.5 mbar (injection volume of ca. 6 nL) and the separation voltage was 30 kV. The capillary was thermostated at 25 °C and absorbance detection of proteins was carried out at 214 nm.CE –MS experiments were conducted using a PrinCE CE system from Prince Technologies B.V. (Emmen, The Netherlands) applying capillaries with a length of 80 cm. Samples were injected for 12 s at 35 mbar (ca. 9 nL). Injection conditions were such that the sample zone was about 0.5% of the capillary volume to window. During sample injection, the nebulizer gas flow and the electrospray voltage of the CE –MS interface were turned off. The separation voltage was 30 kV in all CE –MS experiments. Capillaries were provisionally thermostated during CE –MS experiments by placing them inside a plastic tube of ca. 2 mm ID with a length of ca. 60 cm that comprised most of the capillary, but not the parts inside the auto-injector of the PrinCE and the sheath–liquid interface. In the middle of the tube a tee piece was placed through which a constant flow of air (approx. 2 L min–1) of ambient temperature was led along the CE capillary in order to achieve effective heat dissipation. In order to minimize hydrodynamic flow in the CE capillary induced by the nebulizer gas, a reduced pressure in the range of –10 to –60 mbar was applied at the inlet vial during CE –MS analysis.23MS infusion experiments were carried out by continuously leading the solution of interest through the CE capillary at ca. 250 nL min–1 into the interface and MS ion source with no separation voltage being applied.
Capillary coating procedure
New bare fused-silica capillaries were successively rinsed at 1400 mbar with 20 capillary volumes of water, 30 capillary volumes of 1 M NaOH, and 20 capillary volumes of water. After this treatment, capillaries were coated by subsequently rinsing with 1.5 capillary volumes of a 1% PB solution at 35 mbar, 10 capillary volumes of water at 1400 mbar, 1.5 capillary volumes of a 1% PVS solution at 35 mbar, and 10 capillary volumes of water at 1400 mbar. The capillary was then ready for CE analysis with the BGE of choice. Between runs, coated capillaries were flushed at 1400 mbar with 4 capillary volumes of a 1% PVS solution. During rinses, PVS was prevented from entering the ion source by opening the spray chamber and turning off the electrospray voltage.MS system
CE was coupled to an Agilent Technologies 1100 Series LC/MSD XCT ion-trap mass spectrometer (Waldbronn, Germany) equipped with an electrospray ion source via a coaxial sheath-flow sprayer (Agilent Technologies). The CE capillary outlet was positioned at 0.1–0.2 mm from the tip of the interface. Several types of sheath liquid (methanol–water, isopropanol–water and acetonitrile–water) in several ratios (25 : 75, 50 : 50 and 75 : 25, v/v) and with various concentrations of formic acid (0, 0.5, 1, 5 and 10 vol%) were tested in order to find stable spray conditions and optimum MS responses for the tested proteins. The optimal sheath liquid appeared to be acetonitrile–water–formic acid (75 : 25 : 5, v/v/v) and was used throughout this study. The sheath liquid was supplied by a syringe pump at 4 µL min–1. The nebulizer-gas pressure was 700 mbar and the flow and temperature of the drying gas were 5 L min–1 and 200 °C, respectively. The electrospray voltage was 4.5 kV. MS detection was carried out in the positive ion mode and each spectrum was an average of 2 scans. The ion charge control (ICC) was set to 100000 and the scanned mass range was 1000–2000 m/z. Plate numbers of analyzed proteins were calculated using full width at half height as measured from peaks observed in extracted-ion electropherograms. The instrument was controlled by a PC running the LC -MSD data acquisition software from Agilent Technologies. Charge assignment in the MS spectra was performed with the ‘charge deconvolution’ utility available in the DataAnalysis V. 2.1 program of the Agilent software.
Results and discussion
Influence of BGE on separation and MS detection performance
In a previous study, we have demonstrated the suitability of PB–PVS coated capillaries for reproducible and efficient analysis of proteins by CE –UV using BGEs of Tris phosphate (pH 7–8.5).25 In order to allow electrospray-mass spectrometric (ESI-MS ) detection of proteins, we investigated the performance of PB–PVS coated capillaries in combination with volatile BGEs of medium pH. For this purpose, a test mixture of insulin (50 µg mL–1), α-lactalbumin, and carbonic anhydrase (200 µg mL–1 each) was analyzed by CE –UV with various concentrations (25–150 mM) of ammonium formate (pH 7.5). An improvement of separation efficiency (plate numbers) was observed with the increase of the BGE concentration . However, repeated analyses with each BGE showed that the migration-time of the proteins consecutively decreased. This was most probably caused by the lack of buffer capacity of the ammonium formate BGE at pH 7.5, as was also indicated by a significant pH difference between the BGEs present in the inlet and outlet vial after only three runs.The possibility of using a BGE of ammonium formate at pH 8.5 was then investigated in the concentration range of 25–150 mM. As the ammonium ion has a pKa of 9.3, the BGE provides a much better buffering system at pH 8.5. Indeed, between runs no pH change of the BGE in the CE vials was observed, and protein migration-time were stable. CE –UV of the protein test sample using the PB–PVS coated capillary showed a steady increase in plate numbers when the BGE concentration was raised, reaching a maximum at 75 mM. For example, for α-lactalbumin, plate numbers were 120000 and 200000 at a BGE concentration s of 25 and 75 mM, respectively. This increase most likely is due to the higher ionic strength of the BGE which induces decreased electrodispersion, decreased analytes -capillary wall interactions and enhanced sample stacking. Above a BGE concentration of 75 mM, a gradual decrease in plate numbers was observed for all proteins. At 150 mM ammonium formate, α-lactalbumin showed a plate number of 110000. This decrease was possibly caused by an excess of Joule heating at high BGE concentrations . This was further evidenced by repeated analysis (n = 5) of the test sample at each BGE, which showed that migration-time RSDs increased from ca. 0.8% (BGE concentration of 25–75 mM) to about 3% when higher BGE concentrations (100–150 mM) were used.
Next, the effect of the BGE concentration on the MS signal intensity of the studied proteins was examined. The test proteins α-lactalbumin, insulin and carbonic anhydrase were each dissolved in water and in various concentrations of ammonium formate (pH 8.5). These solutions were infused into the sheath–liquid interface through a PB–PVS coated capillary. Upon increasing the BGE concentration , a decrease in MS signal intensity of all proteins was observed. This trend is illustrated for α-lactalbumin in Fig. 1. For CE –MS analysis at pH 8.5, a BGE of 75 mM ammonium formate was selected as a compromise between plate number, migration-time reproducibility and MS response for the studied proteins. Throughout these experiments, no signals from PB or PVS were observed, confirming that bilayer -coated capillaries can be used in combination with MS detection without any problem.23 Also under CE –MS circumstances, the PB–PVS coating did not cause any background signals.
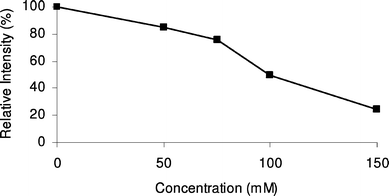 | ||
| Fig. 1 Effect of the concentration of ammonium formate (pH 8.5) on the MS signal intensity of α-lactalbumin obtained during infusion experiments. | ||
In order to be able to perform CE –MS of proteins with a buffering BGE at physiological pH, the possibility of using of N-methyl morpholine (pKa 7.4) instead of ammonium formate was examined. The protein test mixture was analyzed by CE –UV using bilayer -coated capillaries with various concentrations (25–200 mM) of N-methyl morpholine adjusted to pH 7.5 with formic acid. Plate numbers for the proteins were quite unsatisfactory at 25 mM N-methyl morpholine, but increased for higher concentrations reaching a plateau of about 150000 at 100 mM. Migration-time reproducibilities were quite favorable (RSDs < 1%), especially in the 25–100 mM range. However, MS infusion experiments revealed that the N-methyl morpholine caused serious suppression of the protein ionization. A decrease in MS signal intensity of 25–80% was observed as N-methyl morpholineconcentrations were raised from 25 to 200 mM. In order to come to a BGE at pH 7.5 with sufficient ionic strength and buffer capacity, but still acceptable MS sensitivity, the feasibility of a BGE comprising a low concentration of N-methyl morpholine (20 mM) and a high concentration of ammonium formate (75 mM) was investigated. CE –UV of the protein test sample revealed plate numbers of 100000–300000 for the proteins and excellent migration-time reproducibilities (RSDs < 1.0%). After five runs, no significant alterations of the pH values of the BGE solutions in the inlet and outlet vials were observed. These results show that the tested BGE had the required ionic strength and a good buffer capacity. MS infusion experiments with this BGE showed that the MS signals for the studied proteins were ca. 20% lower than the signal intensities obtained with a BGE of 75 mM ammonium formate (pH 8.5).
Finally, CE –MS of proteins using PB–PVS coated capillaries was evaluated with an ammonium formate BGE (pH 8.5) of 75 mM. A test sample of insulin (50 µg mL–1), carbonic anhydrase (400 µg mL–1) and α-lactalbumin (1 mg mL–1) was analyzed with the CE –MS system (Fig. 2). Plate numbers ranged from 70000 to 100000, which is quite favorable for CE –MS of proteins. Still, especially the plate numbers obtained for carbonic anhydrase and α-lactalbumin were lower than those obtained with CE –UV. These lower plate numbers were found to be caused by the relatively high protein concentrations in the sample which induced some extra band broadening. The higher concentrations were necessary due to the limited detection sensitivity of the ion-trap mass spectrometer obtained for these proteins. Actually, CE –UV analysis of the same sample revealed similar plate numbers for the proteins. Migration-time of the proteins with CE –MS were very reproducible with RSDs of less than 1.0% obtained for five consecutive runs. From the acquired mass spectra (Fig. 2B–D) the molecular masses of the analyzed proteins were determined to be 29022, 5807 and 14187 Da for carbonic anhydrase, insulin and α-lactalbumin, respectively, which nicely matches the expected values. These results demonstrate that PB–PVS coated capillaries can be used for the CE –MS of intact proteins, yielding efficient and reproducible separations. Furthermore, it can be concluded that the sheath–liquid interface does not cause significant broadening of the protein peaks produced by the CE system. This is in line with our previous work in which plate numbers for peptides analyzed by CE –MS were similar to those obtained with CE –UV.23
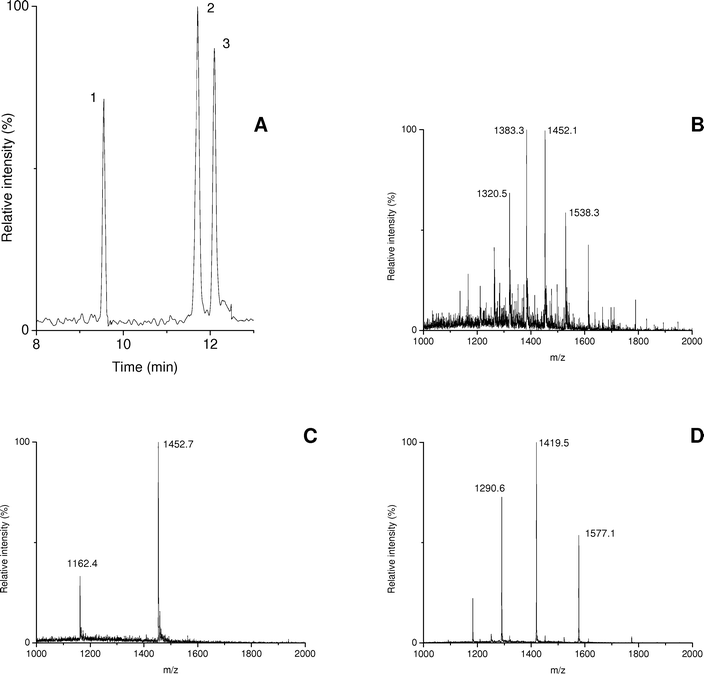 | ||
| Fig. 2 CE –MS of a protein test mixture using a PB–PVS coated capillary and a BGE of 75 mM ammonium formate (pH 8.5). (A) sum of extracted-ion electropherograms obtained at m/z 1383.3, 1452.7 and 1419.5; (B), (C) and (D) average mass spectra of peaks 1 (carbonic anhydrase), 2 (insulin) and 3 (α-lactalbumin), respectively. | ||
CE –MS of recombinant human growth hormone
The potential of the bilayer capillary coating for CE –MS of proteins at medium pH, was further tested by the analysis of the protein recombinant human growth hormone (rhGH). rhGH is a biopharmaceutical that is used in the treatment of retarded growth and dwarfism caused by the inadequate production of the hormone during the growth period. Recently, the European Pharmacopoeia introduced a method for the analysis of this protein by CE with UV detection using bare fused-silica capillaries.12 This method, however, is not MS compatible as it uses high concentrations of nonvolatile phosphate buffer as BGE. Moreover, we have also found that faster and more reproducible CE analysis of rhGH can be achieved with bilayer -coated capillaries;37 these results will be published elsewhere.First, a sample containing 1.5 mg mL–1 rhGH (somatropin CRS) was analyzed by CE –MS using a PB–PVS coated capillary with a BGE of 75 mM ammonium formate (pH 8.5). The sample yielded one symmetric peak at ca. 10.8 min with a plate number of about 90000. Deconvolution of the acquired mass spectrum resulted in an estimated molecular mass for rhGH of 22124 Da, which is in line with reference data. Subsequently, in order to test the feasibility of the CE –MS system for protein degradation studies, a reconstituted commercial rhGH formulation was analyzed. The rhGH solution had been stored for 18 months at 4 °C. Notably, this time is far beyond the storage period (14 days at 2–8 °C) for reconstituted rhGH recommended by the supplier. Fig. 3 depicts the CE –MS result which clearly shows a degradation product. Deconvolution of the mass spectra of the main and minor peak shows that both constituents have virtually identical masses, i.e., 22124 Da. This is an indication that the protein may have undergone deamidation. This degradation route involves the transition of an asparagine residue into an aspartic acid residue leading to a gain of one negative charge on the protein at neutral pH. CE is particularly useful to reveal this kind of charge modifications, as the electrophoretic mobility depends on the charge-to-size ratio of a protein. The change of one charge only already leads to a clear shift in migration-time . However, upon deamidation the overall mass change of the protein is only 1 Da, which explains the detected masses of both compounds to be practically the same. The limited mass resolution of the ion-trap mass spectrometer does not permit the detection of such a small mass difference. Based on the MS responses the percentage of degradation of the rhGH product was estimated to be ca. 25%.
 | ||
| Fig. 3 CE –MS of a formulation of rhGH which after reconstitution was stored for 18 months at 4 °C. A PB–PVS coated capillary was used with a BGE of 75 mM ammonium formate (pH 8.5). (A) sum of extracted-ion electropherograms obtained at m/z 1476.0 and 1581.2; (B) and (C) average mass spectra of peaks 1 (intact hGH) and 2 (degradation product), respectively. | ||
The possibility of using 20 mM N-methyl morpholine with 75 mM ammonium formate (pH 7.5) as BGE for the CE –MS analysis of the degraded rhGH formulation was also examined. The resulting separation profile (Fig. 4) and plate numbers were comparable to those obtained with the BGE with pH 8.5. The analysis time showed to be slightly longer at pH 7.5, which was probably caused by the somewhat higher ionic strength of the morpholine–ammonium formate BGE. As expected, CE –MS using the N-methyl morpholine–ammonium formate BGE presented a lower signal-to-noise ratio for hGH due to an increased ionization suppression and a higher background noise caused by N-methyl morpholine. However, significant signals were still obtained allowing the effective profiling of the sample at pH 7.5. Deconvolution of the mass spectra of each acquired peak, once more revealed the same molecular mass (22124 Da), suggesting that peaks corresponded to intact and desamido rhGH.
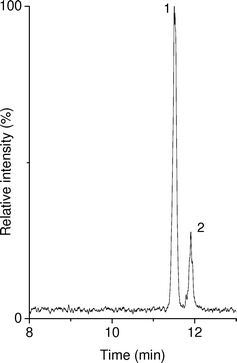 | ||
| Fig. 4 CE –MS of a formulation of rhGH which after reconstitution was stored for 18 months at 4 °C. A PB–PVS coated capillary was used with a BGE of 20 mM N-methyl morpholine with 75 mM ammonium formate (pH 7.5). The depicted trace is the sum of extracted-ion electropherograms obtained at m/z 1476.0 and 1581.2. Peaks: 1, intact hGH; 2, degradation product. | ||
Conclusion
The usefulness of PB–PVS coated capillaries for CE –MS analysis of acidic proteins using volatile BGEs of medium pH is demonstrated. Favorable plate numbers and very good migration-time reproducibilities were achieved for the studied proteins in relatively short analysis times. The use of BGEs with sufficient buffer capacity and relatively high ionic strength were important in order to obtain good separation performance. A BGE of ammonium formate alone appeared to be suitable for analysis at pH 8.5. However, the addition of N-methyl morpholine to the BGE was required in order to achieve both appropriate buffering and MS compatibility at pH 7.5. The potential of the PB–PVS system for protein CE –MS was indicated by the analysis an degraded formulation of rhGH using the BGEs of pH 7.5 and 8.5. The obtained CE and MS data suggest suggests that the rhGH has undergone deamidation. The favorable migration-time reproducibilities induced by the bilayer coating can be of great importance for the comparison of CE –MS profiles obtained in time, e.g., during stability studies of acidic protein pharmaceuticals. Currently, we are studying the potential of triple layer coatings (e.g.PB–PVS–PB) for the efficient CE –MS analysis of basic proteins at medium pH. Further characterization proteins by CE –MS would profit from the use of mass spectrometers , such as time-of-flight (TOF ) instruments, as they can provide a higher mass resolution than the used ion-trap mass spectrometer .Acknowledgements
The authors thank Peter M. J. M. Jongen from the Center for Biological Medicines and Medical Technology of the National Institute for Public Health and the Environment (Bilthoven, The Netherlands) for useful advice and stimulating discussions.References
- K. Hutterer and V. Dolník, Electrophoresis, 2003, 24, 3998–4012 CrossRef CAS.
- S. Hu and N. J. Dovichi, Anal. Chem., 2002, 74, 2833–2850 CrossRef CAS.
- P. G. Righetti, Biopharm. Drug Dispos., 2001, 22, 337–351 CrossRef CAS.
- V. Dolník, Electrophoresis, 1999, 20, 3106–3115 CrossRef CAS.
- V. Dolník, Electrophoresis, 2006, 27, 126–141 CrossRef CAS.
- D. C. Simpson and R. D. Smith, Electrophoresis, 2005, 26, 1291–1305 CrossRef CAS.
- Y. Shen and R. D. Smith, Electrophoresis, 2002, 23, 3106–3124 CrossRef CAS.
- A. Cifuentes, Electrophoresis, 2006, 27, 283–303 CrossRef CAS.
- J. R. Catai, G. J. de Jong and G. W. Somsen, in Methods for structural analysis of protein pharmaceuticals, ed. W. Jiskoot and D. J. A. Crommelin, American Association of Pharmaceutical Scientists, Arlington, VA, 2005, ch. 9, pp. 331–377 Search PubMed.
- J. S. Patrick and A. L. Lagu, Electrophoresis, 2001, 22, 4179–4196 CrossRef CAS.
- S. Ma and W. Nashabeh, Chromatographia, 2001, 53, S75–S89.
- Ph. Eur., 2005, 5.3, 3494–3498 Search PubMed.
- Ph. Eur., 2005, 5.3, 3619–3621 Search PubMed.
- Y. Wang and P. L. Dubin, Anal. Chem., 1999, 71, 3463–3468 CrossRef CAS.
- E. Córdova, J. Gao and G. M. Whitesides, Anal. Chem., 1997, 69, 1370–1379 CrossRef CAS.
- M. X. Li, L. Liu, J. T. Wu and D. M. Lubman, Anal. Chem., 1997, 69, 2451–2456 CrossRef CAS.
- F. B. Erim, A. Cifuentes, H. Poppe and J. C. Kraak, J. Chromatogr., A, 1995, 708, 356–361 CrossRef.
- H. Katayama, Y. Ishihama and N. Asakawa, Anal. Chem., 1998, 70, 2254–2260 CrossRef CAS.
- H. Katayama, Y. Ishihama and N. Asakawa, Anal. Chem., 1998, 70, 5272–5277 CrossRef CAS.
- T. W. Graul and J. B. Schlenoff, Anal. Chem., 1999, 71, 4007–4013 CrossRef CAS.
- L. Bendahl, S. H. Hansen and B. Gammelgaard, Electrophoresis, 2001, 22, 2565–2573 CrossRef CAS.
- K. D. Altria, J. Pharm. Biomed. Anal., 2003, 31, 447–453 CrossRef CAS.
- J. R. Catai, J. Sastre Toraño, G. J. de Jong and G. W. Somsen, Electrophoresis, 2006, 27, 2091–2099 CrossRef CAS.
- J. R. Catai, G. W. Somsen and G. J. de Jong, Electrophoresis, 2004, 25, 817–824 CrossRef CAS.
- J. R. Catai, H. A. Tervahauta, G. J. de Jong and G. W. Somsen, J. Chromatogr., A, 2005, 1083, 185–192 CrossRef CAS.
- V. H. Wysocki, K. A. Resing, Q. F. Zhang and Q. L. Cheng, Methods, 2005, 35, 211–222 Search PubMed.
- A. J. R. Heck and R. H. H. van den Heuvel, Mass Spectrom. Rev., 2004, 23, 368–389 CrossRef CAS.
- S. D. Maleknia and K. Downard, Mass Spectrom. Rev., 2001, 20, 388–401 CAS.
- A. P. Jonsson, Cell. Mol. Life Sci., 2001, 58, 868–884 CrossRef CAS.
- H. F. Alomirah, I. Alli and Y. Konishi, J. Chromatogr., A, 2000, 893, 1–21 CrossRef CAS.
- A. Zuberovic, S. Ullsten, U. Hellman, K. E. Markides and J. Bergquist, Rapid Commun. Mass Spectrom., 2004, 18, 2946–2952 CrossRef CAS.
- P. Schmitt-Kopplin and M. Frommberger, Electrophoresis, 2003, 24, 3837–3867 CrossRef CAS.
- J. Hernandez-Borges, C. Neusüss, A. Cifuentes and M. Pelzing, Electrophoresis, 2004, 25, 2257–2281 CrossRef CAS.
- H. Stutz, Electrophoresis, 2005, 26, 1254–1290 CrossRef CAS.
- C. Simó, C. Elvira, N. González, J. S. Román, C. Barbas and A. Cifuentes, Electrophoresis, 2004, 25, 2056–2064 CrossRef CAS.
- T. He, N. Chandramouli, E. Fu, A. Wu and Y. K. Wang, Anal. Biochem., 1999, 271, 189–192 CrossRef CAS.
- J. R. Catai, J. Sastre Toraño, P. M. J. M. Jongen, G. J. de Jong and G. W. Somsen, J. Chromatogr. B, submitted Search PubMed.
| This journal is © The Royal Society of Chemistry 2007 |