The living test-tube: imaging of real-time gene expression
Yaron
Shav-Tal
Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan 52900, Israel. E-mail: shavtaly@mail.biu.ac.il; Fax: +972-3-535-1824; Tel: +972-3-531-8589
First published on 28th February 2006
Abstract
Cells are dynamic entities. Not only are some cells motile but there is constant motion of organelles, proteins, nucleic acids and other molecules within every living cell. These complex molecular pathways control the life cycle of a cell and all come down to the basic players of the gene expression pathway: DNA, RNA and protein. It is therefore imperative to study biological processes as they naturally occur—in living cells, and to unravel the biophysical rules that govern intracellular dynamics. Towards this end, genetically encoded fluorescent proteins have become one of the major tools available for the study of kinetic processes taking place in real-time. This review will focus on the technical developments available for the study of gene activity in living cells and will summarize the novel biological information extracted from these approaches.
 Yaron Shav-Tal | Yaron Shav-Tal received his PhD in Biology at The Weizmann Institute of Science, Israel, in August 2000. He performed postdoctoral research in the group of Prof. Dov Zipori at the Weizmann Institute from 2001 to 2002, and then with Prof. Robert H. Singer at Albert Einstein College of Medicine in New York from 2002 to 2005. He joined Bar-Ilan University in Israel in 2005. The current interests in his laboratory are the study of gene expression dynamics and nuclear organization using live-cell imaging in single living cells. |
Introduction
Scientists have been peering into the inner parts of cells for decades. Light microscopy and electron microscopy have portrayed the most intricate details of sub-cellular organization and architecture. Yet, these studies were conducted mostly on fixed non-viable cells, therefore focusing mostly on the spatial aspects of cellular structures or proteins. For many years no ample techniques were available to allow the integration of both ‘space’ and ‘time’ into high resolution visual studies of intracellular dynamics. Recent developments, both in experimental techniques and in microscope infrastructure, have revolutionized the means by which we now study biological processes. Using live-cell microscopy it is now possible to visualize and analyze dynamic processes occurring in vivo. These methods have had a major impact on the fields of cell biology, biochemistry and molecular biology, and have encouraged the integration of biophysical and mathematical analyses to the study of cell dynamics. Visual approaches are now applied for the examination of real-time processes taking place in single cells, on single genes and even at the level of single molecules. In fact, single living cells have become the new generation test-tube in which many biological questions are investigated just as they unfold before our eyes. It is expected that these approaches will ultimately allow the probing of the most fundamental molecular pathways taking place during normal and diseased cell states. While live-cell imaging is now applied to many fields of research, this review will focus on the in vivo dynamics of major players in the gene expression pathway, namely DNA, RNA and their associated proteins, and will discuss how different methodologies for the tagging of nucleic acids for in vivo experiments have revolutionized our outlook on intracellular dynamics.Fluorescent tools for the visualization of molecules in vivo
The basic molecular elements in the gene expression pathway are DNA, RNA and protein. DNA resides within the cell nucleus. Genes contained within the DNA molecules are activated in the cell nucleus, they transcribe messenger RNAs (mRNA) that are subsequently exported from the nucleus to the cytoplasm to be translated into proteins. The pre-requisite for conducting live-cell experiments in which one can specifically follow any one of the above elements during a time-course experiment, is the ability to intracellularly detect the molecules of interest without interference from the multitude of other molecules/complexes/structures in their vicinity. The field of bio-imaging therefore strives to develop techniques for specific fluorescent tagging of the molecules of interest. In this manner the fluorescent light emitted from the tagged molecules is detectable with a fluorescent microscope while the other untagged cellular components remain ‘in darkness’. Fluorescent molecules can then be followed spatially and temporally in living cells using time-lapse microscopy.The green fluorescent protein (GFP) and the many variants of fluorescent proteins later discovered or engineered, have become the cornerstone of live-cell imaging experiments.1 The DNA coding sequence of the fluorescent protein of choice (e.g. GFP) is molecularly fused to the DNA coding sequence of the protein of interest. This engineered fused DNA (or gene) is introduced into cells, is expressed and results in a fluorescently-tagged protein that can be followed in time-lapse experiments. For instance, SF2 is a nuclear protein that functions as a splicing factor and is found to accumulate in sub-nuclear domains termed ‘speckles’.2 Fusing of the DNA gene sequence of SF2 with the DNA sequence of GFP resulted in a GFP-SF2 protein that correctly localized to speckles.3 The mobility of these structures could then be examined in live-cell experiments and was found to be rather immobile although a flux of GFP-SF2 proteins entering and exiting them could be structurally detected. This procedure of XFP-tagging of proteins is now common practice in many laboratories and is used for the real-time visualization of proteins, structures and organelles in cells and tissues.
Not only changes in location can be tracked, but the kinetics of fluorescently-labeled proteins can be extracted from time-lapse movies. In fact, the ability to track and analyze mobile populations of molecules, at times even at the single molecule level, has been one of the major breakthroughs brought about by live-cell imaging. A variety of kinetic techniques are now available,4 many of which have been integrated into the software capabilities of confocal fluorescent microscopes. They include: fluorescence recovery after photobleaching (FRAP),5 fluorescence loss in photobleaching (FLIP),6 inverse FRAP (iFRAP)7 and photoactivation8 (Fig. 1). Generally speaking, these procedures are based on the photobleaching or removal of the fluorescent signal in one designated area of the cell, and the subsequent following of the movement of the remaining fluorescent signal in either that area or in other areas of the cell. The kinetics of the exchange of unbleached molecules with the bleached ones in the specified region of interest are measured to provide the basis for calculations of protein mobility, diffusion coefficients, rates of exchange and interactions with other proteins or structures. Other available methods utilize the measurements of fluorescence lifetimes (FLIM) or anisotropy decay of fluorescent molecules,9 however, these are yet to be extensively implemented in the study of nuclear dynamics. Fluorescence-correlation spectroscopy (FCS) is another powerful tool for measuring molecular dynamics in living cells.10 This sensitive method measures the dynamics of single molecules within extremely small volumes using many short acquisition times, therefore requiring less laser power in comparison to the photobleaching methods. FCS uses autocorrelation analysis to determine the correlation between changes in intensity at different time points within the experiment and thereby measures diffusion rates and binding. Although the implementation of FCS in the study of nuclear dynamics is still wanting, several groups have made use of this method for measurements of RNA and protein dynamics in the nucleus.11–14
 | ||
| Fig. 1 Techniques for measuring the mobility of molecules within living cells using photobleaching and photoactivation techniques. In this diagram a nuclear protein (X) is fused to a GFP protein and is diffusely distributed in the nucleoplasm (green), while the cytoplasm is devoid of any fluorescence and therefore depicted in black. FRAP (fluorescence recovery after photobleaching): the fluorescent signal of the GFP-X nuclear protein is bleached at a discrete region of interest (ROI) in the nucleus (black hole). The recovery of the fluorescence in the ROI, as a result of the mobility of this protein, is followed over time until a steady state of distribution is reached once again. In this manner the diffusion coefficient of this molecule can be measured and can be compared to other known proteins or complexes thereby providing information on the dynamics within this biological system. An immobile fraction of the molecules would be detected in the case of a recovery level at the ROI that does not reach the initial levels pre-bleach. iFRAP (inverse FRAP): in contrast to a FRAP experiment, in iFRAP the total nuclear fluorescence is bleached (which requires repeated bleach pulses) except for the fluorescence in an ROI. The disappearance of the fluorescent signal in the ROI is measured over time and provides information about dissociation kinetics. The repeated bleaching does not allow measurements of rapid processes. FLIP (fluorescence loss in photobleaching): repeated bleaching of the ROI during the whole experiment gradually depletes GFP-X fluorescence in the nucleus. The kinetics of fluorescence loss at an additional ROI in the nucleus are measured and can provide information about subpopulations of the GFP-X molecules moving at different rate constants. This technique can also be used to examine the dynamic connection between different domains or regions in the cell. Photoactivation: in this case the nucleus contains a photoactivatable GFP-X protein that does not fluoresce until activated. After photoactivation of PAGFP-X at the ROI, the GFP signal is detected and its disappearance is measured over time. While this resembles iFRAP, the advantages of photoactivation are a short laser pulse in comparison to repeated photobleaching in iFRAP, and the ability to measure fast processes. | ||
While the fluorescent tagging of proteins has proven to be rather straightforward, the tagging of nucleic acids (DNA and RNA) was more challenging. Since fluorescent moieties cannot be directly introduced into the complex molecular chains of nucleic acids without disruption of their biological integrity, indirect approaches for tagging of DNA or RNA have been devised. Fluorescent In Situ Hybridization (FISH)15 is a common protocol for the labeling of DNA or RNA in fixed cells that has also been adapted for live cell experiments (Fluorescent In Vivo Hybridization – FIVH).11,16 A DNA oligonucleotide probe that is complementary to the DNA or RNA sequence of interest is synthesized. The DNA probe contains several modified nucleotides that are covalently linked with fluorescent dyes. These fluorescent probes are then introduced into cells and bind to their endogenous counterparts (Fig. 2). In this fashion the dynamics of the total mRNA population were followed in living cells.11 Direct observation of microinjected specific fluorescently-labeled RNAs has also been possible.17–20 Several problems accompany these approaches. The probes must be actively delivered into living cells using techniques such as microinjection or membrane permeabilization that can be strenuous to the cells.21,22 In many cases the cells are saturated with fluorescent probe and it is not trivial to separate the specific hybridized signal from the non-hybridized signal. Furthermore, the formation of double-stranded nucleic acid hybrids can induce enzymatic degradation pathways in the cells, while the probes themselves are sensitive to intracellular nucleases. The latter problem has been circumvented using modified probes such as peptide nucleic acid (PNA) probes, phosphorothioate oligodeoxynucleotides (PS-ODNs) or 2′-O-methyl probes that are less sensitive to degradation processes.23–29 An important advancement in this field is the development of molecular beacons.30,31 These are hairpin DNA structures that contain fluorophores at each of their ends. When the DNA sequence is closed (hairpin form) the fluorophores quench each other and there is no fluorescent signal. The hybridization of the probe with its complementary sequence in the cell causes the hairpin to open, resulting in the removal of the quenching effect (Fig. 2). This technique therefore provides a specific fluorescent signal only upon melting of the hairpin and binding to the target.
 | ||
| Fig. 2 Methods for the detection of mRNA molecules in living cells. FIVH: fluorescent in vivo hybridization involves the binding of a fluorescently-labeled probe to the target mRNA sequence. To enhance detection, multiple probes that are specific for different regions of the mRNA can be used. Molecular beacons: the beacon probe contains sequences that hybridize intramolecularly thereby bringing the fluorescent molecule in close proximity with a quencher molecule. The quenching effect is released only upon hybridization with the mRNA molecule and a fluorescent signal is obtained. This approach circumvents the problem of detection of hybridized and non-hybridized probes in FIVH. RNA-binding proteins: proteins that bind to specific sequences in the mRNA are utilized for indirect tagging of the mRNA. For example, GFP-polyA binding protein 2 (GFP-PABP2) can be used to follow all nuclear mRNA since this proteins binds specifically to polyA tails found in all mRNAs. MS2 labeling: in order to obtain labeling of one mRNA species, MS2 sequences are inserted into the gene giving rise to MS2 stem-loop structures in the mRNA. These are then bound specifically by GFP-MS2 dimers thereby tagging each mRNA molecule with a multitude of fluorescent proteins. | ||
The above techniques require the synthesis, purification, fluorescent-dye coupling of oligonucleotide probes and at times the laborious delivery of the probes into living cells. Another experimental avenue for tagging of nucleic acids is based on the simple expression of XFP proteins that can specifically bind to DNA or RNA molecules (Fig. 2). XFP-labeled RNA-binding proteins have been used for the indirect binding of mRNAs. For instance, since all mammalian mRNAs contain a poly-A tail, a GFP-poly-A-binding protein 2 (GFP-PABP2) was used to monitor the movement of the total population of cellular mRNA.32 In contrast, a specific RNA-binding protein GFP-exuperantia in Drosophila was employed to follow the travels of a specific RNA species, bicoid RNA, in developing oocytes.33 Unfortunately, most RNA-binding proteins do not bind specifically to one mRNA species. Therefore they cannot serve as a unique tag to a specific mRNA, especially due to the inability to detect a specific signal amongst the overall signal found in the rest of the cell or the nucleus. A way to increase the detectability of a specific signal, or in other words to significantly increase the signal-to-noise ratio, is to insert many repeats of the same RNA-binding sequence into the mRNA of interest, thus allowing the tethering of many fluorescent proteins to one mRNA message.34 This approach has been used successfully in the tagging of specific mRNAs in living yeast, mammalian cells and Drosophila oocytes. In this approach the gene of interest contains an additional series of DNA repeats, termed MS2 repeats. These are bacteriophage sequences that do not interfere or interact with cellular components. When transcribed into mRNA they fold into stem-loop structures and specifically bind dimers of GFP-MS2 proteins (Fig. 2). For example, when 24 MS2 repeats are inserted into the mRNA, the mRNA molecule is theoretically coated with 48 GFP-MS2 proteins which yield a strong and specific signal with a high signal-to-noise ratio.
In a similar manner, a specific DNA region can be visualized in living cells.35 This has been performed by inserting a series of lac operator (lacO) repeats into the DNA of interest. These are bacterial repeats that bind the lac repressor protein (lacI). Fluorescent tagging of lacI provides the means by which the DNA sequence is tagged with many copies of the protein. If the DNA of interest is then integrated into the genome, the XFP-lacI tagging will detect the chromosomal locus of integration. DNA has also been labeled by fluorescent nucleotides36 and fluorescently-tagged chromosome binding proteins, as will be discussed below. This approach labels the entire DNA in cells and not specific regions, and has been applied in studies that have provided novel information on chromosome dynamics during the cell cycle.
Dynamics of DNA
The ability to tag and visualize DNA in vivo has provided the high-resolution means by which to probe the motion of specific chromatin regions during normal cell growth and during transcriptional activation.35 A fundamental outcome of this approach was the understanding that chromatin domains can be highly dynamic.37–40 Chromatin is far from being a rigid or static nuclear structure, but rather chromatin regions are in constant motion, which follows the characteristics of constrained Brownian motion.41 Studies of tagged chromatin loci in yeast cells using the lacO/lacI system have shown fast short-range jumps of small chromatin domains within confined areas, but also slower and long-range motion of larger chromosome regions. Telomeres, which are structures at the ends of eukaryotic chromosomes required for the maintenance of chromosome stability and integrity, have been labeled in yeast using the lacO/LacI system42 or by fluorescent PNA probes in mammalian cells25 and were shown to be dynamically moving structures in interphase cells (see example of telomere tracking in Fig. 3). Despite a certain degree of dynamics, telomere or chromatin movements in general are constrained, thereby preserving the territorial organization of chromosomes.43 Interestingly, chromatin motion is suppressed under conditions of metabolic stress due to the inhibition of ATP synthesis in the cell, and might be the consequence of the inactivation of ATP-dependent enzymes and complexes that are involved in active transcription and chromatin remodeling.39,44 | ||
| Fig. 3 Quantitative analysis of movement of telomeres and centromeres in a single nucleus. Telomeres and centromeres were visualized in living cells using CFP-TRF2 (A) and GFP-hCENPA (B), respectively. The overlay of these images is shown in (C). Quantitative movement analysis of telomeres and centromeres was performed and a selection of trajectories is shown in (D). Average mean square displacement (MSD) values of both telomeres (n = 16) and centromeres (n = 16) in one nucleus plotted against Δt revealed that the average MSD of centromeres is similar to that of the population of constrained telomeres (E). Reprinted by permission from Macmillan Publishers Ltd: EMBO Journal (Molenaar et al., Ref. 25), copyright (2003). | ||
Chromosomes are organized in the nucleus in specific domains termed chromosome territories, which on the global nuclear scale are very large and regarded immobile.45,46 There is probabilistic 3D order of chromosome territories in the nucleus. Moreover, gene-poor chromatin regions are found in proximity of the nuclear envelope, while gene-dense chromatin is enriched in the nuclear interior.47 Kinetic measurements of core histone DNA-binding proteins (H2B, H3, H4) that stabilize chromatin structure, using XFP-tagged core histone proteins, have portrayed extreme immobility of these proteins, indicating tight association with the chromatin (see example of analysis of the dynamics of H1 histone linker protein on chromatin in Fig. 4).48–50 Other histone-related proteins exhibit faster dynamics of exchange.13,51,52 In contrast to global chromosome immobility during interphase, active chromosome motion is a crucial step during cell division. Directional movement of condensed chromosomes takes place by motor-proteins on microtubuli, thereby translocating the segregated chromatids to the cell poles in mitotic cells. Real-time imaging of whole labeled chromosomes during the cell cycle showed minimal movement during interphase, and as mentioned above, non-random 3D arrangement of chromosomes. Surprisingly, after mitosis, the spatial organization of the chromosomes in interphase daughter nuclei tended to be very similar to the original positional order in the mother cell (see example of chromosome dynamics during mitosis in Fig. 5).53–55 These studies have depicted the spatial arrangements of chromosomes in the nucleus and have indicated that chromosome positioning can be transmitted through cell generations, thereby adding another level of complexity to our understanding of ordered nuclear structure in view of intra-nuclear dynamics.
 | ||
| Fig. 4 FRAP analysis of H1 linker histone protein dynamics. (A) A cell expressing H1c-GFP was imaged before and during recovery after bleaching of a nucleoplasmic area. Images were taken at the indicated times after the bleach pulse (yellow circle). The indicated area is shown enlarged in pseudocolour in the lower panel. Scale bars: 2.6 µm (top); 1.2 µm (bottom). (B) Quantitative analysis of FRAP recovery of wild-type H1-GFP variants. The absence of rapid recovery demonstrates the absence of a freely mobile pool of H1-GFP variants. (C) FRAP analysis of H1-GFP variants lacking the globular domain (ΔG) or the C-terminal domain (ΔC) and wild-type (wt) H1-GFP. Mutant recovery was significantly faster than that of wild-type H1-GFP. Values represent means ± s.d. from at least 10 cells from 3 experiments. Reprinted by permission from Macmillan Publishers Ltd: Nature (Misteli et al., Ref. 48), copyright (2000). | ||
 | ||
| Fig. 5 Positioning of labeled chromosome subsets in mitotic cells. The chromatin in NRK cells was labeled using the co-expression of H2B-CFP and H2B-YFP. The YFP signal was bleached in half of the nucleus of prophase cells, resulting in a merged-channel image depicting either H2B-CFP labeled chromatin (red) or H2B-CFP and H2B-YFP labeled chromatin (yellow). The cells were then imaged in 4D through mitosis. The orientation of the labeling boundary in the prophase nucleus was classified after division by the orientation of the metaphase plate. (A) Labeling boundary oriented parallel to the spindle axis. Projections of 5–10 z-slices (upper row) and segmentation of the projections (lower row; red = bleached regions, green = unbleached regions) are shown. (B) Perpendicular orientation. (C) Diagonal orientation. Bars are equal to 10 µm. Reprinted by permission from Elsevier: Cell (Gerlich et al., Ref. 53), copyright (2003). | ||
Gene activity in living cells
We now zoom-in from whole chromosomes to focus on the dynamics of specific gene regions contained within the chromosomal DNA molecule. Gene activation entails structural changes in the DNA strands in conjunction with the recruitment of transcription factors and the RNA polymerase. On the kinetic level, a wide range of dynamic events involving the chromatin plus the disassociation and association of many proteins, are necessary for the productive onset of transcription. Several approaches have been implemented for the interrogation of chromatin dynamics at a specific gene locus during transcriptional activation. In the one system, lacO repeats were integrated together with the dihydrofolate reductase (DHFR) gene,56 and in another system lacO repeats were inserted upstream of an inducible transcription unit (tetracycline induction).57 In both cases the lacO repeats bound by the XFP-lacI proteins serve both as an intra-nuclear marker for the gene locus in living cells and as the DNA structure that is being followed. These experiments showed that in the inactive state, the integrated gene was structurally detected as one tight and condensed locus within the nuclear volume, whereas transcriptional activation caused dramatic decondensation and unwinding of the chromatin structure. Most interestingly, the lacO-DHFR locus was found to move from an inactive nuclear-periphery location to the nuclear interior as a result of transcriptional activation58 and this repositioning effect was also shown for endogenous trans-activation regulatory elements.59 Although at this time we do not fully appreciate the significance of such translocations, we do realize that transitions in the functional state of the cell, be it quiescent of actively transcribing, have an impact on the dynamic structure and positioning of nuclear gene expression elements.Other systems, based on naturally occurring biological interactions in the nucleus, have been devised for the real-time analysis of gene activation. Interactions of ligand-dependent steroid receptors with hormone ligands have been used for live-cell imaging of gene activation. A genomic integration of mouse mammary tumor virus (MMTV) elements was activated by the binding of GFP-glucocorticoid receptor (GR) and the decondensation of the actively transcribing DNA could be followed.60,61 The interactions of GFP-progesterone receptor (PR) with the MMTV array were found to be dynamic and transient.62 In another set of studies the dynamics of a transcriptionally active GFP-estrogen receptor (ER) or CFP-ER-lacI were followed.63,64 Addition of estrogen to the cells reduced ER mobility due to binding of the nuclear receptor to the DNA, and ATP depletion also immobilized the ER. Importantly, these studies have shown that transcription-activating factors associate dynamically with chromatin elements in what has been termed a “hit-and-run” mechanism, and that residence times of different factors on the gene can vary, therefore yielding a kinetic-based regulation mechanism of transcriptional activation.
The RNA polymerase itself has been used as a tool to study live-cell transcription. Since the RNA polymerase II holoenzyme is a multi-subunit protein complex, fluorescent tagging that preserves enzymatic functionality was not straightforward. A GFP-tagged functional version of RNA polymerase II was established65 and used for the analysis of the polymerase transcription cycle.66 This research group expected to detect three kinetic populations of GFP-RNA Pol II: a fast free diffusing fraction (unbound polymerase), a slow elongating fraction and an intermediate pool involved in transcriptional initiation. However, at first only two fractions were detected: about a quarter of GFP-Pol II in the nucleus was found to be engaged in active transcription while the inactive fraction of the protein was not assembled in large stable complexes, meaning that the polymerase complex is disassembled in the inactive state. This study also concluded that probably not more than 1 active polymerase is associated with a typically expressed gene at any given time. In a subsequent study, the authors applied a different experimental approach to this study and indeed a third kinetic population of GFP-Pol II that is probably engaged in the process of transcriptional initiation was identified.67 In a different study, the recruitment of GFP-Pol II to an MMTV array was studied in single living cells.68 Although the peak in transcriptional activity was seen 20 to 30 min post-activation, there was significant variability in recruitment times of GFP-Pol II to the gene array between different cells. Thereafter, transcriptional activity declined rapidly. Cell to cell differences might reflect differences in the particular stage of the cell cycle, but could also indicate that transcription is a stochastic process depending on the availability and concerted assembly of different factors into a transcriptionally active complex.69 Interestingly, induction of metabolic stress and transcriptional inhibition in living cells had a significant slowing down effect on the dynamics of many nuclear proteins or nuclear domains involved in the gene expression pathway.70–72 When the assembly of RNA polymerase I on ribosomal DNA (rDNA) in the nucleolus was followed using a GFP-tagged subunit of this enzyme, an inefficient step-wise assembly of the complex was observed.7 Once again, these results indicated that inactive RNA Pol I is not present as a preassembled holoenzyme, but rather that it assembles only after import of subunits into the nucleolus. In fact, the structural concept emerging from kinetic experiments in the nucleus is that not only are nucleoplasmic proteins in constant flux, but even the building blocks of nuclear domains, the nucleolus for instance, demonstrate dynamic binding and disengagement of proteins from their site of function (see example of nucleolar protein dynamics measured by FLIP in Fig. 6).49,73–76
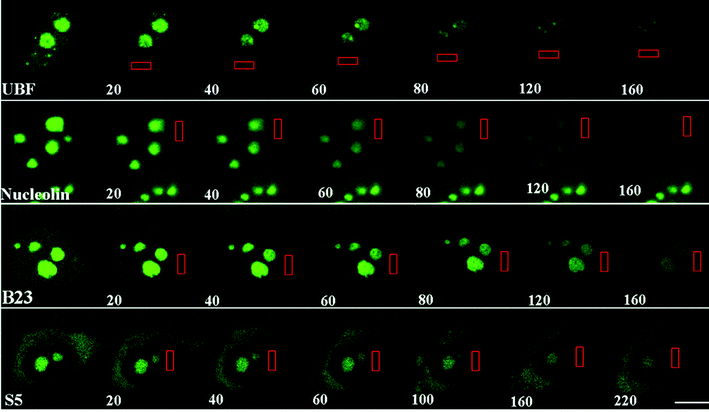 | ||
| Fig. 6 Mobility of nucleolar proteins. Cells were transfected with different nucleolar proteins fused to GFP. FLIP analyses demonstrate that the examined nucleolar components exit the interphase nucleolus rapidly. The rectangles indicate the areas of bleaching, and the numbers represent the cumulative bleaching duration in seconds. Reprinted by permission from the Rockefeller University Press: Journal of Cell Biology (Chen and Huang, Ref. 73), copyright (2001). | ||
RNA mobility in real-time
Messenger RNA must travel to the cytoplasm in order to pass on the genetic information encoded in the DNA. What bio-physical rules govern the movement of RNA in cells? Does the cell invest energy in the moving of RNA cargos, or perhaps RNAs move randomly until they happen to reach their final destination? These and other questions related to RNA mobility can only be answered if RNA movement is probed at real-time. We begin in the nucleus. Nuclear RNAs have been labeled in living cells as described above, using either fluorescent RNA probes or GFP-fused RNA binding proteins. Some studies have addressed the movement of the total population of mRNAs in the nucleus, have measured a variety of diffusion coefficients using different kinetic techniques, and have concluded that diffusion is the mechanism of translocation in the nucleus and that mRNA in transit does not accumulate in sub-nuclear structures.11,12,24,32,77 The MS2 RNA labeling system has allowed the detection of individual mRNA molecules in the nucleus of living cells.78 This study was performed in a cell system that contained both the lacO repeats for the detection of the gene locus, and MS2 repeats for the detection of mRNA in real-time.79 In this fashion individual mRNAs could be followed from their site of production through nucleoplasmic travels. Frame by frame tracking of the movements of single mRNA molecules showed that indeed the motion is diffusion-based although constrained movements could be observed (Fig. 7). The latter emphasizes that mRNA en route to the nuclear envelope must move through the mesh of the nuclear environment,46,80,81 which can probably cause hindering of the progression.82 While mRNA mobility in the nucleus is passive, and directed energy-driven motion towards the nuclear envelope has not been observed, energy depletion has been shown to affect both RNA and DNA movement in the nucleus.32,39,44,78 Although we do not fully understand the mechanism behind the reduction in mobility, it seems that chromatin structure may be sensitive to metabolic stress and suggests that such conditions alter normal intra-nuclear architecture and can potentially abrogate the pathway of diffusing mRNAs.78 | ||
| Fig. 7 Live-cell imaging and single-particle tracking of individual mRNPs. Images from time-lapse movies acquired from a cell co-transfected with (A) CFP-lac repressor detecting the genomic locus of the gene of interest and (B) YFP-MS2 for tagging of the mRNA produced from the actively transcribing gene. (C) Reduction of noise for tracking of mRNPs was obtained by deconvolution. Bar, 2 µm. (D) Tracking of mRNP particles in another transcriptionally active cell induced for only 20 min (arrow, transcription site) (bar, 2 µm) showed (E) diffusing particles and (F) corralled particles. Tracks are marked in green, and time in seconds from the beginning of tracking for each particle appears in each frame. Bars, 1 µm. The mean-square displacement (MSD) of tracked nucleoplasmic particles versus time indicated the presence of three types of characterized movements: diffusive, corralled, and stationary. Directed movement was never detected. Reprinted by permission from the American Association for the Advancement of Science: Science (Shav-Tal et al., Ref. 78), copyright (2004). | ||
After finally reaching the nuclear envelope, the mRNA is transported into the cytoplasm. mRNAs typically associate with ribosome subunits and proceed to translation. However, many RNAs are destined to specific regions in the cell and this process requires an initial step of localization to be followed by translation.83 This process of ‘RNA localization’ occurs in many systems and is based on the shuttling of RNA cargos on cytoskeletal filaments by motor proteins. Cytoplasmic mRNAs tagged with fluorescent MS2 proteins were also found to diffuse. However, rapid directional movements on microtubules could also be detected.84 Interestingly, mRNAs could switch between modes of movements implying a dynamic cycle between diffusion and active transport states. The cell therefore does not actively transport mRNAs from their point of synthesis in the nucleus towards the nuclear envelope, yet in the cytoplasm, energy is invested in the shuttling of many mRNAs probably due to the relatively large distances that must be traversed.
Since the genome of some viruses is RNA-based, the movement of RNA retroviruses could be monitored by tagging the RNA. MS2-GFP labeling of murine leukemia virus (MLV) RNA was used to follow the in vivo route of transport of the virus RNA in vesicles and showed that two possible transport pathways can be used by the virus in order to reach the plasma membrane.85 Poliovirus RNA was labeled using molecular beacons and its dynamic behavior in living cells was analyzed.86 Diffusional movement was detected for half of the cytoplasmic viral RNA population while the other half were immobile. Disruption of the microtubule network indicated its importance in the cytoplasmic transport of this virus. Molecular beacons have now been used to follow a 7 day time-course of infection with respiratory syncytial virus (RSV).87 This virus is an RNA virus too, and by labeling of the viral genome, the formation of inclusion bodies during infection could be detected and monitored.
Now that methods for live-cell imaging are available for following gene expression processes in single cells, we aspire to follow these processes in the full context of a living organism. This has yet to be accomplished in living animals, but RNA in transit has been tracked in lower organisms. Following are some specific examples. Active transport of mRNA was first observed in budding yeast using the MS2 tagging system.34 GFP-MS2 tagged elements of the Ash1 mRNA translocated from the mother yeast cell to the budding daughter cell at a speed that correlates with that of the myosin V motor, and localized at the bud tip. Real-time imaging of nanos mRNA localization in Drosophila oocytes found that this mRNA reaches the posterior region of the oocyte through diffusion enhanced by microtubule-dependent cytoplasmic movements, where it remained anchored by the actin cytoskeleton.88 This finding is in contrast to most other localizing mRNAs that require a motor protein for their translocation. The anterior localization of bicoid mRNA in Drosophila oocytes was followed using either tagging of the mRNA by GFP-exuperantia or with injected fluorescent probes and showed that this process requires the assembly of an exuperantia-bicoid mRNA complex for translocation on the microtubule network.33,89oskar mRNA localization was followed in Drosophila oocytes as it moved from nurse cells to the posterior cortex using either injected fluorescent probes90 or molecular beacons.91
Conclusions and future directions
Live-cell imaging has transformed the way we study cells. The ability to follow kinetic processes as they occur in real-time incorporates a new level of complexity to the biological picture we strive to assemble. We now utilize not only biochemical and molecular tools, but require the integration of bio-physical and mathematical expertise for the analysis of kinetic visual data. Indeed, much novel information on the gene expression pathway has been gained using such interdisciplinary approaches. Functional nuclear architecture appears to be defined by the formation of distinct structures and complexes that provide both the scaffold and the functional components for the assembly of active gene expression. Yet, this vision is a dynamic one encompassing the constant flux of proteins in and out of nuclear domains; the constrained motion of chromatin; the reversible binding of DNA-binding proteins to the chromatin; and the traveling of mRNA from sites of synthesis to the nuclear membrane through inter-chromatin regions. How these processes come together to produce a functional nucleus is still an open question. Future challenges will be to track multiple processes occurring simultaneously on different genes and to understand the physical rules that govern the activation and shut-off of gene expression in living cells. The development of tools for the analysis of real-time processes will ultimately allow the progression from studies performed in single cells towards the understanding of the complex gene expression pathways occurring in whole organisms.Acknowledgements
Yaron Shav-Tal is a Stern-Lebell Family Fellow in Life Sciences.References
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS.
- A. I. Lamond and D. L. Spector, Nat. Rev. Mol. Cell Biol., 2003, 4, 605–612 CrossRef CAS.
- T. Misteli, J. F. Caceres and D. L. Spector, Nature, 1997, 387, 523–527 CrossRef CAS.
- J. Lippincott-Schwartz and G. H. Patterson, Science, 2003, 300, 87–91 CrossRef.
- D. Axelrod, D. E. Koppel, J. Schlessinger, E. Elson and W. W. Webb, Biophys. J., 1976, 16, 1055–1069 CrossRef CAS.
- N. B. Cole, C. L. Smith, N. Sciaky, M. Terasaki, M. Edidin and J. Lippincott-Schwartz, Science, 1996, 273, 797–801 CrossRef CAS.
- M. Dundr, U. Hoffmann-Rohrer, Q. Hu, I. Grummt, L. I. Rothblum, R. D. Phair and T. Misteli, Science, 2002, 298, 1623–1626 CrossRef CAS.
- G. H. Patterson and J. Lippincott-Schwartz, Science, 2002, 297, 1873–1877 CrossRef CAS.
- R. M. Clegg, O. Holub and C. Gohlke, Methods Enzymol., 2003, 360, 509–542 CAS.
- D. Magde, E. L. Elson and W. W. Webb, Biopolymers, 1974, 13, 29–61 CrossRef CAS.
- J. C. Politz, R. A. Tuft, T. Pederson and R. H. Singer, Curr. Biol., 1999, 9, 285–291 CrossRef CAS.
- J. C. Ritland Politz, R. A. Tuft, K. V. Prasanth, N. Baudendistel, K. E. Fogarty, L. M. Lifshitz, J. Langowski, D. L. Spector and T. Pederson, Mol. Biol. Cell, 2005 Search PubMed.
- L. Schmiedeberg, K. Weisshart, S. Diekmann, G. Meyer Zu Hoerste and P. Hemmerich, Mol. Biol. Cell, 2004, 15, 2819–2833 CrossRef CAS.
- M. Wachsmuth, W. Waldeck and J. Langowski, J. Mol. Biol., 2000, 298, 677–689 CrossRef CAS.
- J. M. Levsky and R. H. Singer, J. Cell Sci., 2003, 116, 2833–2838 Search PubMed.
- J. C. Politz, K. L. Taneja and R. H. Singer, Nucleic Acids Res., 1995, 23, 4946–4953 CrossRef CAS.
- K. Ainger, D. Avossa, F. Morgan, S. J. Hill, C. Barry, E. Barbarese and J. H. Carson, J. Cell Biol., 1993, 123, 431–441 CrossRef CAS.
- M. R. Jacobson and T. Pederson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7981–7986 CrossRef CAS.
- M. R. Jacobson, L. G. Cao, Y. L. Wang and T. Pederson, J. Cell Biol., 1995, 131, 1649–1658 CrossRef CAS.
- G. S. Wilkie and I. Davis, Cell, 2001, 105, 209–219 CrossRef CAS.
- E. L. Barry, F. A. Gesek and P. A. Friedman, Biotechniques, 1993, 15, 1016–1018, 1020 CAS.
- P. L. McNeil and E. Warder, J. Cell Sci., 1987, 88(Pt 5), 669–678 Search PubMed.
- M. Carmo-Fonseca, R. Pepperkok, B. S. Sproat, W. Ansorge, M. S. Swanson and A. I. Lamond, EMBO J., 1991, 10, 1863–1873 CAS.
- C. Molenaar, A. Abdulle, A. Gena, H. J. Tanke and R. W. Dirks, J. Cell Biol., 2004, 165, 191–202 CrossRef CAS.
- C. Molenaar, K. Wiesmeijer, N. P. Verwoerd, S. Khazen, R. Eils, H. J. Tanke and R. W. Dirks, EMBO J., 2003, 22, 6631–6641 CrossRef CAS.
- S. Agrawal, Z. Jiang, Q. Zhao, D. Shaw, Q. Cai, A. Roskey, L. Channavajjala, C. Saxinger and R. Zhang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2620–2625 CrossRef CAS.
- T. L. Fisher, T. Terhorst, X. Cao and R. W. Wagner, Nucleic Acids Res., 1993, 21, 3857–3865 CrossRef CAS.
- P. Lorenz, B. F. Baker, C. F. Bennett and D. L. Spector, Mol. Biol. Cell, 1998, 9, 1007–1023 CAS.
- P. Lorenz, T. Misteli, B. F. Baker, C. F. Bennett and D. L. Spector, Nucleic Acids Res., 2000, 28, 582–592 CrossRef CAS.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
- N. Nitin, P. J. Santangelo, G. Kim, S. Nie and G. Bao, Nucleic Acids Res., 2004, 32, e58 CrossRef.
- A. Calapez, H. M. Pereira, A. Calado, J. Braga, J. Rino, C. Carvalho, J. P. Tavanez, E. Wahle, A. C. Rosa and M. Carmo-Fonseca, J. Cell Biol., 2002, 159, 795–805 CrossRef CAS.
- W. E. Theurkauf and T. I. Hazelrigg, Development, 1998, 125, 3655–3666 CAS.
- E. Bertrand, P. Chartrand, M. Schaefer, S. M. Shenoy, R. H. Singer and R. M. Long, Mol. Cell, 1998, 2, 437–445 CrossRef CAS.
- A. S. Belmont, Trends Cell Biol., 2001, 11, 250–257 CrossRef CAS.
- E. M. Manders, H. Kimura and P. R. Cook, J. Cell Biol., 1999, 144, 813–821 CrossRef CAS.
- W. F. Marshall, A. Straight, J. F. Marko, J. Swedlow, A. Dernburg, A. Belmont, A. W. Murray, D. A. Agard and J. W. Sedat, Curr. Biol., 1997, 7, 930–939 CrossRef CAS.
- J. Vazquez, A. S. Belmont and J. W. Sedat, Curr. Biol., 2001, 11, 1227–1239 CrossRef CAS.
- P. Heun, T. Laroche, K. Shimada, P. Furrer and S. M. Gasser, Science, 2001, 294, 2181–2186 CrossRef CAS.
- J. R. Chubb, S. Boyle, P. Perry and W. A. Bickmore, Curr. Biol., 2002, 12, 439–445 CrossRef CAS.
- S. M. Gasser, Science, 2002, 296, 1412–1416 CrossRef CAS.
- F. Hediger, F. R. Neumann, G. van Houwe, K. Dubrana and S. M. Gasser, Curr. Biol., 2002, 12, 2076–2089 CrossRef CAS.
- K. Bystricky, T. Laroche, G. van Houwe, M. Blaszczyk and S. M. Gasser, J. Cell Biol., 2005, 168, 375–387 CrossRef CAS.
- V. Levi, Q. Ruan, M. Plutz, A. S. Belmont and E. Gratton, Biophys. J, 2005, 89, 4275–4285 CrossRef CAS.
- D. Zink, T. Cremer, R. Saffrich, R. Fischer, M. F. Trendelenburg, W. Ansorge and E. H. Stelzer, Hum. Genet., 1998, 102, 241–251 CrossRef CAS.
- T. Cremer and C. Cremer, Nat. Rev. Genet., 2001, 2, 292–301 CrossRef CAS.
- A. Bolzer, G. Kreth, I. Solovei, D. Koehler, K. Saracoglu, C. Fauth, S. Muller, R. Eils, C. Cremer, M. R. Speicher and T. Cremer, PLoS Biol., 2005, 3, e157 Search PubMed.
- T. Misteli, A. Gunjan, R. Hock, M. Bustin and D. T. Brown, Nature, 2000, 408, 877–881 CrossRef CAS.
- R. D. Phair and T. Misteli, Nature, 2000, 404, 604–609 CrossRef CAS.
- H. Kimura and P. R. Cook, J. Cell Biol., 2001, 153, 1341–1353 CrossRef CAS.
- M. A. Lever, J. P. Th'ng, X. Sun and M. J. Hendzel, Nature, 2000, 408, 873–876 CrossRef CAS.
- R. D. Phair, P. Scaffidi, C. Elbi, J. Vecerova, A. Dey, K. Ozato, D. T. Brown, G. Hager, M. Bustin and T. Misteli, Mol. Cell. Biol., 2004, 24, 6393–6402 CrossRef CAS.
- D. Gerlich, J. Beaudouin, B. Kalbfuss, N. Daigle, R. Eils and J. Ellenberg, Cell, 2003, 112, 751–764 CrossRef CAS.
- J. Walter, L. Schermelleh, M. Cremer, S. Tashiro and T. Cremer, J. Cell Biol., 2003, 160, 685–697 CrossRef CAS.
- E. M. Manders, A. E. Visser, A. Koppen, W. C. de Leeuw, R. van Liere, G. J. Brakenhoff and R. van Driel, Chromosome Res., 2003, 11, 537–547 CrossRef CAS.
- T. Tumbar, G. Sudlow and A. S. Belmont, J. Cell Biol., 1999, 145, 1341–1354 CrossRef CAS.
- T. Tsukamoto, N. Hashiguchi, S. M. Janicki, T. Tumbar, A. S. Belmont and D. L. Spector, Nat. Cell Biol., 2000, 2, 871–878 CrossRef CAS.
- T. Tumbar and A. S. Belmont, Nat. Cell Biol., 2001, 3, 134–139 CrossRef CAS.
- S. Dietzel, K. Zolghadr, C. Hepperger and A. S. Belmont, J. Cell Sci., 2004, 117, 4603–4614 Search PubMed.
- J. G. McNally, W. G. Muller, D. Walker, R. Wolford and G. L. Hager, Science, 2000, 287, 1262–1265 CrossRef CAS.
- W. G. Muller, D. Walker, G. L. Hager and J. G. McNally, J. Cell Biol., 2001, 154, 33–48 CrossRef CAS.
- G. V. Rayasam, C. Elbi, D. A. Walker, R. Wolford, T. M. Fletcher, D. P. Edwards and G. L. Hager, Mol. Cell. Biol., 2005, 25, 2406–2418 CrossRef CAS.
- D. L. Stenoien, A. C. Nye, M. G. Mancini, K. Patel, M. Dutertre, B. W. O'Malley, C. L. Smith, A. S. Belmont and M. A. Mancini, Mol. Cell. Biol., 2001, 21, 4404–4412 CrossRef CAS.
- D. L. Stenoien, K. Patel, M. G. Mancini, M. Dutertre, C. L. Smith, B. W. O'Malley and M. A. Mancini, Nat. Cell Biol., 2001, 3, 15–23 CrossRef CAS.
- K. Sugaya, M. Vigneron and P. R. Cook, J. Cell Sci., 2000, 113(Pt 15), 2679–2683 Search PubMed.
- H. Kimura, K. Sugaya and P. R. Cook, J. Cell Biol., 2002, 159, 777–782 CrossRef CAS.
- M. Hieda, H. Winstanley, P. Maini, F. J. Iborra and P. R. Cook, Chromosome Res., 2005, 13, 135–144 CrossRef CAS.
- M. Becker, C. Baumann, S. John, D. A. Walker, M. Vigneron, J. G. McNally and G. L. Hager, EMBO Rep., 2002, 3, 1188–1194 Search PubMed.
- J. M. Levsky and R. H. Singer, Trends Cell Biol., 2003, 13, 4–6 CrossRef CAS.
- M. Muratani, D. Gerlich, S. M. Janicki, M. Gebhard, R. Eils and D. L. Spector, Nat. Cell Biol., 2002, 4, 106–110 CrossRef CAS.
- M. Platani, I. Goldberg, A. I. Lamond and J. R. Swedlow, Nat. Cell Biol., 2002, 4, 502–508 CrossRef CAS.
- Y. Shav-Tal, J. Blechman, X. Darzacq, C. Montagna, B. T. Dye, J. G. Patton, R. H. Singer and D. Zipori, Mol. Biol. Cell, 2005, 16, 2395–2413 CrossRef CAS.
- D. Chen and S. Huang, J. Cell Biol., 2001, 153, 169–176 CrossRef CAS.
- S. Snaar, K. Wiesmeijer, A. G. Jochemsen, H. J. Tanke and R. W. Dirks, J. Cell Biol., 2000, 151, 653–662 CrossRef CAS.
- K. E. Handwerger, C. Murphy and J. G. Gall, J. Cell Biol., 2003, 160, 495–504 CrossRef CAS.
- J. E. Sleeman, L. Trinkle-Mulcahy, A. R. Prescott, S. C. Ogg and A. I. Lamond, J. Cell Sci., 2003, 116, 2039–2050 Search PubMed.
- J. C. Politz, E. S. Browne, D. E. Wolf and T. Pederson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6043–6048 CrossRef CAS.
- Y. Shav-Tal, X. Darzacq, S. M. Shenoy, D. Fusco, S. M. Janicki, D. L. Spector and R. H. Singer, Science, 2004, 304, 1797–1800 CrossRef CAS.
- S. M. Janicki, T. Tsukamoto, S. E. Salghetti, W. P. Tansey, R. Sachidanandam, K. V. Prasanth, T. Ried, Y. Shav-Tal, E. Bertrand, R. H. Singer and D. L. Spector, Cell, 2004, 116, 683–698 CrossRef CAS.
- Z. Zachar, J. Kramer, I. P. Mims and P. M. Bingham, J. Cell Biol., 1993, 121, 729–742 CrossRef CAS.
- J. M. Bridger, C. Kalla, H. Wodrich, S. Weitz, J. A. King, K. Khazaie, H. G. Krausslich and P. Lichter, Exp. Cell Res., 2005, 302, 180–193 CrossRef CAS.
- M. Carmo-Fonseca, M. Platani and J. R. Swedlow, Trends Cell Biol., 2002, 12, 491–495 CrossRef CAS.
- Y. Shav-Tal and R. H. Singer, J. Cell Sci., 2005, 118, 4077–4081 Search PubMed.
- D. Fusco, N. Accornero, B. Lavoie, S. M. Shenoy, J. M. Blanchard, R. H. Singer and E. Bertrand, Curr. Biol., 2003, 13, 161–167 CrossRef CAS.
- E. Basyuk, T. Galli, M. Mougel, J. M. Blanchard, M. Sitbon and E. Bertrand, Dev. Cell, 2003, 5, 161–174 CrossRef CAS.
- Z. Q. Cui, Z. P. Zhang, X. E. Zhang, J. K. Wen, Y. F. Zhou and W. H. Xie, Nucleic Acids Res., 2005, 33, 3245–3252 CrossRef CAS.
- P. Santangelo, N. Nitin, L. Laconte, A. Woolums and G. Bao, J. Virol., 2006, 80, 682–688 CrossRef CAS.
- K. M. Forrest and E. R. Gavis, Curr. Biol., 2003, 13, 1159–1168 CrossRef CAS.
- B. J. Cha, B. S. Koppetsch and W. E. Theurkauf, Cell, 2001, 106, 35–46 CrossRef CAS.
- J. B. Glotzer, R. Saffrich, M. Glotzer and A. Ephrussi, Curr. Biol., 1997, 7, 326–337 CrossRef CAS.
- D. P. Bratu, B. J. Cha, M. M. Mhlanga, F. R. Kramer and S. Tyagi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13308–13313 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |