DNA polymerization on the inner surface of a giant liposome for synthesizing an artificial cell model
Koh-ichiroh
Shohda
and
Tadashi
Sugawara
*
Department of Basic Science, Graduate School of Arts and Sciences, The University of Tokyo, Komaba, Meguro-ku, Tokyo 153-8902, Japan. E-mail: suga@pentacle.c.u-tokyo.ac.jp; Fax: +81 3 5454 6997; Tel: +81 3 5454 6742
First published on 24th March 2006
Abstract
We have designed a new artificial cell model consisting of a giant liposome, an enzyme, and DNA conjugated with a cholesterol tag by a poly(ethylene glycol) spacer to characterize the model system. The cholesterol tag of the conjugated molecule was anchored to the inner surface of the giant liposome and the single-stranded DNA unit hybridized with a 100-mer template DNA that was added to the water pool inside the liposome. We found that the DNA unit acted as a primer, DNA polymerization proceeded on the inner surface of the liposome. This reaction was a key step of our cell model. Production of a full-length strand was proved by selective cleavage of the polymerized DNA by a restriction enzyme.
Introduction
Studies on the origins of life and the synthesis of artificial cellular life have drawn considerable attention from many scientists. These topics require multi-disciplinary approaches not only from biology but also from chemistry, physics, mathematics, etc. An informational substance, catalytic activity, and a compartment which is separated from the outer world are indispensable elements of the cellular life.1 Liposomes, which are an assembly of amphiphilic molecules, are regarded as a suitable compartment in an artificial cell model because of their resemblance to living cells in shape and structure.2–8 Recently, we reported on a couple of replicating membrane systems composed of artificial amphiphilic molecules.9,10 To synthesize a more advanced cell model, replication of an informational substance (RNA or DNA) inside the liposome is required. Although RNA synthesis inside liposomes has been reported in earlier investigations, the informational substance and the catalyst are uniformly dissolved in the aqueous medium inside liposomes.6,11–13 To closely correlate the replication of membranes with replication of the informational substances in a well defined manner, we recently designed an informational amphiphile (1) consisting of an oligodeoxyribonucleotide (DNA 15-mer) with a cholesterol tag, a poly(ethylene glycol) (PEG) unit inserted between these two parts as a spacer (Fig. 1).14 We reported that the cholesterol unit of conjugate molecule 1 was embedded itself in the membrane of a phospholipid liposome and that the 15-mer DNA of 1 was able to hybridize with a complementary DNA on the surface of the liposome. | ||
| Fig. 1 Ester-linked informational amphiphile (cholesterol–PEG–DNA conjugate 1). | ||
In the present study, we have developed a more stable informational amphiphile 2 (Scheme 1) and a novel artificial cell model consisting of informational amphiphile 2, a single-stranded 100-mer DNA template (as a gene model), DNA polymerase, and a giant liposome (Fig. 2). Furthermore, we found that a polymerizing enzyme could generate a new DNA strand on the gene-modeled DNA template located on the inner surface of the giant liposome. The DNA-directed DNA polymerization in the cell-sized compartment is a key reaction toward the perfection of our artificial cell model.
 | ||
| Fig. 2 Artificial cell model. Ribbon: 100-mer template DNA (gene model); solid circle: DNA polymerase. | ||
 | ||
| Scheme 1 Synthesis of amide-linked cholesterol–PEG–DNA conjugate 2. (a) Phthalimide, diethylazodicarboxylate (DEAD), triphenylphosphine, DMF, 97%; (b) hydrazine monohydrate, pyridine, 81%; (c) succinic anhydride, pyridine–CH2Cl2 (1 ∶ 1, v/v), 98%; (d) water-soluble carbodiimide, N-hydroxysuccinimide, CH2Cl2; (e) 5′-aminohexyl oligodeoxyribonucleotide, NaHCO3, DMF–H2O (1 ∶ 1, v/v), 33% (based on 5′-modified DNA). | ||
Results and discussion
Synthesis of cholesterol–PEG–DNA conjugate 2
Although earlier we had synthesized cholesterol–PEG–DNA conjugate 1, in which cholesterol and DNA are linked by an ester group,14 we noticed that the ester group of 1 is gradually hydrolyzed even in a mild alkaline aqueous solution. To be sure of observing events occurring purely on a membrane surface and not in solution, we therefore designed a new informational amphiphile, cholesterol–PEG–DNA conjugate 2 which had an amide linkage instead of the ester linkage (Scheme 1).First, the end hydroxyl group of commercially available PEGylated cholesterol 3 (the average degree of polymerization and molecular weight of the PEG unit were ca. 110 and 4900, respectively) was converted into a phthalimide group by the Mitsunobu reaction.15 Product 4 was treated with hydrazine monohydrate to remove the phthaloyl group.16 Succinic anhydride was added to the resulting amino derivative 5, giving rise to compound 6 with a carboxyl group at the end. Following our previous protocol,14 we transformed compound 6 into activated ester 7 by using a water-soluble carbodiimide.17 Coupling of activated ester 7 to 5′-aminohexyl DNA 15-mer, which was prepared by standard phosphoramidite chemistry,18,19 provided the desired amide-linked cholesterol–PEG–DNA conjugate 2.
DNA polymerization using the informational amphiphile 2 under liposome free conditions
A Klenow fragment (Escherichia coli) was used as the DNA-polymerizing catalyst.20,21 We carried out chain-elongation of the primer (the DNA part of conjugate 2) on the 100-mer template DNA under liposome-free conditions to check whether the elongation reaction proceeds even in the presence of the bulky cholesterol–PEG unit.The starting materials and a product were analyzed by polyacrylamide gel electrophoresis (PAGE) under non-denaturing conditions (Fig. 3). First, the conjugate 2 showed an extremely smeared band (lane 1 in Fig. 3). This result was not due to degradation of conjugate 2 or to contamination with an impurity, but was attributed to the PEG unit which had a broad range of molecular weights and multiple conformations.22 The 100-mer template DNA in lane 2 (Fig. 3) showed also a smeared band due to the conformational diversity of single-stranded DNA under non-denaturing conditions. Strangely, this single-stranded 100-mer template DNA had a similar mobility to the 100-“bp” duplex of the ladder marker. This means that the single-stranded DNA which constitutes a secondary structure like mRNA is more difficult to pass through the gel matrix than the DNA duplex with the same weight. After the treatment with the Klenow fragment, most of the partial duplex in lane 3 disappeared and a new band appeared (lane 4 in Fig. 3). The mobility of the product was greater than that of the initial partial duplex in spite of increasing the molecular weight. Since the partial duplex (lane 3) also contained a long single-stranded region (85-mer) its mobility should be considerably retarded like the 100-mer template.
 | ||
| Fig. 3 DNA polymerization in the presence of cholesterol–PEG–DNA conjugate under lipid-free conditions. Lanes: (1) cholesterol–PEG–DNA conjugate 2, (2) 100-mer DNA template, (3) partial duplex which consisted of the primer (conjugate 2) and the template DNA (5 ∶ 1), (4) the product of Klenow fragment treatment, (5) Hind III treatment of the product in lane 4, (M) 25-bp-step ladder of double-stranded markers. | ||
To verify that the product in lane 4 was actually a full-length DNA duplex, the product was treated with the restriction enzyme Hind III. A central position of the 100-mer template DNA was the 5′-AAGCTT-3′ sequence which is cleaved by Hind III.23 The Hind III treatment provided two new bands which appeared in lane 5 (Fig. 3). Since one band was broad, we determined that this was a 50-bp DNA with the cholesterol–PEG unit, and the other was an unmodified 50-bp DNA. This result clearly indicates that the product in lane 4 must be a full-length (100-bp) DNA duplex with a cholesterol–PEG unit. Namely, the sequence information on the 100-mer template was perfectly transferred to the informational amphiphile 2.
DNA polymerization on the inner surface of a liposome
Preparation of the liposome here is somewhat complicated. Liposomes containing the DNA fragments and the enzyme were prepared by the lyophilization method (Fig. 4).24 This method is suitable for constructing giant liposomes that contain macromolecules such as enzymes and polynucleotides.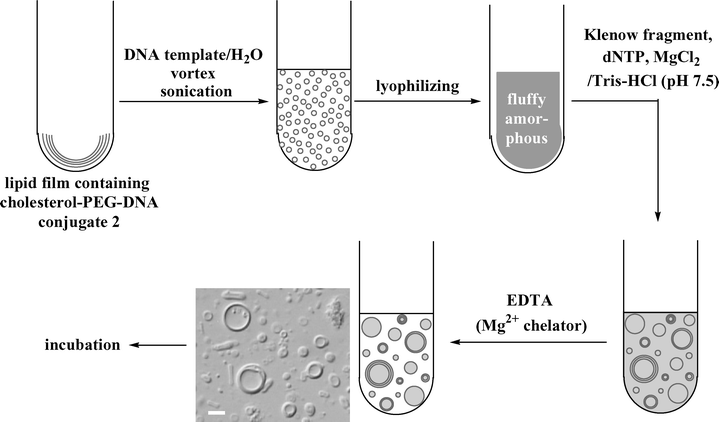 | ||
| Fig. 4 Preparation of liposomes containing DNA molecules and the enzyme. The photograph is a differential interference microscopy image of the prepared liposomes (scale bar; 20 µm). | ||
A solution of informational amphiphile 2 in methanol was mixed with a mixture of lipids consisting of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphatidylethanolamine-N-[poly(ethylene glycol)-5000] (DSPE–PEG5000) in chloroform. The third lipid was added to inhibit the unfavorable interaction between the enzyme and the membrane.11 The gene-modeled template 100-mer DNA was added in the first preparation of the liposome which was then lyophilized to form an amorphous mixture of the components. Swelling the amorphous mixture by a buffer solution containing a Klenow fragment generated many giant liposomes (Fig. 4, differential interference microscopy image). Immediately, EDTA was added to the liposome suspension in order to halt the DNA polymerization on the external surface of the liposome. Since EDTA can scarcely permeate the lipid bilayer due to its polyionic character, its addition to the external water phase should not affect the enzymatic reaction inside the liposome.25
We have already reported that the cholesterol–PEG–DNA conjugate 1 was able to hybridize with a complementary DNA strand on the surface of the liposomes.14 Therefore, the Klenow fragment would produce a full-length DNA duplex on the inner surface of the liposomes.
After incubation for 24 h, the usual procedure—the phenol–chloroform extraction and the ethanol precipitation—for isolating DNA or RNA from living cells did not yield any DNA fragments from these liposomes. Because of the presence of a lipophilic cholesterol group, the product would be aggregated between organic and aqueous layers of the phenol–chloroform extraction. Therefore, the product was purified by a C18 reversed-phase cartridge column. All UV-absorbing materials at 260 nm were retrieved as a mixture except for dNTPs, which did not bind to the C18 column. On the other hand, all the lipids which were the most hydrophobic components of the liposome system were thoroughly removed by this column chromatography.
The product was analyzed by PAGE (Fig. 5). The roughly purified product (lane 4 in Fig. 5) does not clearly show a band of a full-length DNA duplex due to an overlap with the intact conjugate 2 which is widely smeared. Therefore, the mixture containing the product was treated with Hind III. As in the corresponding experiment in the absence of liposomes (Fig. 3), a new band with similar mobility to the 50-bp duplex of the marker appeared, although the other 50-bp duplex having the cholesterol–PEG unit still overlapped with the smeared conjugate 2. These results suggested that the delivery of information from a template to a newly produced DNA strand occurred on the inner surface of the liposomes.
 | ||
| Fig. 5 DNA polymerization in the presence of cholesterol–PEG–DNA conjugate on the inner layer of liposomes. Lanes: (1) cholesterol–PEG–DNA conjugate 2, (2) 100-mer DNA template, (3) partial duplex which consisted of the primer (conjugate 2) and the template (5 ∶ 1 molar ratio), (4) roughly purified mixture of Klenow fragment treatment, (5) Hind III treatment of the mixture in lane 4, (M) 25-bp-step ladder of double-stranded markers. | ||
Direct visualization of DNA polymerization on the inner surface of the liposome
A fluorescent-labeled dUTP (TexasRed-L12-dUTP: dUTP*) was added to a buffer solution containing polymerase, and the resulting solution swelled the amorphous lipid as described in the previous section. If all the labeled dUTP in the water pool inside the giant liposome is incorporated into the primer (the DNA part of conjugate 2) on the inner surface of liposome, it must be observable in an optical microscopy image in which only the membrane of the liposome fluoresces. In order to obtain such a microscopy image, the amount of dUTP*–TTP mixture was reduced in this experiment. Although most of the liposomes emitted the fluorescence from the entire body, some liposomes emitted fluorescence from the membrane, rather than from the water pool inside (Fig. 6b). This observation strongly suggests that the duplication of DNA did occur on the inner surface of the liposome. The product of polymerization was roughly purified, treated with Hind III, and analyzed by the PAGE. However, the 50-bp band which should fluoresce in red did not reach a detectable amount. On the other hand, a generation of TexasRed-labeled DNA duplex was confirmed by a blank experiment without the liposome (data not shown). | ||
| Fig. 6 Optical microscopy images of the liposome encapsulating the reaction mixture of DNA polymerization containing cholesterol–PEG–DNA and TexasRed-labeled dUTP. (a) Phase contrast and (b) fluorescence images. Scale bar; 5 µm. | ||
The above results indicate that a combination of three conditions [(i) the reaction on the surface of membrane, (ii) the modification of the primer by the sterically-hindered cholesterol–PEG unit, and (iii) the modification of nucleoside triphosphate by the bulky fluorophore] strongly inhibited DNA polymerization, in particular, the incorporation of dUTP*. Originally, the reactivity of DNA polymerization on the surface of the liposome is reduced by the restriction of molecular motion, and the plausible influence from the electric double layer. In these circumstances, the modification of dNTP would strongly restrict the polymerization by the Klenow fragment.
From the above experiences using dUTP*, it may be concluded that the cholesterol–PEG–DNA–template complex is firmly anchored on the inner surface of the liposome throughout polymerization. Therefore the above result is indirect evidence for the interpretation that the DNA polymerization proceeds on the inner surface of the liposome in the non-labeled experiment.
According to Ogden et al., immediately after the origin of replication of chromosome oriC in E. coli , the two new oriCs bind to the cell membrane.26 Consequently, the newly synthesized and the enormous genomic DNAs are effectively distributed to the daughter cells. A primitive model of living cells is required to partition its informational substance equally into its offspring. Whatever style of division is adopted for artificial cell systems, the attachment of informational substance to the inner surface of the membrane guarantees the partition of genomic information to the divided daughter artificial cells.
Conclusion
We have demonstrated that genomic information that is attached to the inner surface of a liposome can be transferred to the informational amphiphile by a Klenow fragment. Although DNA polymerization described here is not the replication but the copying of information on the template, this achievement brings scientists one step closer to the realization of synthesis of artificial cellular life. It seems likely that this type of minimal cell system will be able to distribute the genomic material to daughter liposomes safely, because the DNA having a cholesterol anchor should move along with the membrane.Experimental
General
The 100-mer template DNA sequence dCAA GAC TAT CAT AGA CAT ACT CGA ATT CAG CGT TGA GTG TCG CAC GTA AGC TTC ATA GCT AAG TGA GTC AAA CTG TTC AAT CGG CAT GCG TCC ATC ACG A (bold indicates the Hind III site) was purchased from Hokkaido System Sciences Co. (Japan). This template DNA was purified by the polyacrylamide gel electrophoresis (PAGE). The Klenow fragment and Hind III were purchased from Takara Bio Inc. (Japan). Phosphoramidite units and the 5′-amino-modifier [N-MMTr-aminohexyl-(2-cyanoethyl-N,N-diisopropyl)-phosphoramidite] were purchased from Glen Research Co. (USA). The cholesterol–PEG-OH unit and DSPE-PEG5000 were purchased from NOF Co. (Japan). Sep-Pak® Plus reverse-phase cartridge columns (C18) were purchased from Waters Co. (USA). All other reagents and solvents were purchased from Wako Pure Chemicals Co. (Japan) and Tokyo Kasei Kogyo Co. (Japan).Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OH)O-d(TCGTGATGGACGCAT)-3′ was synthesized by the standard β-cyanoethyl phosphoramidite protocol (1 µmol scale).10 The oligonucleotide was purified by the trityl-ON procedure with a Sep-Pak® Plus reversed-phase cartridge column. Detritylation was performed on the column with 2% CF3COOH in water. The fractions containing the 5′-aminohexyl DNA fragment were collected, evaporated, and lyophilized (88 OD260, 55% yield). The oligonucleotide was characterized by MALDI–TOF mass spectrometry (Voyager-DE™, Applied Biosystems Co., USA), using a matrix solution of 3-hydroxypicolinic acid (50 mg), citric acid diammonium salt (2.5 mg), and CF3COOH (1 µL) in 1 mL of acetonitrile-H2O (1 ∶ 1, v/v) (calcd. for [M + H]+: 4785.9; found: 4786.0).
O)(OH)O-d(TCGTGATGGACGCAT)-3′ was synthesized by the standard β-cyanoethyl phosphoramidite protocol (1 µmol scale).10 The oligonucleotide was purified by the trityl-ON procedure with a Sep-Pak® Plus reversed-phase cartridge column. Detritylation was performed on the column with 2% CF3COOH in water. The fractions containing the 5′-aminohexyl DNA fragment were collected, evaporated, and lyophilized (88 OD260, 55% yield). The oligonucleotide was characterized by MALDI–TOF mass spectrometry (Voyager-DE™, Applied Biosystems Co., USA), using a matrix solution of 3-hydroxypicolinic acid (50 mg), citric acid diammonium salt (2.5 mg), and CF3COOH (1 µL) in 1 mL of acetonitrile-H2O (1 ∶ 1, v/v) (calcd. for [M + H]+: 4785.9; found: 4786.0).
PAGE analysis
The polymerized DNA strands were analyzed by electrophoresis on 15% polyacrylamide gels with Tris (25 mM)-glycine (193 mM) buffer (pH 8.8) under non-denaturing condition. After electrophoresis, the gel was stained with SYBR-Gold dye (Molecular Probes, USA), and photographed under UV illumination (302 nm) through a cutoff filter (>550 nm, Olympus, Japan).Optical microscopy
The prepared liposome was sampled on a glass plate with a frame-sealed chamber as a spacer. The sample suspension was covered with a glass cover slip and observed using a differential interference (BX51, objective lens; ×20, Olympus Co., Japan), phase contrast or fluorescent microscope (IX70, objective lens; ×100 (N. A. = 1.35) with immersion oil, (Olympus Co., Japan). The fluorescent image of the liposome was photographed with a cooled CCD camera (ORCA-ER, Hamamatsu, Japan) using a WIG filter set (ex; 560–590 nm, em; >615 nm, Olympus, Japan).Liposome-free DNA polymerization using informational amphiphile 2 as the primer
The 100-mer template DNA (20 pmol) and informational amphiphile 2 (40 pmol) were dissolved in a buffer solution containing 10 mM Tris-HCl (pH 7.5), 7 mM MgCl2, 0.1 mM dithiothreitol, and 0.2 mM each of dATP, dGTP, dCTP, and TTP (total volume 100 µL). Klenow fragment (4 U, 1 µL) was added, and the mixture was incubated at 25 °C. After 12 h, Hind III (6 U, 0.5 µL) was added to an aliquot (20 µL) of the mixture, followed by incubation at 25 °C for 12 h. The products were analyzed by PAGE.DNA polymerization on the inner surface of liposomes
To the informational amphiphile 2 (1 nmol) in 0.2 mL of CH3OH was added 0.5 mL of 10 mM lipid mixture (POPC–cholesterol–DSPE-PEG5000 in proportions 58 ∶ 39 ∶ 3) in CHCl3. By blowing nitrogen gas over the mixed solution, a lipid film was obtained coating the inside of the glass vessel, and solvent remaining in the lipid film was completely removed under reduced pressure.27 Addition of 1 mL of aqueous solution containing 100-mer template DNA (0.5 nmol) caused the lipid film to swell. Vortexing (for 5 min) and sonication (for 5 min) resulted in the formation of many liposomes containing informational amphiphile 2 and the 100-mer template DNA.28 The resulting liposome suspension was lyophilized and a fluffy amorphous lipid was obtained. To the lyophilized materials on ice was added a buffer solution containing 10 mM Tris-HCl (pH 7.5), 7 mM MgCl2, 0.1 mM dithiothreitol, 0.2 mM each of dATP, dGTP, dCTP, and TTP, and 25 U Klenow fragment (total volume is 0.5 mL). Gentle pipetting on ice yielded many liposomes containing the Klenow fragment, the template, and primer DNA (this is part of conjugate 2) in the interior water pool. Immediately, additional buffer solution containing 2 mM EDTA (ethylenediamine tetraacetate), 10 mM Tris-HCl (pH 7.5), 9 mM NaCl, and 0.1 mM dithiothreitol (total volume is 4.5 mL) was added to the liposome suspension in order to chelate the essential magnesium ion and to halt DNA polymerization occurring in the exterior water phase (final concentrations of lipids, Mg2+ and EDTA were 1 mM, 0.7 mM and 1.8 mM, respectively). The resulting liposome suspension was incubated at 25 °C for 24 h. Then the liposome suspension was loaded on a Sep-Pak® Plus reversed-phase column (C18) that was equilibrated with 50 mM NH4OAc aq., the column was washed with 50 mM NH4OAc aq. and then water to remove EDTA and dNTPs. Remaining UV-absorbing materials (monitored at 260 nm) were eluted with CH3CN–water (2 ∶ 3, v/v), and the fractions were collected, evaporated, and lyophilized. The lyophilized materials were dissolved in water (0.2 mL); and to an aliquot (50 µL) was added 6 µL of 10 × M buffer [100 mM Tris-HCl (pH 7.5), 100 mM MgCl2, 10 mM dithiothreitol, 500 mM NaCl] and Hind III (25 U, 2.1 µL). The mixture was incubated at 25 °C for 24 h, and the products were analyzed by PAGE.Direct visualization of DNA polymerization on the inner surface of the liposome using fluorescent labeled dUTP
To the polymerization system in the previous section, 15 µM of TexasRed-L12-dUTP (λex = 596 nm, λex = 611 nm) (dUTP*, Chromatide series, Invitrogen Inc., USA) was added. The concentration of TTP was reduced from 200 µM to 15 µM, the other parameters and the procedure were not altered. Before the reversed phase column chromatography, an aliquot (5 µL) of liposome suspension was diluted by 45 µL of the buffer solution containing EDTA (10 mM Tris-HCl (pH 7.5), 9 mM NaCl, 2 mM EDTA, and 0.1 mM dithiothreitol) in order to reduce the fluorescence of dUTP* in the exterior water phase of the liposome suspension. This diluted sample was observed by the phase contrast and the fluorescent microscopes as described above.Acknowledgements
This work was supported by a grant-in-aid from the 21st Century COE (Center of Excellence) program (Research Center for Integrated Science) of the Ministry of Education, Culture, Sports, Science, and Technology, Japan.References
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387 CrossRef CAS.
- P. Hinkle, Biochem. Biophys. Res. Commun., 1970, 41, 1375 CAS.
- W. Ford, J. W. Otvos and M. Calvin, Nature, 1978, 274, 507 CAS.
- I. Tabushi, T. Nishiya, M. Shimomura, T. Kunitake, H. Inokuchi and T. Yagi, J. Am. Chem. Soc., 1984, 106, 219 CrossRef CAS.
- Y. Kobuke and I. Hamachi, J. Chem. Soc., Chem. Commun., 1989, 1300 RSC.
- P. Walde, A. Goto, P.-A. Monnard, M. Wessicken and P. L. Luisi, J. Am. Chem. Soc., 1994, 116, 7541 CrossRef CAS.
- G. Steinberg-Yfrach, P. A. Liddell, S.-C. Hung, A. L. Moore, D. Gust and T. A. Moore, Nature, 1997, 385, 239 CrossRef CAS.
- M. M. Hanczyc, S. M. Fujikawa and J. W. Szostak, Science, 2003, 302, 618 CrossRef CAS.
- K. Takakura, T. Toyota and T. Sugawara, J. Am. Chem. Soc., 2003, 125, 8134 CrossRef CAS.
- K. Takakura and T. Sugawara, Langmuir, 2004, 20, 3832 CrossRef CAS.
- W. Yu, K. Sato, M. Wakabayashi, T. Nakaishi, E. P. Ko-Mitamura, Y. Shima, I. Urabe and T. Yomo, J. Biosci. Bioeng., 2001, 92, 590 Search PubMed.
- S. M. Nomura, K. Tsumoto, T. Hamada, K. Akiyoshi, Y. Nakatani and K. Yoshikawa, ChemBioChem, 2003, 4, 1172 CrossRef CAS.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669 CrossRef CAS.
- K.-i. Shohda, T. Toyota, T. Yomo and T. Sugawara, ChemBioChem, 2003, 4, 778 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- T. Sasaki, K. Minamoto and H. Itoh, J. Org. Chem., 1978, 43, 2320 CrossRef CAS.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303 CrossRef CAS.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859 CrossRef CAS.
- S. L. Beaucage and R. P. Iyer, Tetrahedron, 1992, 48, 2223 CrossRef CAS.
- D. Brutlag, M. R. Atkinson, P. Setlow and A. Kornberg, Biochem. Biophys. Res. Commun., 1969, 37, 982 CAS.
- H. Klenow and I. Henningsen, Proc. Natl. Acad. Sci. U. S. A., 1970, 65, 168 CAS.
- H. Matsuura and K. Fukuhara, J. Mol. Struct., 1985, 126, 251 CrossRef CAS.
- R. Old and K. Murray, J. Mol. Biol., 1975, 92, 331 CrossRef CAS.
- H. Jizomoto, E. Kanaoka and K. Hirano, Chem. Pharm. Bull., 1989, 37, 1895 CAS.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 4th edn., 2002, pp. 616–617 Search PubMed.
- G. B. Ogden, M. J. Pratt and M. Schaechter, Cell, 1988, 54, 127 CrossRef CAS.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660 CrossRef CAS.
- D. Papahadjopoulos and J. C. Watkins, Biochim. Biophys. Acta, 1967, 135, 639 CAS.
| This journal is © The Royal Society of Chemistry 2006 |