Interactions between different solar UVB/UVA filters contained in commercial suncreams and consequent loss of UV protection
Daniele
Dondi
a,
Angelo
Albini
*a and
Nick
Serpone
*ab
aDipartimento di Chimica Organica, Università di Pavia, Via Taramelli 10, Pavia, 27100, Italia. E-mail: angelo.albini@unipv.it; nick.serpone@unipv.it; Fax: (+39) 0382-987323; Tel: (+39) 0382-987316
bProfessor Emeritus, Concordia University, Montreal, Canada. E-mail: nickser@alcor.concordia.ca
First published on 3rd August 2006
Abstract
A systematic investigation of two well-known and popular commercial suncreams reveals significant degradation when exposed to simulated UV sunlight at an irradiance corresponding to natural sunlight. We have examined the photochemistry of two widely used sunscreen active agents in pure solvents separately and together (in solution), and in neat form, as well as their photochemistry when present in the actual suncream emulsion (as thin films on a glass substrate) since their combination typically produces suncreams with high sun protection factors (SPF): (1a) octyl methoxycinnamate (OMC; octinoxate) and (2a) 4-tert-butyl-4′-methoxydibenzoylmethane (also known as avobenzone and Parsol 1789), present in the two suncream formulations in combination with others (one also contained TiO2). Intermediates and/or photoproducts were identified by UV/visible spectroscopy, HPLC and liquid chromatographic/mass spectral methods, and by both 1H and 13C-NMR techniques. Structural assignments of the substrates produced were aided by examining model systems {viz. ethyl cinnamate (1b) and dibenzoylmethane (2b)} of the two sunscreen active agents. Irradiation of the cinnamates and the diketones together led to a [2 + 2] photocycloaddition process yielding cinnamate dimers and cyclobutylketone photoadducts that subsequently fragmented into substituted oxopentanoates and oxobutanoates. Similar findings were observed when the two active agents were simultaneously present in the same suncream emulsion.
Introduction
Harmful effects suffered by one's exposure to solar UV radiation are an ever increasing concern. They include sunburn cell formation, basal and squamous cell carcinomas, the deadly melanoma, cataracts, photoaging of the skin and suppression of the immune system.1 Although photochemoprevention is a topic of increased attention, use of sunscreen lotions has become the principal first line protection against harmful UV radiation. Consumers have indeed come to rely on the belief that such cosmetic products (in the United States considered as over-the-counter OTC drugs) prevent skin damage, while permitting gradual tanning.Sunscreens typically contain ‘chemical filters’, that is organic compounds that absorb strongly in the UV (most often, UVB) and ‘physical filters’, such as TiO2 and ZnO that block UVB and UVA sunlight through reflection and scattering;2 however, they also absorb significant UV radiation as attested in recent studies by Serpone and Emeline.3 The active ingredients are typically incorporated in an oil-in-water emulsion (sometimes in a water-in-oil emulsion) that, given the diverse chemical nature of the ingredients, serves an essential function.
Ideally, the UVB/UVA filters should be photostable; that is, they should degrade light to heat when they absorb UV radiation, or simply reflect and scatter the radiation when a metal–oxide physical filter is used. In actual fact, most chemical filters exhibit some photoreactivity, even when this is minimal, or otherwise lead to formation of a photoproduct(s) that might still act as a filter(s) albeit in a different spectral region, as may be the case for some isomerization reactions.4–6 Moreover, it is also possible for the UV filter itself to display some phototoxic action,7 as appears to be the case for TiO2, which when photo-activated by UV radiation generates the highly oxidizing hydroxyl radicals.8
Evaluation of the potential hazard(s) associated with the use of a chemical as a sunscreen necessitates an examination of its photochemistry. Typically, this is usually done in dilute solutions, with results of the study extended to conditions reproducing those prevalent under actual use by consumers. This notwithstanding, however, there is an obvious possibility that new photoreactions may also take place under more practical conditions.
In most sunscreens, at least two organic filters are used in formulations displaying high SPF (sun protection factor) numbers, one with optimal screening in the UVB region (290–320 nm) and the other in the UVA region (320–400 nm) in addition to, in many instances, a ‘physical filter’. Two widely used representatives of such classes of chemical UV filters are the cinnamates (UVB) and the dibenzoylmethanes (UVA). They are often associated with many commercial suncream formulations available extensively in many countries, including the European Union and the United States. Two key questions are then the persistence of such filters under UV irradiation and, when several components are present in the same formulation, whether the photostability of the active UV filters is significantly jeopardized relative to the photostability of the single components.
Several reports have appeared on the photochemistry of representative sunscreen active components, usually in dilute solutions of a single component9–11 or in the presence of TiO2 dispersions.12 Some workers13 have contended that such studies have no bearing on the reactions occurring when sunscreens are used by consumers for UV radiation protection, because several filters are typically present at relatively high concentrations in an apolar phase. To clarify this issue, we have herein examined the photochemistry of the most widely used representative UVB and UVA filters, viz. 2-ethylhexyl-4′-methoxycinnamate (1a; OMC, also known as octinoxate) and 4-ter-butyl-4′-methoxydibenzoylmethane (2a; also known as avobenzone and as Parsol 1789) under conditions that parallel their individual application, as well as when they are both present in commercial samples of suncreams.
Experimental
Materials and reagents
Octyl methoxycinnamate and avobenzone were a kind gift from BASF, Ludwigshafen, Germany. Ethyl cinnamate and dibenzoylmethane were of commercial origin (Aldrich) and were used as received.Analytical methods
High pressure liquid chromatographic analyses were carried out on a HPLC system consisting of a Jasco PU980 dual pump and a Jasco UV-975 UV-Vis detector. The column was an Aquasil C-18 reverse phase column (25 × 46 mm, particle size 5 µm); the eluent consisted of a gradient acetonitrile/water mixture used from 7 : 3 to 9 : 1 in 50 min. The flow rate was 1.2 mL min−1; the chromatograms were recorded at the three wavelengths 240, 310 and 360 nm for better sensitivity. The solar simulator was a Solarbox 1500e (CO.FO.ME.GRA s.r.l., Milan) equipped with an outdoor filter with IR treatment to filter out infrared radiation. NMR spectra were obtained in CDCl3 by means of a Brucker 300 MHz instrument, whereas mass spectra were recorded using a Finnigan instrument and UV/Visible absorption spectra by a Jasco spectrophotometer.Irradiation in solutions
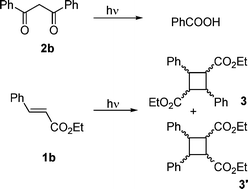
The procedure is exemplified by the reaction between ethyl cinnamate and dibenzoylmethane, with 0.5 g of the cinnamate 1b and 0.25 g of the dibenzoylmethane 2b dissolved in 20 mL methanol. Chromatographic separations revealed cinnamate dimers (3) and cross adducts. Structure assignments of the latter systems are based on distortionless enhancement polarization transfer (DEPT) experiments and appropriate two-dimensional NMR experiments. A characteristic CH–CH2–CH2 fragment was present in all of the isomers appearing at δca. 3.2 and 3.4 ppm (CH2), and 4.3 and 5.9 ppm in the 1H-NMR spectra, and at δca. 40 ppm (CH2), 42 and 57 ppm in 13C-NMR spectra. These data accord with assignments indicated later. The spectra of 4b, and 5b are in keeping with those reported previously by Christoffers.14 Particularly diagnostic was the correlation between the carboxyethyl group with all the protons of the CH–CH2–CH2 group in 6b.The (R, S) isomer (5b) was identified in a following fraction admixed with 4b. 1H-NMR spectra revealed peaks at 1.20 (t, 3H), 3.35 (dd, 1H, J = 7, 18 Hz, H-4), 3.45 (dd, 1H, J = 3, 18 Hz, H-4), 3.80 (q, 2H), 4.10 (ddd, 1H, J = 3,7,12 Hz, H-3), 5.32 (d, 1H, J = 12 Hz, H-2), 7.2–8.1 ppm (m, 15H); 13C-NMR spectra gave 13.8 (CH3), 37.5 (CH), 43.4 (CH2), 52.5 (CH), and 60.4 ppm (CH2). Aromatic signals could not be clearly distinguished from those of the isomer.
The (R, S) isomer (7b) was identified in a following fraction admixed with 6b. 1H-NMR spectra displayed signals at 1.1 (t, 3H), 2.9 (dd, 1H, J = 4, 17 Hz, Hα), 3.45 (dd, 1H, Hα), 4.2(ddd, 1H, H-2), 5.32 (d, 1H, J = 10, H-3), 7.2–8.1 ppm (m, 15H); in 13C-NMR spectra, 13.7 (CH3), 37.5 (CH), 43.4 (CH2), 52.5 (CH), and 60.5 ppm (CH2). The aromatic signals were not assigned.
Photoinduced reaction between octyl methoxycinnamate (1a) and methoxydibenzoylmethane (2a) in organic solvent media
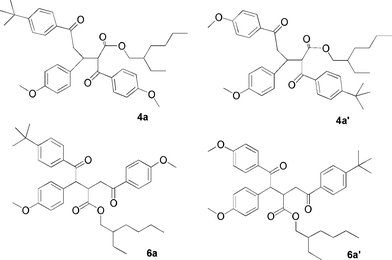
Cinnamate 1a (0.5 g) and 0.25 g of dibenzoylmethane 2a were dissolved in either 20 mL of methanol or 20 mL of cyclohexane. The solutions were then poured into a cylindrical vessel and exposed for 10 h to the radiation of the solar simulator that was equipped with an outdoor filter and emitting a total UV irradiance of 27 W m−2. Subsequently, the solvent was evaporated in vacuo and the resulting residue was purified by column chromatography. A liquid chromatographic/mass spectral analysis (HPLC-MS) showed formation of the Z isomer of the methoxycinnamate, of photodimers of the same compound {3a, 3a′; three peaks, m/z = 581 (M + H+) 90–100% intensity} and of further partially superimposed peaks with m/z 601 (M + H+, 90–100%) corresponding to 1a/2a adducts.Chromatographic separation as done above gave two main fractions containing 4a/4a′ and 6a/6a′, respectively, as the main components. Most signals in the 1H- and 13C-NMR spectra were broad or otherwise split because of the presence of two regioisomers (see e.g. structures 4a and 4a′ in lieu of the single product 4b obtained from the symmetric reagents 1b, 2b), as well as the presence of diastereomers arising from the asymmetric ethylhexyl ester chain.
Photoinduced reaction between octyl methoxycinnamate and methoxydibenzoylmethane in a solvent-free mixture
A solution was prepared by mixing 2 g of the octyl methoxycinnamate 1a and 1 g of the dibenzoylmethane 2a until formation of a homogeneous oil. The latter was poured onto a glass plate and covered with a thin glass plate to give, on squeezing the two plates, a UV absorption at 366 nm of ca. 1. The mixture was then irradiated in the solar simulator equipped with an outdoor filter for ca. 10 h (total UV irradiance in the 290–400 nm range: 27 W m−2), after which it was chromatographed on silica gel using cyclohexane as the eluent. The products were the octyl methoxycinnamate dimers (30 mg) and the cross-photoadducts (4a-7a, 80 mg overall) described in some detail below.Photodegradation of thin films of commercial suncreams
The suncream emulsion containing the active agents was layered onto a glass plate forming a homogeneous thin film with UV absorption at 360 nm of about A = 1, and subsequently was UV-irradiated with the solar simulator (power: 500 W). Loss of weight and decrease of absorption were monitored at regular time intervals. At the end of irradiation, the residue was suspended in methanol, filtered through a 0.2 µm PTFE filter and analyzed by HPLC-MS techniques. The UV irradiation intensity was measured by means of a radiometer with a total UV probe giving an irradiance of 44 W m−2.HPLC/MS analysis showed formation of the photodimers of methoxycinnamate (3a, 3a′) as well as the adducts 4–7, analogous to those reported above from the irradiation of compounds 1a and 2a in solution.
Results and discussion
As alluded to earlier, most commercial sunscreen formulations typically consist of a combination of physical (TiO2 and ZnO) and chemical filters to provide broadband protection over the UVA and UVB regions.15,16 The US Federal Register on OTC drugs17 does not recommend the combination octyl methoxycinnamate (OMC) and butylmethoxydibenzoylmethane (Parsol 1789; avobenzone) in suncream formulations because of an inherent photoinstability. Apparently, some photoadducts form between OMC and photogenerated fragments of Parsol 1789.18 Indeed, it seems typical of sunscreens that subsequent to absorption of UVB and UVA sunlight radiation, the sunscreen active agents undergo photo-fragmentation and/or photoisomerization as well as energy and/or electron transfer.10a,10d,19,20 Photoisomerizations yield species that could be less light absorbing than the parent species, and thus less useful as sunscreen agents, not to mention that any reactive intermediate or photoproduct could also be potentially toxic to DNA.21,22 Photofragmentation processes cause the absorbing molecule to dissociate into reactive fragments (e.g., free radicals) or reactive intermediates. Formation of photoadducts between active agents and thymine and thymidine bases has been demonstrated.23,24 In addition, some active agents could increase the rate of formation of potentially carcinogenic DNA photoproducts on irradiation (e.g. the cyclobutane-type pyrimidine dimers25), or otherwise undergo photochemical changes that might result in loss of UVB filtering ability.The photostability of commercial sunscreen lotions can be ascertained by exposure of real sunscreen lotions to simulated UVB/UVA sunlight (290–400 nm) using a solar simulator that can deliver a light irradiance that corresponds closely to natural sunlight whose irradiance ranges between ca. 10 W m−2 (cloudy day) and about 60 W m−2 (sunny day);26 as well, the temperature should also reflect the temperatures suncream consumers may be exposed to (ca. 35–40 °C).
Fig. 1 displays the fate of two well-known and popular commercial suncreams (one contains TiO2, the other not, in addition to chemical filters) after being exposed to simulated sunlight (wavelength region, 290–400 nm; light irradiance, 27 W m−2) for up to 4 h as thin films whose thickness was in keeping with the quantity typically used by consumers (some tens of µm). After this time period, both suncreams have undergone significant degradation (about 40–50%). It was imperative, therefore, to examine the photochemical fate of some of the active agents used in these two sunscreen formulations. Specifically we have focused our studies to address the photochemistry/photostability of two widely used chemical UV filters: octyl methoxycinnamate (1a) and Parsol 1789 (2a).
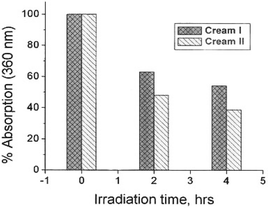 | ||
| Fig. 1 Photostability of two well-known commercial sunscreen lotions (labelled cream I and cream II) under exposure to UV radiation in a solar simulator (see text for experimental details). | ||
Photochemistry of pure sunscreen active agents in organic solvent media and in neat form
The absorption spectra of 2-ethylhexyl-4′-methoxycinnamate (1a) and 4-tert-butyl-4′-methoxydibenzoylmethane (2a) in acetonitrile are reported in Fig. 2, which also illustrates the spectral range of the UVB and UVA regions where they have screening capability. The former is a UVB filter; the latter is a UVA sunscreen.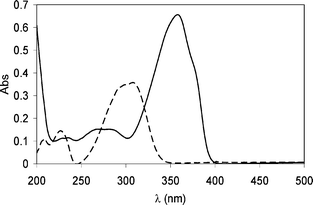 | ||
| Fig. 2 Absorption spectra of acetonitrile solutions of octinoxate (1a; dashed line) and avobenzone (2a; solid line). | ||
Irradiation of dilute 3 × 10−5 M air-equilibrated solutions of 1a in cyclohexane, acetonitrile and methanol at 310 nm (absorbed flux, 1.3 × 10−5 Einstein min−1; 1a does not absorb at 360 nm) produced conspicuous changes in the UV spectra as a result of E/Z isomerization of the cinnamate that was confirmed by HPLC analysis, and which also demonstrated that equilibrium had been reached. This isomerization was investigated no further, however. Similarly, irradiation of 3 × 10−5 M of the diketone 2a in the same solvents caused a decrease of the long-wavelength component of the spectrum and an increase in the absorption around 260 nm (Fig. 3). This corresponds to the ketonization of the intramolecularly hydrogen-bonded enol form, largely predominant in many solvents (> 97%).10,27 The quantum yield of isomerization (irradiation at 360 nm) was Φ = 1.6 × 10−4 in cyclohexane. Near complete recovery of the original spectrum was achieved after ca. 10 h in the dark, with the rate of recovery depending on solvent polarity,10e being much faster in CH3CN for example (Φ = 1 × 10−4; recovery, about 15 min). The quantity (in %) of the keto tautomer accumulated when irradiating a more concentrated solution was rather low (same photon flow): for example, ≪ 0.5% with both 2 × 10−3 M and 2.5 × 10−2 M solutions owing to a reversible isomerization process.
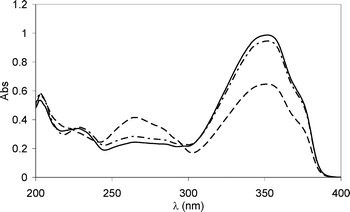 | ||
| Fig. 3 UV spectra of a cyclohexane solution 3 × 10−5 M of 2a before irradiation (solid line), after 15 min of irradiation (dashed line), followed by 10 h in the dark to allow for recovery (dash–dot–dash line). See text for details. | ||
Table 1 compares the key results after irradiation of relatively concentrated solutions (0.025 M) of compounds 1a and 2a for a fixed time period. They suggest that the main reaction from cinnamate 1a remains the E/Z isomerization, although some dimerization also takes place. During this time span, the diketone 2a is essentially photostable (Φ < 0.001) under irradiation in a polar solvent such as acetonitrile (identical results were obtained in methanol, but not reported in Table 1), whereas it undergoes significant decomposition in cyclohexane (Φ ≈ 0.02) yielding methoxybenzoic acid and tert-butylbenzoic acid as the main products.
| Initial concentration/M | Conditions | Consumed/M | Yield of cross adducts/M | ||
|---|---|---|---|---|---|
| 1a | 2a | 1a | 2a | 4–7 | |
| a Isomerization of compound 1a is not included. The Z : E ratio is ca. 1 : 1 on irradiation at 360 nm, and 3 : 1 on irradiation at 310 nm. Dimerization of 1a to 3 is also not included. | |||||
| 0.025 | — | C6H12, 360 nm | < 0.001 | — | — |
| 0.025 | — | C6H12, 310 nm | < 0.001 | — | — |
| — | 0.025 | C6H12, 360 nm | — | 0.022 | — |
| — | 0.025 | C6H12, 310 nm | — | 0.014 | — |
| 0.025 | 0.025 | C6H12, 360 nm | 0.0125 | 0.019 | 0.0024 |
| 0.0125 | 0.025 | C6H12, 360 nm | 0.009 | 0.022 | 0.001 |
| 0.025 | 0.025 | C6H12, 310 nm | 0.013 | 0.023 | 0.0028 |
| 0.025 | 0.0125 | C6H12, 310 nm | 0.0095 | 0.011 | 0.0028 |
| 0.025 | 0.00625 | C6H12, 310 nm | 0.0165 | 0.006 | 0.0023 |
| 0.00625 | 0.025 | C6H12, 310 nm | 0.0033 | 0.025 | 0.0007 |
| 0.05 | 0.05 | MeCN, 360 nm | < 0.001 | < 0.001 | — |
| 0.05 | 0.05 | MeCN, 310 nm | < 0.001 | < 0.001 | — |
| 0.05 | 0.05 | MeCN, 360 nm, TiO2 | 0.0125 | 0.0175 | — |
Next, we examined the possibility of a synergistic effect between the two compounds 1a and 2a. When a solution of both components (each 0.025 M) was irradiated, the result was essentially the same as with the separate compounds in acetonitrile. However, subsequent to irradiation at 310 and 360 nm some new products (the photoadducts 4a/4a′, and 6a/6a′ being the major components) were obtained in cyclohexane, in addition to cinnamate dimers 3a and 3a′ (Scheme 1). The chemical yield of such adducts was essentially independent of the initial concentration of the diketone in the range 0.00625–0.05 M, but decreased proportionately at lower concentrations of the cinnamate. The quantum yield of conversion of 2a in cyclohexane was ca. 0.004 (see Table 3 below), identical to the quantum yield obtained in the presence of 0.025 M of 1a.
| Initial concentration/mmol | Conditions (Power) | Consumed/mmol | Total amount of products 4–7/mmol | ||
|---|---|---|---|---|---|
| 1a | 2a | 1a | 2a | ||
| 0.035 | 0.035 | melt, 360 nm, | 0.0175 | 0.007 | 0.0009 |
| 0.007 | 0.0018 | Cream 1, 360 nm | 0.006 | 0.0012 | 0.00036 |
| 0.005 | 0.001 | Cream 2, 360 nm | 0.005 | 0.0008 | 0.0003 |
| 0.0035 | 0.0009 | Cream 1, Solarbox (500 W) | 0.0034 | 0.0008 | 0.0002 |
| 0.00125 | 0.00025 | Cream 2, Solarbox (500 W) | 0.00122 | 0.00012 | 0.0001 |
| 0.0016 | 0.0004 | Cream 1, Solarbox (250 W) | 0.0015 | 0.0033 | 0.0001 |
| Initial concentration/M | Conditions | Consumed | Products | |
|---|---|---|---|---|
| 1a | 2a | |||
| a Photon flow at 360 nm, 1.3 × 10−5 Einstein min−1. Full recovery, 15 h in cyclohexane, and 15 min in MeCN. Conditions for the Solarbox: power, 500 W; irradiation time, 5 h; surface area of film, 5 cm2; light irradiance, 44 W m−2; photon flow, 3.7 × 10−6 Einstein min−1. | ||||
| — | 3 × 10−5 | C6H12, 360 nm | Φ r (2a) < 1 × 10−5 | Φ isom = 0.00016 |
| — | 3 × 10−5 | MeCN, 360 nm | Φ r (2a) < 1 × 10−5 | Φ isom = 0.0001 |
| — | 0.002 | C6H12, 360 nm | Φ r (2a) = 0.0016 | — |
| — | 0.025 | C6H12, 360 nm | Φ r (2a) = 0.0037 | — |
| 0.025 | 0.025 | C6H12, 360 nm | Φ r (2a) = 0.0037 | — |
| 0.4 | 0.1 | Cream I, Solarbox | Φ r (2a) = 0.007 | — |
| 0.33 | 0.067 | Cream II, Solarbox | Φ r (2a) = 0.007 | — |
| ∼3 | ∼3 | Neat, Solarbox | Φ r (2a) = 0.05 | — |
 | ||
| Scheme 1 | ||
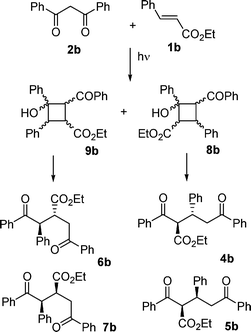
A mass spectral analysis of the products from the reaction between 1a and 2a suggested formation of mixed adducts. To test the generality of this reaction, derivatives that bear no hydrophobic ester chain were examined since they could provide means to simplify the separation and could facilitate the structural assignments of the products. Accordingly, the photochemistry of the simpler analogs ethyl cinnamate (1b) and dibenzoylmethane (2b) was also investigated—see Scheme 1. The HPLC-MS analysis showed that the products could be classified into two groups: dimers of 1b (3 and 3′ species) and the mixed adducts 1b + 2b (Scheme 2). Moreover, column chromatography permitted the separation of each component, or at least pairs of components. In particular, the NMR spectral analysis of a fraction of the reacting system 1b/2b revealed that the expected cyclobutane dimers of 1b (i.e., 3b and 3b′) were present as a mixture by comparison with literature results.28 These dimers were examined no further. The two most abundant fractions, however, contained a species whose mass spectra at m/z 400 corresponded to the 1b + 2b adduct. Each fraction contained a major product along with a minor component. NMR also showed that each of the components contained two methoxyphenyl groups, a tert-butyl-phenyl fragment and a carboxyethyl group, as well as two ketones and a characteristic A2MX system.
Ultimately, structure assignment was accomplished on evidence from double irradiation NMR experiments. One of the product pairs was identified as the benzoyldiphenyloxopentanoates 4b (R, R, major isomer) and 5b (R, S, minor isomer) as also suggested by comparison with the characteristics of the products synthesized previously by Christoffers.14 The product pair 6b and 7b was identified as isomeric benzoylmethyldiphenyloxobutanoates and was distinguished by the correlation of the carboxyl C with all the H of the A2MX system, for which the M component was shifted considerably upfield with respect to the previous pair (Scheme 2). These diketones derive from the hydroxylcyclobutylketones 8b and 9b, respectively, through a retro-aldol C–C bond cleavage. In turn, these cyclobutanes resulted from a [2 + 2] photocycloaddition between the enolic form of the diketone 1b and the cinnamate 2b, in keeping with the photoinduced cycloaddition of dibenzoylmethane to electrophilic alkenes and the ensuing opening of the four-membered ring (the de Mayo reaction), which had been reported previously for quinones.29
 | ||
| Scheme 2 | ||
Irradiation of the mixed solution of 1a and 2a, under otherwise identical conditions as for the model systems 1b and 2b, led to an analogous albeit more complicated product pattern as observed by HPLC methods. Two fractions containing cross adducts were obtained from the chromatographic separation with each fraction containing a major component. Apart from the ring substituent and the ester chain, these fractions displayed NMR spectra virtually identical to those of 4b and 6b, respectively. However, many signals were split, as expected from the formation of regioisomers because of the use of an asymmetric dibenzoylmethane and from the formation of diastereomers as a result of the presence of a stereogenic center in the ester chain. Indeed, NMR spectra showed that two components with very similar spectra were present in roughly the same amount in lieu of single (main) components of the previous case. Extensive NMR examination supported the structure assignment 4a and 4a′, and 6a and 6a′ displayed in Scheme 3. Minor stereoisomers, presumably bearing structure types 5a and 7a (equivalent to 5b and 7b in Scheme 2) were present in low concentrations.
 | ||
| Scheme 3 | ||
The photochemistry of mixtures of 1b and 2b compounds (both liquids) was also examined in the absence of solvents (i.e., in neat form). Under these conditions, addition of the two components was by far the main process. Admixing the two compounds in a 1 : 1 molar ratio produced a clear liquid. Subsequent irradiation of this liquid as a thin film (thickness, ca. 75 µm) in the solar simulator resulted in a mixture, which on chromatographic separation gave the adducts illustrated in Scheme 2. The analogous derivatives 4a and 6a were likewise the major products formed when irradiation was carried out on the clear mixture obtained by mixing compound 2a (a solid) and 1a (a liquid). Under such conditions, the quantum yield of reaction for compound 2a (wavelength, 360 nm; light absorbed mostly by this component) was Φ = 0.05 (see Table 3 below).
In summary then, irradiation of the diketones 2 and the cinnamates 1 in neat form lead to a cycloaddition process producing the cyclobutylketones 8 and 9 as revealed by the rearranged resulting products 4–7.
Photochemistry of the chemical UV filters 1a and 2a in the presence of TiO2
An acetonitrile solution of 1a and 2a (0.025 M in each) was added to a 1 mg L−1 dispersion of the physical filter Degussa P25 titanium dioxide. The resulting dispersion was irradiated as above (Solarbox) with continuous stirring. Under these conditions, both reagents were completely consumed and all of the peaks corresponding to adduct 3, and compounds 4, 5, 6, and 7 were less than 10% of the area observed upon irradiation of a solution in the absence of TiO2. Several small peaks at short HPLC retention times suggested the formation of lower molecular weight polar fragments.Photochemistry of commercial sunscreen formulations (suncreams)
In order to link the above results of the photochemistry of the sunscreen active agents 1a and 2a and TiO2, alone or in combination, to practical applications, commercial sunscreen samples of high SPF (i.e, SPF = 30) containing 1a and 2a (concentration determined by HPLC) were examined. The first commercial widely used suncream (cream I; Fig. 1) also contained titanium dioxide and ethylhexyltriazone, whereas cream II, also a widely used commercial suncream, further contained octyl salicylate and benzophenone-3. Irradiation of a thin film (10–50 µm) of these creams led to a conspicuous spectral change within a few hours (see Fig. 1). HPLC analysis showed that the dimers of 1a (that is, 3a and 3a′) and cross adducts 4a–7a noted above also formed in both cases, with a similar product distribution (Table 2). The quantum yield for the consumption of the active agent 2a was Φ = 0.007 for both suncreams (Table 3).Finally, in order to compare previous literature data obtained mainly from the use of solar simulators, thin films of the suncreams were also irradiated using a similar light source with a combined UVA/UVB output irradiance of 44 W m−2, which corresponds to a photon flow of about 3.7 × 10−6 Einstein min−1. The ensuing photochemical reactions occurring on these films were once again similar to those obtained with irradiation from phosphor-coated lamps.
Mechanistic scenario
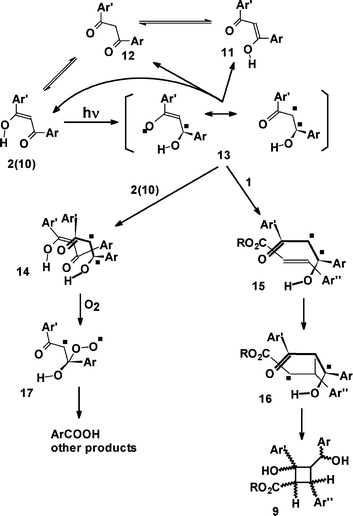
A detailed kinetic analysis of the various photochemical reactions occurring in the suncreams proved a formidable challenge because of the varying contribution to the absorption spectra by the E/Z isomerization of 1a, by the presence of conformers of 2a and by the photoproducts. Nonetheless, the presently available data are sufficient to infer a reasonable mechanistic proposition.Dibenzoylmethanes such as 2a are present in solution in the chelated enol form 10 (see Scheme 4) that is responsible for the strong absorption in the UVA spectral region. Flash photolysis experiments10c have shown bleaching of such absorption and suggested that this was due to cleavage of the intramolecular hydrogen bond to give the non-chelated enol form 11 rather efficiently, (Φ = 0.27). Thermal equilibration of this enol to the starting form as well as to the keto form 12 occurs within a few microseconds. Our results suggest that the path to the keto form is only a minor one (Φ ≈ 1.6 × 10−5). Thus, with an absorbed incident photon flow of the order of 10−6–10−5 Einstein min−1, compounds 2a and 2b can accumulate in the keto form only when using dilute solutions in an apolar solvent. Note that thermal equilibration back to the starting conformation is in most cases non-negligible (see above, and Fig. 3). For concentrations 0.01 M or greater, ketonization is all but insignificant. The keto forms absorb at shorter wavelengths, with the spectra displaying only a weak tail in the UVB/UVA region. Taking acetophenone as a model system, in cyclohexane λmax = 320 nm and log ε = 1.6 to be compared with λmax = 341 nm and log ε = 4.49 for the predominant enolic form (96.5%) in the case of the 2a substrate. Accordingly, the keto form plays a minimal role in the photochemistry at 360 nm irradiation.
 | ||
| Scheme 4 | ||
In the case of irreversible photoreactions, Schwack and coworkers10b,30 noted that 2a is photostable in alcohols but reactive in hydrocarbon solutions, undergoing a Norrish type I cleavage of the CO–C bond, a reaction that was attributed to the keto form of the molecule. We have confirmed that the pattern of the photodecomposition of 2a does indeed correspond to a Norrish type I fragmentation, although the quantum yield is rather low (Φ = 0.004). However, decomposition was somewhat slower on irradiation at 310 nm than 360 nm, which therefore cannot be attributed to a small fraction of the keto form 12 since this form does not absorb appreciably at 360 nm. Although the quantum yield is low, it is nonetheless greater than the quantum yield of the ketonization reaction observed at the lower concentrations. This suggests formation of some complex (Scheme 4) that may involve an intermediate in the fragmentation process (see further below for interaction with 1).
Insofar as the reaction with cinnamate is concerned, it appears unlikely that ketonization plays a role in it because a significant accumulation of short-lived 12 (Scheme 4) under continuous irradiation is too low and the quantum yield of this reaction is much greater (Φ = 0.0016–0.0037) than the isomerization quantum yield (Φ = 0.00016). Thus, it is reasonable to suggest that an excited state of 2a in the enolic form, or an intermediate derived from it, undergoes an intermolecular reaction with 1a, a significant process when the concentration of the latter is greater than 0.005 M. Also, this reaction occurs only in an apolar medium. It is tempting to hypothesize that the same species is involved both in ketonization and in the addition to 1a.
A mechanistic proposal that takes into account the above observations is depicted in Scheme 4. The planarity of the conjugated system of 2 is lost during the photochemical ketonization of β-dicarbonyl derivatives.31 Thus, a rotation of the C–C bond or pyrimidalization in excited 2* may lead to an intermediate with a diradical structure (13), as indicated in Scheme 4. The intermediacy of such species is not inconceivable since both radical sites are stabilized (α-keto and α-hydroxy-α-benzylic). This leads to either species 11 or 12, or decays back to ground state 2. At reasonable concentrations of enol 2, this may form a complex with 13 (species 14) that prolongs its lifetime, thereby making reaction with dissolved molecular oxygen O2 possible to give the peroxy diradical species 17. This in turn leads to the ultimate observed cleavage of the latter to benzoic acids. In the presence of a conjugated ester such as 1, however, interaction of 1 with the diradical 13 is much stronger and a covalent bond is formed to yield structure 16, subsequently initiating the well-known two-step diradical mechanism of [2 + 2] cycloaddition of unsaturated diketones that leads to the cyclobutane 9. This explains the dramatic concentration dependence of both the irreversible reactions observed from 2, fragmentation and addition to 1, and reconciles the reactions with a common photochemical primary step.
From a practical application viewpoint, it is relevant to note that mixing two sunscreen active agents leads to a photochemistry different from the photochemistry of each component alone,32 and that the reaction of combined sunscreens is strongly concentration- and medium-dependent. Moreover, in an apolar medium—similar in properties to that used in suncreams—and at a concentration of 2–3% by weight, as commonly used in commercial suncreams, formation of adducts is rather efficient leading to more than 50% decomposition of the UV filters in a 10 µm film on exposure to a simulated UV sunlight flux of 32 J cm−2, which corresponds to ca. 3 MED (for skin type I). This shows that the simultaneous presence of dibenzoylmethanes and other sunscreens such as methoxycinnamates does not lead to an enhancement of photostability (durability) of the chemical filters, but rather causes loss of protection when the suncreams are used under actual applicative conditions.
In addition to the above, both fragmentation and addition reactions of 2a led to the formation of aromatic ketones. Such products are known for their high photoreactivity, in particular toward hydrogen abstraction leading to a Norrish type I photo-oxygenation. Accordingly, we cannot preclude that, while photoreaction may lead to an increase in the UV filtering effect in the UVB at the expense of UVA, it is not a positive effect since such an increase may be linked to increased photoactivity.
Our conclusions are consistent with results from solar-simulated irradiation of various sunscreens, which have shown that the seemingly appealing combination of octyl methoxycinnamate (1a) and Parsol 1789 (2a) generally lead to a rapid loss of UV protection,4 and with the reported protective effect by naphthyl ketones33 that conceivably quench the triplet state of diketones 2 and thus of the initiating isomerization. Moreover, the photoreactivity of diketones 1 with α,β-unsaturated esters suggests that such compounds may react, for example, with DNA pyrimidines if the sunscreen agents indeed penetrate the skin as some recent studies have indicated.34
Concluding remark
4-tert-Butyl-4′-methoxydibenzoylmethane (also known as avobenzone and Parsol 1789) and octyl methoxycinnamate (OMC; octinoxate), the two most widely used UV filters in high SPF solar creams, undergo an irreversible photochemical reaction in apolar solvents, neat form and commercial suncream formulations. This reaction is a [2 + 2] cycloaddition process, which ultimately yields a mixture of diketones. The reaction is relatively efficient as evidenced by Φ = 0.007 in the suncreams and Φ = 0.05 in the neat mixture, indeed much more efficient than when each of the filter is irradiated separately, and leads to rapid loss of UV protection. A consistent mechanistic picture has been described that is consistent with experimental observations.Acknowledgements
We are grateful to the Ministero dell'Istruzione, dell'Università della Ricerca (MIUR - Rome) for support of our work in Pavia. We also wish to thank Prof. W. Schwack for communicating their results of ref. 30.References
- N. A. Cridland, and R. D. Saunders, Cellular and Molecular Effects of UVA and UVB, HMSO Publication Centre, London, 1994 Search PubMed.
- (a) World Health Organization, Sunscreens, IARC Handbooks of Cancer Prevention, Oxford University Press, Oxford, UK, 2001, vol. 5. Address: International Agency for Research on Cancer, 150 cours Albert Thomas, 69372 Lyon, France Search PubMed; (b) F. P. Gasparro, Sunscreen Photobiology - Molecular, cellular and physiological aspects, Springer-Verlag, New York, 1997. Search PubMed.
- N. Serpone and A. V. Emeline, Modeling heterogeneous photo-catalysis by metal-oxide nanostructured semiconductor and insulator materials: Factors that affect the activity and selectivity of photocatalysts, Res. Chem. Intermed., 2005, 31, 391–432 CrossRef CAS.
- (a) H. Maier, G. Schauberger, K. Brunnhofer and H. Hönigsmann, Change of ultraviolet absorbance of sunscreens by exposure to solar-simulated radiation, J. Invest. Dermatol., 2001, 117, 256–262 CrossRef CAS; (b) N. Tarras-Wahlberg, G. Stenhangen, O. Larkö, A. Rosén, A. M. Wennberg and O. Wennerström, Changes in ultraviolet absorption of sunscreens after ultraviolet irradiation, J. Invest. Dermatol., 1999, 113, 547–553 CrossRef CAS.
- (a) W. Schwack and T. Rudolph, Photostability and photoreactivity of UVA-filters in cosmetics, GIT Lab. J., 1996,(4), 373–377 Search PubMed; (b) W. Schwack and T. Rudolph, Photoreactions of chemicals UVA filters in cosmetics, GIT Lab. J., 1997,(1), 17–20 Search PubMed.
- C. A. Bonda, The photostability of organic sunscreen actives: A review, Cosmet. Sci. Technol. Ser., 2005, 28, 321–349 Search PubMed.
- (a) S. T. Butt and T. Christensen, Toxicity and phototoxicity of chemical sun filters, Radiat. Prot. Dosim., 2000, 91, 283–286; (b) F. Journe, M. C. Marguery, J. Rakotondrazafy, F. El. Sayed and J. Bazex, Sunscreen sensitization: a 5 year study, Acta Derm. Venereol., 1999, 79, 211–213 CrossRef CAS; (c) N. Cook and S. Freeman, Report of 19 cases of photoallergic contact dermatitis to sunscreens seen at the Skin and Cancer Foundation, Aust. J. Dermatol., 2001, 42, 257–259 Search PubMed; (d) A. Darway, I. R. White, R. J. Rycroft, A. B. Jones, J. L. Hawk and J. P. McFadden, Photoallergic contact dermatitis is uncommon, Br. J. Dermatol., 2001, 145, 597–601 CrossRef; (e) F. Afaq, V. M. Adhami and H. Mukhtar, Photochemoprevention of ultra-violet B signalling and photocarcinogenesis, Mutat. Res., 2005, 571, 153–173 CrossRef CAS.
- R. Dunford, A. Salinaro, L. Cai, N. Serpone, S. Horikoshi, H. Hidaka and J. Knowland, Chemical oxidation and DNA damage catalyzed by inorganic sunscreen ingredients, FEBS Lett., 1997, 418, 87–90 CrossRef CAS.
- N. Serpone, A. Salinaro, A. V. Emeline, S. Horikoshi and H. Hidaka, An in vitro systematic spectroscopic examination of the photostabilities of a random set of commercial sunscreen lotions and their chemical UVB/UVA active agents, Photochem. Photobiol. Sci., 2002, 1, 970–981 RSC.
- (a) M. Dubois, P. Gilard, P. Tiercet, A. Deflandre and M. A. Lefebvre, Photoisomerisation of the sunscreen filter Parsol 1789, J. Chim. Phys. Phys.-Chim. Biol., 1996, 95, 388–394; (b) W. Schwack and T. Rudolph, Photochemistry of dibenzoylmethane UVA filters. Part 1, J. Photochem. Photobiol., B, 1995, 28, 229–234 CrossRef CAS; (c) A. Cantrell and D. J. McGarvey, Photochemical studies of 4-tert-butyl-4′-methoxydibenzoylmethane (BM-DBM), J. Photochem. Photobiol., B, 2001, 64, 117–122 CrossRef CAS; (d) I. Andrae, A. Bringhen, F. Böhm, H. Gonzenbach, T. Hill, L. Mulroy and T. G. Truscott, A UVA filter (4-tert-butyl-4′-methoxydibenzoylmethane): photoprotection reflects photo-physical properties, J. Photochem. Photobiol., B, 1997, 37, 147–150 CrossRef CAS; (e) S. Tobita, J. Ohba, K. Nakagawa and H. Shizuka, Recovery mechanism of the reaction intermediate produced by photoinduced cleavage of the intramolecular hydrogen bond of dibenzoylmethane, J. Photochem. Photobiol., A, 1995, 92, 61–67 CrossRef CAS.
- (a) P. Morlière, O. Avice, T. Sa, e Melo, L. Dubercret, M. Giraud and R. Santus, A study of the photochemical properties of some cinnamate sunscreens by steady state and laser flash photolysis, Photochem. Photobiol., 1982, 36, 395–399 CrossRef CAS; (b) H. U. Gonzenbach and P. Schudel, Spectral stability - a meaningful term?, Int. J. Cosmet. Sci., 1988, 9, 287–292.
- A. Ricci, M. N. Chretien, L. Maretti and J. C. Scaiano, TiO2-promoted mineralization of organic sunscreens in water suspension and sodium dodecyl sulfate micelles, Photochem. Photobiol. Sci., 2003, 2, 487–492 RSC.
- R. M. Sayre, J. C. Dowdy, A. Ricci, M. N. Chrétien and J. C. Scaiano, Mineralization of organic sunscreens: interesting, but relevant? Comment and response, Photochem. Photobiol. Sci., 2002, 2, 1050–1051 Search PubMed.
- J. Christoffers, Novel chemoselective and diastereoselective iron-(III)-catalysed Michael reactions of 1,3-dicarbonyl compounds and enones, J. Chem. Soc., Perkin Trans. 1, 1997, 3141–3150 RSC.
- B. Catlow, Formulating of sunscreen with ultrafine titanium dioxide, Seifen, Oele, Fette, Wachse, 1993, 119, 497–500 Search PubMed.
- Formulators fine-tune TiO2-based screens, Manuf. Chem., 1993, 64, pp. 2633–2635 Search PubMed.
- Sunscreen Drug Products for Over-the-Counter Human Use, Final Monograph, Federal Register 64 27666, U.S. Food, and Drug Administration, Rockville, MD, 2000, http://www.cfsan.fda.gov/%E2%88%BClrd/fr990521.html.
- W. Johncock, Formulations of sunscreens: favorable and unfavorable interactions, Cosmet. Toiletries, 1999, 114, 75–80 Search PubMed.
- (a) A. Deflandre and G. Lang, Photoisomerization of benzylidine camphor and derivatives, Cosmet. Toiletries, 1988, 103, 69–75 Search PubMed; (b) A. Deflandre and G. Lang, Photostability assessment of sun-screens. Benzylidine camphor and dibenzoylmethane derivatives, Int. J. Cosmet. Sci., 1988, 10, 53–62 CrossRef CAS.
- (a) I. Beck, A. Deflandre, G. Lang, R. Arnaud and J. Lemaire, Study of the photochemical behavior of sunscreens - benzylidine camphor and derivatives, Int. J. Cosmet. Sci., 1981, 3, 139–152 CrossRef CAS; (b) I. Beck, A. Deflandre, G. Lang, R. Arnaud and J. Lemaire, Study of the photochemical behavior of sunscreens - benzylidine camphor and derivatives. II. Photosensitized, isomerization by aromatic ketones and deactivation of the 8-methoxypsoralen triplet state, J. Photochem., 1985, 30, 215–227 CrossRef CAS.
- B. Epe, Genotoxicity of singlet oxygen, Chem.-Biol. Interact., 1991, 80, 239–260 CrossRef CAS.
- (a) J. M. Allen, C. J. Gossett and S. K. Allen, Photochemical formation of singlet molecular oxygen in illuminated aqueous solutions of several commercially available sunscreen active agents, Chem. Res. Toxicol., 1996, 9, 605–609 CrossRef CAS; (b) J. M. Allen, C. J. Gossett and S. K. Allen, Photochemical formation of singlet molecular oxygen (1O2) in illuminated aqueous solutions of p-aminobenzoic acid (PABA), J. Photochem. Photobiol., B, 1996, 32, 33–37 CrossRef CAS.
- (a) A. A. Shaw, L. A. Wainschel and M. D. Shetlar, The photochemistry of p-aminobenzoic acid, Photochem. Photobiol., 1992, 55, 647–656 CAS; (b) A. A. Shaw, L. A. Wainschel and M. D. Shetlar, Photoaddition of p-aminobenzoic acid to thymine and thymidine, Photochem. Photobiol., 1992, 55, 657–663 CAS.
- B. S. Martincigh, J. M. Allen, and S. K. Allen, Sunscreens: The molecules and their photochemistry, in Sunscreen Photobiology, ed. F. P. Gasparro, Springer-Verlag: New York, 1997, pp. 11–45 Search PubMed.
- (a) S. Y. Wang, Ed., Photochemistry and Photobiology of Nucleic Acids, Academic Press, New York, 1976, vol. I Search PubMed; (b) J. M. Allen, S. K. Allen and B. Lingg, Chemical compounds used as active ingredients in sunscreens, Spec. Publ. – R. Soc. Chem., 1998, 225, 171–181 Search PubMed.
- H. Hidaka, H. Kubota, M. Graetzel, E. Pelizzetti and N. Serpone, Photodegradation of surfactants II. Degradation, of sodium dodecylbenzenesulfonate catalyzed by titanium dioxide particles, J. Photochem., 1986, 35, 219–230 CrossRef CAS.
- (a) P. Yankov, S. Saltiel and I. Petkov, Photoketonization and excited state relaxation of dibenzoylmethane in non-polar solvents, J. Photochem. Photobiol., A, 1988, 41, 205–214 CrossRef CAS; (b) M. Moriyasu, A. Kato and Y. Hashimoto, Kinetic studies of fast equilibrium by means of high-performance liquid chromatography. Part II. Keto-enol, tautomerism of some β-dicarbonyl compounds, J. Chem. Soc., Perkin Trans. 2, 1986, 515–520 RSC; (c) A. J. Villa, C. M. Lagier and A. C. Olivieri, Proton transfer in solid 1-phenylbutane-1,3-dione and related 1,3-diones as studied by carbon-13 CPMAS NMR spectroscopy and AM1 calculations, J. Phys. Chem., 1991, 95, 5069–5073 CrossRef CAS.
- (a) M. D'Auria, Regio- and stereochemical control in the photo-dimerization of methyl 3-(2-furyl)acrylate, Heterocycles, 1996, 43, 959–968 CrossRef CAS; (b) F. D. Lewis, S. L. Quillen, P. D. Hale and J. D. Oxman, Lewis acid catalysis of photochemical reactions. 7. Photodimerization and cross-cycloaddition of cinnamic esters, J. Am. Chem. Soc., 1988, 110, 1261–1267 CrossRef CAS.
- (a) S. S. Kim, J. S. Lim, J. M. Lee and S. C. Shim, Photo-chemical formation of 1,5-diketones from dibenzoylmethane and some quinones, Bull. Korean Chem. Soc., 1999, 20, 531–534 CAS; (b) G. Kornis and P. de Mayo, Photochemical synthesis. IX. The, conversion of dibenzoylmethane to tribenzoylethane, Can. J. Chem., 1964, 42, 2822–2827 CrossRef CAS.
- At near completion of our work, we learned that formation of adducts from substrates 1a and 2a were also reported by M. Köhhnlein, in “Untersuchungen zum photochemischen Verhalten des UV-B Filters Octylmethoxycinnamat in Modellsystemen, Sonnenschutzmitteln sowie auf der Haut”, Ph.D. Thesis, Universitat Hohenheim, Germany, 2000, and in a short communication; see for example M. Köhnlein and W. Schwack, Photo-reaktionen von UV-Filtern in Modellsystemen, in Sonnen-schutzmitteln sowie auf der Haut, Lebensmittelchemie, 2000, 54, 33 Search PubMed.
- G. Nikolov and P. Markov, Photochemical hydrogen abstraction as a radiationless transition in the photoketonization of b-dicarbonyls, J. Photochem., 1981, 16, 93–104 CrossRef CAS.
- The increased photoreaction of 1a in the presence of 2a was observed previously and was attributed to energy transfer from the latter component. See for example R. M. Sayre, J. C. Dowdy, A. J. Gerwig, W. J. Sheieds and R. V. Lloyd, Unexpected photolysis of the sunscreen octinoxate in the presence of the sunscreen avobenzone, Photochem. Photobiol., 2005, 81, 452–456 Search PubMed.
- L. R. Robinson (Procter & Gamble Co), US Patent 99–264139 199990305. Search PubMed See, for example, Chem. Abst., 1999, 131, 314104.
- (a) F. Pflücker, H. Hohenberg, E. Hölzle, T. Will, S. Pfeiffer, R. Wepf, W. Diembeck, H. Wenck and H. Gers-Barlag, The outer-most stratum corneum layer is an effective barrier against dermal uptake of topically applied micronized titanium dioxide”, Int. J. Cosmet. Sci., 1999, 21, 399–405 CrossRef CAS; (b) F. Menzel, T. Reinert, J. Vogt and T. Butz, Investigations of percutaneous uptake of ultrafine TiO2 particles at the high energy ion nanoprobe LIPSION, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 219–220, 82–86 CrossRef CAS; (c) Bennat and Müller-Goymann, Skin penetration and stabilization of formulations containing microfine titanium dioxide as physical UV filter, Int. J. Cosmet. Sci., 2000, 22, 271–283 CrossRef; (d) J. Lademann, H.-J. Weigmann, C. Rickmeyer, H. Barthelmes, H. Schaefer, G. Mueller and W. Sterry, Penetration of titanium dioxide microparticles in a sunscreen formulation into the horny layer and the follicular orifice, Skin Pharmacol. Appl. Skin Physiol., 1999, 12, 247–256 Search PubMed; (e) R. M. Brand, J. Pike, R. M. Wilson and A. R. Charron, Sunscreen containing physical UV blockers can increase trans-dermal absorption of pesticides, Toxicol. Ind. Health, 2003, 19, 9–16 CrossRef CAS; (f) R. Jiang, M. S. Roberts, D. M. Collins and M. S. Benson, Absorption of sunscreens across human skin: an evaluation of commercial products for children and adults, Brit. J. Clin. Pharmac., 1999, 48, 635–637 Search PubMed; (g) V. K. Gupta, J. L. Zatz and M. Rerek, Percutaneous absorption of sunscreens through micro-Yucatan pig skin in vitro, Pharmacol. Res., 1999, 16, 1602–1608 CrossRef CAS; (h) V. Sarveiya, S. Risk and H. A. E. Benson, Liquid chromatographic assay for common sunscreen agents: application to in vivo assessment of skin penetration and systemic absorption in human volunteers, J. Chromatogr., B, 2004, 803, 225–231 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2006 |