SELEX and dynamic combinatorial chemistry interplay for the selection of conjugated RNA aptamers
Anthony
Bugaut†
*ab,
Jean-Jacques
Toulmé
abc and
Bernard
Rayner
ab
aINSERM U386, 146 rue Léo Saignat, 33076 BORDEAUX Cedex, France
bUniversité Victor Segalen Bordeaux 2, 146 rue Léo Saignat, 33076 BORDEAUX Cedex, France
cInstitut Européen de Chimie et Biologie, 2 rue Robert Escarpit, 33607 PESSAC Cedex, France
First published on 6th October 2006
Abstract
SELEX (for Systematic Evolution of Ligands by Exponential enrichment) has proven to be extraordinarily powerful for the isolation of DNA or RNA aptamers that bind with high affinity and specificity to a wide range of molecular targets. However, the modest chemical functionality of nucleic acids poses some limits on the versatility of aptamers as binders and catalysts. To further improve the properties of aptamers, additional chemical diversity must be introduced. The design of chemical modifications is not a trivial task. Recently, dynamic combinatorial chemistry (DCC) has been introduced as an alternative to traditional combinatorial chemistry. DCC employs equilibrium shifting to effect molecular evolution of a dynamic combinatorial library of molecules. Herein, we describe an original process that combines DCC and SELEX for the in vitro selection of modified aptamers which are conjugated to chemically diverse small-molecules. Its successful application for the selection of small-molecule conjugated RNA aptamers that bind tightly to the transactivation-response (TAR) element of HIV-1 is presented.
Introduction
During the past decade, molecular evolution-based combinatorial approaches have received considerable attention.1 They contrast with conventional synthesis and screening methods by allowing the simultaneous evaluation of a large number of molecules and requiring only small quantities of material. However, such approaches can only be applied to molecules that can be amplified, i.e. copied and multiplied. Recently, dynamic combinatorial chemistry (DCC) has been introduced as an alternative to traditional combinatorial chemistry (CC) combining synthesis and screening into a single step.2 DCC is a convergence of concepts taken from both CC and molecular evolution.3 It is based on a reversible exchange process that make use of non-covalent interactions between a mixture of compounds at equilibrium (a dynamic combinatorial library, DCL) and a molecular target to template the preferential covalent bond formation of the strongest target binders. This method has been applied to a variety of combinatorial systems, ranging from material science to drug discovery.4 DCC experiments have been performed by using various biological targets,5 including nucleic acids.6 However, all published examples so far have been limited to relatively small libraries. Large DCLs include numerous products that bind weakly to the target and compete with the hit(s), severely limiting the amplitude of the equilibrium shift.7 This limitation of DCC may be overcome by refining the selection using an iterative process of selection and amplification.8 To date, only one such evolutionary system, making use of a photochemical isomerization reaction, has been reported.9In contrast, the use of replicable biopolymers, such as nucleic acids, and the development of amplification techniques, such as the polymerase chain reaction (PCR),10 allows molecular biologists to screen libraries containing up to 1015 individual molecules. Hence, SELEX (for Systematic Evolution of Ligands by Exponential enrichment)11 has been developed as a method for the in vitro selection of aptamers,12i.e. structured DNA or RNA oligonucleotides that display specific target-binding or catalytic properties.1a Nevertheless, the modest chemical functionality of DNA and RNA oligomers still poses some limits on the versatility of aptamers, and their poor cellular uptake, as well as their sensitivity to nucleases, restricts their use as therapeutic or diagnostic agents. To further enhance functionality and/or to improve properties of interest for in vivo use, chemical modifications of aptamers need to be introduced.13 So far, there are two strategies for producing chemically modified aptamers. The first, known as post-SELEX approach, consists in modifying a posteriori a selected DNA or RNA aptamer via chemical synthesis.14 This approach generally requires a detailed structural knowledge of the complex formed between the aptamer and its target. Otherwise, modified nucleotides and/or small-molecule appended groups are randomly introduced both in and around the core functional domain, trying to identify chemical modifications that provide extra properties without affecting binding. The disadvantage of this method is that elucidation of a successful combination remains a significant screening challenge.14 The second approach rests on the use of modified deoxy- or ribo-nucleoside triphosphates (dNTPs or rNTPs) during the selection process. The range of useful chemical modifications is rather restricted by the ability of polymerases commonly used for SELEX to accurately and efficiently incorporate modified NTPs. To date, only a few modified NTPs have been used for SELEX experiments. They essentially consist in 2′-amino- and 2′-fluoro-pyrimidines, in phosphorothioate nucleotides, or in some pyrimidine nucleobases modified in position C5 and some purine nucleobases substituted in position N6 or C8.13
Herein, we describe an original process that combines DCC and SELEX for the in vitro selection of modified aptamers which are conjugated to chemically diverse small-molecules (Scheme 1). We report its application to the selection of conjugated RNA aptamers that bind tightly to the transactivation-responsive (TAR) element of HIV-1.
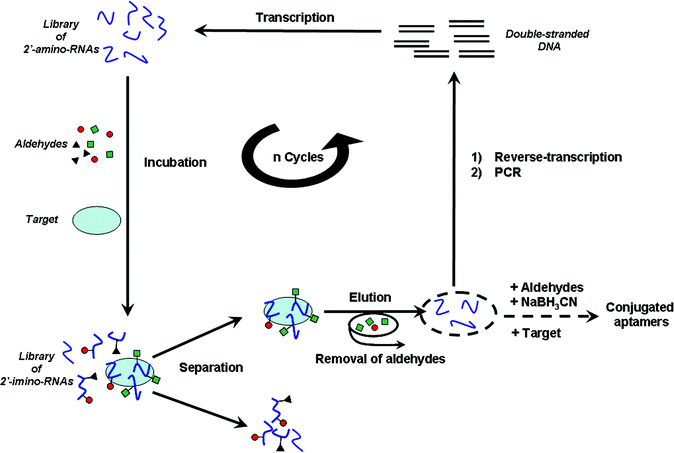 | ||
| Scheme 1 Schematic representation of the in vitro selection process. | ||
Results
Design of the in vitro selection process
We have recently established that, due to its favourable pKa value (6.2) and its higher nucleophilicity compared to those present in nucleobases, the sugar primary amine of 2′-amino-nucleotides can specifically react with a set of aldehydes at near physiological conditions to produce a DCL of 2′-imino conjugated oligonucleotides.6d,e Reversible imine formation has been reported to be suitable for the formation of responsive DCLs.15 We have demonstrated that the reversible exchange between 2′-imino conjugated oligonucleotide ligands is shifted by the presence of a nucleic acid target towards the preferential formation of the strongest binders.6d,e Furthermore, the 2′-amino-nucleotides of pyrimidines are among the few rNTP analogues that can be efficiently incorporated by the T7 RNA polymerase.16Taking advantage of these properties of 2′-amino-pyrimidines, we have designed an original directed evolution process that combines SELEX and DCC for the selection of small-molecule conjugated aptamers. Our process rests on the use of a library of random RNA sequences containing 2′-amino-pyrimidines instead of their “natural” counterparts. In a first round of selection, the random library of 2′-amino-RNAs is incubated with a set of aldehydes and a target molecule (Scheme 1). At the thermodynamic equilibrium, the mixture is composed of a large number of conjugated 2′-imino-RNAs, some of them being associated with the target molecule. Partitioning of ligand–target complexes from unbound candidates is performed. Ligands are then eluted from the target, causing concomitant hydrolysis of imine linkages. After removal of aldehydes, selected 2′-amino-RNA scaffolds are isolated, reverse-transcribed, and amplified by PCR. Resulting double-stranded DNAs are then transcribed into 2′-amino-RNAs and another round of selection can be carried out. Repetition of this selection and amplification process progressively leads to a population of 2′-amino-RNA scaffolds that have evolved in the presence of the set of aldehydes and the target to furnish high affinity conjugated 2′-imino-RNA ligands. At the end of the selection process, remaining sequences are identified by cloning and sequencing. Selected 2′-amino-RNA scaffolds are then resynthesized and individually incubated with the aldehydes, the target molecule and sodium cyanoborohydride (NaBH3CN), which selectively reduces the imine bonds. Thus, conjugated aptamers displaying the highest affinity for the target are preferentially in situ synthesized and converted into chemically stable analogues.6d,e,15
Validation of the in vitro selection process
In order to validate our in vitro selection process, we applied it to the isolation of conjugated aptamers directed against an RNA hairpin, MiniTAR (Fig. 1c), which is a 27-nucleotide truncated form of the TAR element of HIV-1. This target was chosen because it is an important element of the replication cycle of the virus and, moreover, it has been previously used for several in vitro selections of DNA and RNA aptamers performed in our lab.17 The MiniTAR sequence was 3′-biotinylated to allow separation of ligand–target complexes from unbound candidates by using streptavidin-coated magnetic beads. A random library of 2′-amino-RNAs and a set of three aldehydes, 1 (1 mM), 2 (1.2 mM), and 3 (200 µM) (Fig. 1b) were employed.‡ Oligomers contained a random region of 14 nucleosides, A, C, G or 2′-amino-U (UNH2), flanked by two constant primer binding regions, which did not contain any uridine (Fig. 1a).![(a) Design of the random library of 2′-amino-RNAs, (b) structures of aldehydes: 4-[3-(dimethylamino)propoxyl]benzaldehyde hydrochloride 1, 3-hydroxy-4-methoxybenzaldehyde 2, nalidixic aldehyde 3, and (c) sequence and secondary structure of the MiniTAR target employed for the in vitro selection.](/image/article/2006/OB/b610890c/b610890c-f1.gif) | ||
| Fig. 1 (a) Design of the random library of 2′-amino-RNAs, (b) structures of aldehydes: 4-[3-(dimethylamino)propoxyl]benzaldehyde hydrochloride 1, 3-hydroxy-4-methoxybenzaldehyde 2, nalidixic aldehyde 3, and (c) sequence and secondary structure of the MiniTAR target employed for the in vitro selection. | ||
Two in vitro selections were performed in parallel, at room temperature, in a 20 mM sodium phosphate buffer at pH 6.0 containing 20 mM NaCl, 140 mM KCl and 3 mM MgCl2 (subsequently referred to as buffer 1 × SE), by using the 2′-amino RNA library and MiniTAR 3′-biotin, either in the presence (selection S+) or in the absence (selection S−) of the aldehydes. All other experimental conditions (library and target concentrations, counter-selection step, incubation times, number and volume of washings§, elution conditions) were strictly identical in selections S+ and S−. For both selections S− and S+, seven rounds of selection and amplification were carried out, with the selection pressure being progressively increased: (i) by decreasing oligonucleotide and target concentrations and (ii) by increasing the number of washings (Table 1). Then, selected 2′-amino-RNA candidates were cloned and sequenced.
For selection S−, carried out in absence of the aldehydes, A−15 and A−13 were the most represented sequences (12 out of 32) (Fig. 2a). They are complementary to the 5′-end of the stem of MiniTAR (Fig. 2a). Conversely, we found only twice such an antisense-like sequence, similar to A−15, from selection S+. Selection S+ led to sequence A+30 as the most represented sequence (7 out of 18 sequences) (Fig. 2b). A+30 exhibits a sequence complementary to the top part of MiniTAR and can possibly form a hairpin structure displaying the interacting region into the loop (Fig. 2b). The dissimilarity between sequences originating from selections S− and S+ indicates that the aldehydes have influenced the outcome of selection S+. The 2′-amino-RNA population from selection S+ might have evolved to furnish a particular 2′-amino-RNA scaffold (A+30) with which the aldehydes react for producing conjugated aptamers with high affinity for MiniTAR.
 | ||
| Fig. 2 Most represented sequences obtained for (a) selection S−, carried out in absence of the aldehydes, and (b) selection S+, performed in the presence of the aldehydes. Watson–Crick complementarity between the target MiniTAR and the selected sequences is indicated (nucleotides in italic bold). For aptameric sequences, U indicates 2′-amino-uridine (UNH2) and the fixed regions are denoted in lower case. Stem forming sequences are underlined with arrows. | ||
Then, a 19-nucleotide truncated form of A+30 (A+30sl, Fig. 3) was employed. A+30sl consists of the 2′-amino-RNA hairpin (Tm = 64.0 ± 0.5 °C, in buffer 1 × SE) that retains the affinity of the originally selected sequence for the target MiniTAR (Kd (A+30–MiniTAR) = 38 ± 5 nM; Kd (A+30sl–MiniTAR) = 23 ± 3 nM; determined by electrophoresis mobility gel shift assays (EMSA), in buffer 1 × SE). It contains three UNH2 residues at positions 6, 7 and 9 (Fig. 3); and thus three reactive 2′-amino groups, which can potentially lead to the formation of 63 mono-, bi- or tri-conjugated aptamers in the presence of the three aldehydes. When A+30sl was incubated with the set of aldehydes 1, 2, and 3 in buffer 1 × SE and in presence of NaBH3CN, a complex mixture of products was obtained after a 24 hour reaction (Fig. 4a). However, this mixture contained one major product, corresponding to peak p4 (Fig. 4a). When the same reaction was carried out in the presence of MiniTAR, three products strongly emerged (peaks p1, p2 and p3, Fig. 4b). Amplification of these particular products occurs at the expense of the other products of the reaction. In particular, the product corresponding to peak p4, which is predominant in the absence of MiniTAR, is almost suppressed (Fig. 4).
 | ||
| Fig. 3 Structures of 2′-amino-RNA scaffold (A+30sl), amplified products (4, 5, and 6) and deselected product (7). | ||
 | ||
| Fig. 4 UV-RP-HPLC traces showing component composition of the mixtures after a 24 h-reaction between A+30sl (10 µM) and aldehydes 1 (1 mM), 2 (1.2 mM) and 3 (200 µM) in buffer 1 × SE containing 5 mM NaBH3CN: (a) in the absence or (b) in the presence of MiniTAR 3′-biotin (10 µM). Reaction mixtures were dialyzed prior RP-HPLC in order to remove free aldehydes that could overlap with peak products. | ||
These products were collected and analysed by MALDI-ToF mass spectrometry and time-dependent snake venom phosphodiesterase digestion followed by MALDI-ToF, as previously reported.6e The amplified products, 4 (peak p1 in Fig. 4b), 5 (peak p2 in Fig. 4b) and 6 (peak p3 in Fig. 4b), were identified as mono-conjugated products between A+30sl and 1, 2, and 3, respectively (Fig. 3). They all derived from reductive amination reactions with the 2′-amino group in position 9. The product corresponding to peak p4 in Fig. 4 is the tri-nalidixic conjugate 7 (Fig. 3).
Affinities of these products for MiniTAR were then determined by EMSA and UV-monitored melting experiments (Table 2). All amplified products exhibit comparable high affinities for the target; whereas the tri-nalidixic conjugate 7, which is “under-expressed” in the presence of MiniTAR, binds very poorly to the target.
Discussion
Nucleic acid structures termed aptamers can readily be selected in vitro to tightly and specifically bind to diverse small and macromolecular targets.1a Nevertheless, due to the limited chemical arsenal, the sensitivity to nucleases and the poor cellular uptake of pure DNA- and RNA-based aptamers, their use as in vitro tools or diagnostic and therapeutic agents remains limited. Enhancement of the scope of applications of aptamers requires the introduction of additional chemical functionalities to the natural nucleotides.13 A wide variety of nucleotide analogues is available for the chemical synthesis of modified aptamers from selected DNA and RNA sequences.13,14 In general, many compounds must be individually synthesized and evaluated before identifying a hit, i.e. a chemically modified aptamer with conserved or improved binding properties.14,18,19 Recently, there have also been considerable efforts directed toward the enzymatic incorporation of DNA- and RNA-monomers bearing chemical modifications.13,20 Nonetheless the efficient incorporation of additional chemical diversity by the polymerases commonly used for SELEX still remains a limiting factor.20 Herein, we have proposed a novel methodology that bypasses these limitations by displaying various reversibly appended groups to a random library of oligonucleotides during the in vitro selection.Labelling of oligonucleotides with lipophilic, positively charged and/or intercalating small-molecule appended groups have been demonstrated to influence cellular uptake, nuclease resistance and binding affinity.21 Small-molecule appended groups have also been used for transducing aptamer–target interactions into electrochemical, mechanical or fluorescent signals in vitro.19,22 Notably, Weeks and Merino have recently reported the generation of target sensitive 2′-ribose-derivatized aptamers by incorporation of a 2′-amino-nucleotide into previously selected DNA aptamers and subsequent reaction of the 2′-amine with a carboxylic acid fluorophore.19
We have carried out in parallel two in vitro selections by using a single 2′-amino RNA library and a single target, MiniTAR, either in the presence (selection S+) or in the absence (selection S−) of a set of three aldehyde molecules (1, 2 and 3). The aldehydes could reversibly react with 2′-amino groups, thus producing a DCL of 2′-imino RNA conjugates containing virtually2b up to 714 candidates at the start. For both selection S+ and S−, seven rounds of selection and amplification have been performed, before the remaining oligonucleotide populations were cloned and sequenced. Selection S+ and S− led to two different populations of sequences. This suggests that the reversibly appended small-molecule residues have changed the outcome of the in vitro evolution, by guiding the selection S+ towards particular 2′-amino-RNA scaffolds that are suitable for both incorporating the modifications and binding to the target. It is noteworthy that the outcome of previous in vitro selections performed in our laboratory using pools of DNA and RNA sequences and the TAR element of HIV-1 as the target did not lead either to A+30 or A−15, the most represented sequences from selection S+ and S−, respectively.17 Moreover, subsequent chemical modifications of the selected RNA aptamer has been a challenging task.18
A + 30sl, a 19-nucleotide truncated form of A+30, was then employed for the DCC resynthesis of the selected conjugated aptamers. A+30sl consists of a 2′-amino-RNA hairpin that is likely to form a loop–loop complex with MiniTAR. It contains three UNH2 residues at positions 6, 7 and 9 that can potentially lead to the formation of 63 conjugated aptamers in the presence of three aldehydes. In the absence of MiniTAR, reaction of A+30sl with aldehydes 1, 2 and 3, in the salt and pH conditions of the in vitro selection, and in the presence of NaBH3CN, gave rise to a complex mixture of products, the major species being a tri-conjugated product (7). In contrast, the same reaction resulted in the preferential formation of only three products (4, 5 and 6) in the presence of MiniTAR. Each product originates from the reductive amination reaction between an aldehyde and the 2′-amino group at position 9 within A+30sl. It is worth mentioning that the UNH2 at position 9 is contained in the region complementary to MiniTAR; whereas the other UNH2, at positions 6 and 7, can be located in a highly constrained region within the complex, at the junction between the stem of the aptamer and the loop–loop duplex. This might indicate that UNH2 at position 6 and 7 have been selected as part of an optimized “linker”, whereas UNH2 at position 9 has been selected to incorporate appended residues.
Affinities of isolated conjugates for the target, determined by EMSA and UV-melting, correlate well with the equilibrium shift observed during the DCC resynthesis. Indeed, amplified conjugates (4, 5, and 6) all exhibit strong binding properties; while the “under-expressed” product (7) binds very poorly to the target.
In summary, our in vitro selection has led to the identification of a unique 2′-amino-RNA scaffold. This sequence has been selected in the presence of reversibly reacting aldehydes and readily provides chemically stable conjugated aptamers that bind tightly to the target when incubated with the aldehydes and the target, in the same conditions of the in vitro selection and in presence of NaBH3CN.
Conclusion
In this proof-of-principle study we have shown that reversibly attached small-molecules can be used during the SELEX process to drive the selection towards a particular nucleic acid scaffold that is appropriate for the subsequent DCC resynthesis of high affinity conjugated aptamers. This original selection process represents a promising approach to overcome some of the major drawbacks associated with the existing methods for generating chemically modified aptamers. We have applied this methodology by using the imine reversible exchange between a pool of RNA oligomers containing 2′-amino-uridine and a set of aldehydes. However, we believe it could be applied by using other enzymatically incorporable DNA- or RNA-monomers, such as amino-modified nucleobases for example, and/or other reversible reactions, such as disulfide formation for example. This process is thus of general interest for expanding the applications of aptamers for diagnostics, therapeutics and nanobiotechnologies. Particularly, the use of reporter groups with particular fluorescence or electrochemical properties should give rise to interesting original biosensors.Experimental
Enzymatic reactions
In vitro selection protocols
Selection S−. The 2′-amino-RNA library (or selected sequences in the successive rounds of selection) was incubated for 30 min at room temperature with 50 µg (10 µL of a 5 mg mL−1 solution) of streptavidin-coated magnetic beads (pre-washed several times with binding buffer) in 100 µL of binding buffer 1 × SE (20 mM sodium phosphate buffer at pH 6.0 containing 20 mM NaCl, 140 mM KCl, and 3 mM MgCl2). The mixture was manually stirred every 5 minutes to resuspend the beads. The supernatant was then collected, and used for the selection step.
Selection S+. The 2′-amino-RNA library (or selected sequences in the successive rounds of selection) was incubated for 5 min at room temperature with aldehydes in 90 µL of buffer 1 × SE. Then 10 µL of a 5 mg mL−1 solution of streptavidin-coated magnetic beads in buffer 1 × SE were added and the mixture was incubated for 30 min at room temperature. The mixture was manually stirred every 5 min to resuspend the beads. The supernatant was then collected, and used for the selection step.
Selection S−. 3′-Biotinylated MiniTAR was added to the supernatant (100 µL) recovered after the counter-selection step. The mixture was incubated at room temperature for 25 min, and 10 µL of a 5 mg mL−1 solution of streptavidin-coated magnetic beads in buffer 1 × SE were added for 5 min. The supernatant was then removed, and beads were washed with buffer 1 × SE. Oligonucleotides bound to the target were eluted two times in 60 µL of water at 75–80 °C for 1 min. Fractions were pooled, and ethanol precipitated.
Selection S+. Supernatant (100 µL) recovered after the counter-selection step was used in the same conditions as those described for selection S−, except that beads were washed by using a solution containing the three aldehydes, 1 (1 mM), 2 (1.2 mM) and 3 (200 µM) (Fig. 1), in buffer 1 × SE. Residual aldehydes were removed by selective ethanol precipitation of oligonucleotides.
Cloning and sequencing
Recovered 2′-amino-RNA candidates after the last round of selection were reverse-transcribed and amplified as described above, and an extra 10 min at 72 °C at the end of the PCR was added. PCR products were then directly cloned into the vector of the TOPO TA cloning kit from Invitrogen. Escherichia coli XL1 TOP10 One Shot™ (Invitrogen) cells were transformed according to the manufacturer's instructions. Individual clones were sequenced with Dye ET terminator cycle sequencing kit from Amersham Biosciences, according to the manufacturer's protocol, and analyzed by using an ABI-310 (Perkin Elmer) automatic sequencer.HPLC analyses
HPLC analyses were performed with detection at 260 nm on a DIONEX system equipped with a GP50 gradient pump and a PDA100 photodiode-array detector and using an Uptisphere 5ODB 5 µm C18-column (250 × 4.6 mm, Interchim France). Prior to HPLC analyses, reaction mixtures were dialyzed (Slide-A-Lyzer Mini Dialysis Units, 3500 MW cut-off, Pierce) in 3 L of water for 16 h. Binary solvent gradient (A: 0.1 M triethylammonium acetate pH 6.5; B: 80% acetonitrile in A) was used at a flow rate of 1 mL min−1: isocratic with 9% B for 10 min, linear gradients from 9 to 20% in B for 45 min, then 20 to 100% in B for 10 min.UV-Monitored melting experiments
Thermal denaturation experiments were performed on a Cary 1E spectrophotometer interfaced with a Peltier device that controlled the temperature to within ±0.1 °C. Denaturation of the samples was achieved by increasing the temperature by 0.5 °C min−1 from 5 to 85 °C and was monitored at 260 nm. Samples contained 1 µM of aptamer and 1 µM of MiniTAR in buffer 1 × SE. Crude data were analyzed with KALEIDAGRAPH 3.0 (Abelbeck Software). The melting temperature was taken as the maximum of the first derivative of the UV melting curves.Electrophoretic mobility shift experiments
1 nM of 32P-labelled MiniTAR was incubated for 30 min with increased concentrations of conjugated aptamer in 10 µL of buffer 1 × SE. Mixtures were loaded on a 15% non-denaturing polyacrylamide gel (acrylamide : bisacrylamide 75 : 1) in 50 mM tris-acetate pH 6.0 and 3 mM MgCl2, and run at 4 °C for 9 h at 7.5 V cm−1. Bands were quantified with an Instant Imager (Hewlett-Packard). Kd values were deduced from data point fitting with KALEIDAGRAPH 3.0 (Abelbeck Software), according to: B = Bmax[ligand]0/([ligand]0 + Kd), where B is the proportion of complex, Bmax is the maximum of complex formed, and [ligand]0 is the total concentration of ligand.Acknowledgements
We thank Katel Bathany for mass spectrometry analyses and the Conseil Régional d'Aquitaine for financial support.References
- (a) D. S. Wilson and J. W. Szostak, Annu. Rev. Biochem., 1999, 68, 611–647 CrossRef CAS; (b) S. V. Taylor, P. Kast and D. Hilvert, Angew. Chem., Int. Ed., 2001, 40, 3310–3335 CrossRef; (c) Z. J. Gartner, Pure Appl. Chem., 2006, 78, 1–14 CrossRef CAS.
- (a) A. Ganesan, Angew. Chem., Int. Ed., 1998, 37, 2828–2831 CrossRef CAS; (b) J.-M. Lehn, Chem.–Eur. J., 1999, 9, 2455–2463 CrossRef.
- (a) B. Klekota and B. L. Miller, Trends Biotechnol., 1999, 17, 205–209 CrossRef CAS; (b) C. Karan and B. L. Miller, Drug Discovery Today, 2000, 5, 67–75 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- O. Ramstrom and J. M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26–36 CrossRef CAS.
- (a) B. Klekota, M. H. Hammond and B. L. Miller, Tetrahedron Lett., 1997, 38, 8639–8642 CrossRef CAS; (b) C. Karan and B. L. Miller, J. Am. Chem. Soc., 2001, 123, 7455–7456 CrossRef CAS; (c) A. M. Withney, S. Ladame and S. Balasubramanian, Angew. Chem., Int. Ed., 2004, 43, 1143–1146 CrossRef CAS; (d) A. Bugaut, J.-J. Toulmé and B. Rayner, Angew. Chem., Int. Ed., 2004, 43, 3144–3147 CrossRef CAS; (e) A. Bugaut, K. Bathany, J.-M. Schmitter and B. Rayner, Tetrahedron Lett., 2005, 46, 687–690 CrossRef CAS; (f) S. Ladame, A. M. Withney and S. Balasubramanian, Angew. Chem., Int. Ed., 2005, 44, 5736–5339 CrossRef CAS.
- I. Huc and R. Nguyen, Comb. Chem. High Throughput Screening, 2001, 4, 53–74 CAS.
- K. Severin, Chem.–Eur. J., 2004, 10, 2565–2580 CrossRef CAS.
- (a) A. V. Eliseev and M. I. Nelen, J. Am. Chem. Soc., 1997, 119, 1147–1148 CrossRef CAS; (b) A. V. Eliseev and M. I. Nelen, Chem.–Eur. J., 1998, 5, 825–834 CrossRef CAS.
- K. B. Mullis and F. A. Faloona, Methods Enzymol., 1987, 155, 335–350 CrossRef CAS.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- (a) W. Kusser, Rev. Mol. Biotechnol., 2000, 74, 27–38 CrossRef CAS; J. A. Bittker, K. J. Phillips and D. R. Liu, Curr. Opin. Chem. Biol., 2002, 6, 367–374 CrossRef CAS; (b) S. Verma, S. Jager, O. Thum and M. Famulok, Chem. Rec., 2003, 3, 51–60 CrossRef CAS.
- (a) B. E. Eaton and W. A. Pieken, Annu. Rev. Biochem., 1995, 64, 837–863 CrossRef CAS; (b) B. E. Eaton, L. Gold, B. J. Hicke, N. Janjic, F. M. Jucker, D. P. Sebesta, T. M. Tarasow, M. C. Willis and D. A. Zichi, Bioorg. Med. Chem., 1997, 5, 1087–1096 CrossRef CAS.
- (a) I. Huc and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS; (b) M. Hochgurtel, H. Kroth, D. Piecha, M. W. Hofmann, C. Nicolau, S. Krause, O. Schaaf, G. Sonnenmoser and A. V. Eliseev, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3382–3387 CrossRef; (c) M. Hochgurtel, R. Biesinger, H. Kroth, D. Piecha, M. W. Hofmann, S. Krause, O. Schaaf, C. Nicolau and A. V. Eliseev, J. Med. Chem., 2003, 46, 356–358 CrossRef CAS; (d) S. Zameo, B. Vauzeilles and J.-M. Beau, Angew. Chem., Int. Ed., 2005, 44, 965–969 CrossRef CAS.
- H. Aurup, D. M. Williams and F. Eckstein, Biochemistry, 1992, 31, 9636–9641 CrossRef CAS.
- (a) F. Ducongé and J.-J. Toulmé, RNA, 1999, 5, 1605–1614 CrossRef CAS; (b) C. Boiziau, E. Dausse, L. Yurchenko and J.-J. Toulmé, J. Biol. Chem., 1999, 274, 12730–12737 CrossRef CAS; (c) D. Sekkai, E. Dausse, C. Di Primo, F. Darfeuille, C. Boiziau and J.-J. Toulmé, Antisense Nucleic Acid Drug Dev., 2002, 12, 265–274 CrossRef CAS; (d) F. Darfeuille, D. Sekkai, E. Dausse, G. Kolb, L. Yurchenko, C. Boiziau and J.-J. Toulmé, Comb. Chem. High Throughput Screening, 2002, 5, 313–325 CAS.
- F. Darfeuille, J. B. Hansen, H. Orum, C. Di Primo and J.-J. Toulmé, Nucleic Acids Res., 2004, 32, 3101–3107 CrossRef CAS.
- E. J. Merino and K. M. Weeks, J. Am. Chem. Soc., 2005, 127, 12766–12767 CrossRef CAS.
- (a) S. Jäger and M. Famulok, Angew. Chem., Int. Ed., 2005, 43, 3337–3340; (b) S. Jäger, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef; (c) T. Schoetzau, J. Langner, E. Moyroud, I. Roehl, S. Vonhoff and S. Klussmann, Bioconjugate Chem., 2003, 14, 919–926 CrossRef CAS; (d) R. Kawai, M. Kimoto, S. Ikeda, T. Mitsui, M. Endo, S. Yokoyama and I. Hirao, J. Am. Chem. Soc., 2005, 127, 17286–17295 CrossRef CAS; (e) K. Moriyama, M. Kimoto, T. Mitsui, S. Yokoyama and I. Hirao, Nucleic Acids Res., 2005, 33, e129 CrossRef . For a recent review related to polymerases engineered for incorporating modified nucleotides, see: A. A. Henry and F. E. Romesberg, Curr. Opin. Biotechnol., 2005, 16, 370–377 Search PubMed.
- J. Goodchild, Bioconjugate Chem., 1990, 1, 165–187 CrossRef CAS.
- (a) S. Jhaveri, M. Rajendran and A. D. Ellington, Nat. Biotechnol., 2000, 18, 1293–1297 CrossRef CAS; (b) M. Rajendran and A. D. Ellington, Comb. Chem. High Throughput Screening, 2002, 5, 263–270 CAS; (c) R. Nutiu and Y. Ly, Chem.–Eur. J., 2004, 10, 1868–1876 CrossRef CAS; (d) S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2005, 20, 2424 CrossRef CAS.
Footnotes |
| † Current address: University of Cambridge, Department of Chemistry, Lensfield road, Cambridge, UK CB2 1EW. E-mail: E-mail: ab605@cam.ac.uk |
| ‡ Concentrations of aldehydes were adjusted to compensate for their differences in reactivity with 2′-amino-uridine and to provide comparable proportions of conjugated products. |
| § For selection S+, washings were performed by using a solution containing the three aldehydes, 1 (1 mM), 2 (1.2 mM) and 3 (200 µM), in buffer 1 × SE (see Experimental section). |
| This journal is © The Royal Society of Chemistry 2006 |