Diphenylphosphinoyl-mediated synthesis of ketones†
David J.
Fox
*,
Daniel Sejer
Pedersen
and
Stuart
Warren
Cambridge University, University Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: djf34@cam.ac.uk
First published on 6th July 2006
Abstract
α-Diphenylphosphinoyl ketones are selectively and sequentially alkylated at the α-position. Double lithiation and selective alkylation occurs at the less stabilised γ-position. Dephosphinoylation of the alkylation products gives ketones. Mono-alkylation is selective, highly crystalline intermediates are formed and a one-pot strategy is possible. The method is ideally suited for the preparation of acid-sensitive ketones.
The two-step synthesis of ketones via the addition of a carbon-centred nucleophile to a carboxylic acid, or equivalent, followed by enolate generation and reaction with electrophiles is one of the most important reaction sequences in synthetic chemistry. Generally, the addition of organolithium reagents to simple carboxylate esters or acyl chlorides is complicated by the tendency of the nucleophile to react with the ketone generated if the tetrahedral intermediate decomposes during the reaction.1 Grignard reagents do react with acid chlorides at low temperature to make ketones selectively.2 It is possible to acylate other organometallic species with acid chlorides and thioesters via a C–Cl or C–S bond insertion mechanism: magnesium,3–5 copper,6–8 tin,9 and zinc10,11 organometallics selectively produce ketones, often with palladium catalysis. Alternatively, good yields of ketone can be ensured by trapping12,13 or by stabilisation of the tetrahedral intermediate through chelation of the metal to the leaving group (Weinreb amides,14 or thiopyridyl esters15) or the nucleophile. Sulfoxides, sulfones and sulfonamides,16 sulfinamides,17 phosphinates and thiophosphinates,17 and phosphine oxides18,19 are good chelating groups for hard metal ions and also act as electron-withdrawing groups (EWGs) in the generation of carbon nucleophiles by deprotonation. In the reactions of these stabilised anions over-addition is rarely observed (Scheme 1).
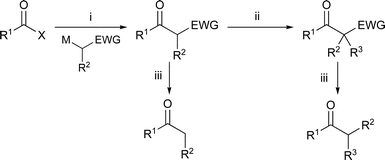 | ||
| Scheme 1 Ketone synthesis: (i) acylation; (ii) alkylation; (iii) deprotection. X = OR; M = Li, Na, K; EWG = CO2R, S(O)R, SO2R, S(O)NR2, SO2NR2, PO(OR)2, P(O)R2. | ||
In many cases the regioselective generation of enolates from near-symmetrical ketones can be problematic, and the yields observed in the alkylations of simple lithium enolates with alkyl halides are not always high.20 The use of other metal counterions (e.g. boron,21 tin,22,23 zinc,24 or manganese25–27) or masked ketone equivalents such as hydrazones28,29 can give synthetically useful yields of alkylated products but many of these methods still require the use of strong bases and/or low temperatures and the difficult drying of metal salts for transmetallation. Alternatively, the activation of ketones with electron-withdrawing groups allows milder and regioselective enolate generation. In particular, β-keto-esters,30,31 β-keto-sulfoxides,32 β-keto-sulfones,33 β-keto-phosphonates34 and β-keto-phosphine oxides35 can all be alkylated (Scheme 1). In these cases the bases used in the alkylation are weaker than the lithium amide bases needed for the alkylation of unactivated ketones or hydrazones. The activating ester,30,31 sulfoxide, sulfone, sulfonamide,16 sulfinamide,17 phosphonate,36 or phosphine oxide35 groups can be removed with a variety of reagents to give simple ketones (Scheme 1). The use of a phosphine oxide as an auxiliary activating group is of particular benefit because β-keto-diphenylphosphine oxides are easy to make, are highly crystalline and are not hydroscopic.
The diphenylphosphinoyl group can facilitate carbon–carbon bond formation and then be removed to give olefins37 and cyclopropanes.38 Both cases involve an intramolecular oxygen nucleophile and a carbon leaving group from phosphorus. The removal of the phosphinoyl group may also occur with an intermolecular nucleophile as long as the leaving group ability is maintained. This suggestion prompted the first experiment: the treatment of β-keto-phosphine oxide383e [Scheme 2, eqn. (1)] with aqueous sodium hydroxide in ethanol, similar conditions to those reported for the analogous intramolecular phosphoryl transfer [Scheme 2, eqn. (2)].39
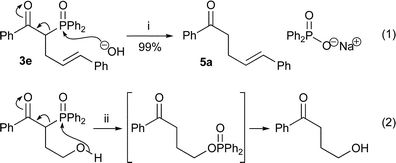
This reaction produced ketone 5a cleanly and in high yield, with the sodium diphenylphosphinate by-product easily removed by aqueous base. Taken in conjunction with the acylation18,19 and alkylation reactions, this promised to be a simple, powerful and general ketone synthesis (Scheme 3).
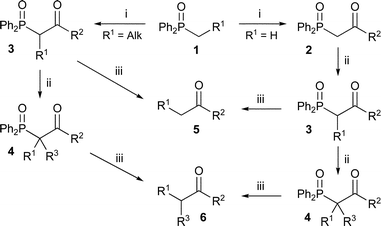 | ||
| Scheme 3 (i) Acylation; (ii) alkylation; (iii) dephosphinoylation. | ||
A series of β-keto-phosphine oxides was synthesised by the method of Warren and Torr (Table 1).19 With the exception of entries 5 and 9, all these β-keto-phosphine oxides are known compounds prepared by a similar method. Typically, these reactions never go to completion. However, it is usually simple to separate unreacted starting material from the product by column chromatography or crystallisation. Bartoli et al. have developed conditions that improve the yield of the acylation of phosphine oxides by using an excess of base and acylating agent.40 However, as it is simple to separate starting material from product, the less wasteful Warren procedure, which employs stoichiometric base and acylating agent, was used.
| Entry | R1 | R2 | Product | Yield (%) |
|---|---|---|---|---|
| a Conditions: THF, −78 °C, n-BuLi (1.05 equiv.), methyl ester (1.1 equiv.) except for entries 1, 6 and 7 where the ethyl ester was used. | ||||
| 1 | H | Me |
2a![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 41 41 |
52 |
| 2 | H | Et |
2b19 |
46 |
| 3 | H | i-Pr |
2c42 |
49 |
| 4 | H | Ph |
2d19 |
77 |
| 5 | H | Furan-2-yl | 2e | 76 |
| 6 | H | PhCH2CH2 |
2f43 |
69 |
| 7 | Me | Me |
3a19 |
62 |
| 8 | Me | Et |
3b19 |
63 |
| 9 | Me | i-Pr | 3c | 65 |
| 10 |

|
Ph |
3d44 |
74 |
Selective mono-alkylation is difficult with, for example, traditional acetoacetate chemistry in particular with reactive electrophiles such as allylic halides. We hoped that our method would provide a more reliable method for this purpose. As can be seen from Table 2, selective mono-alkylation occurs in good yields with sodium methoxide as base and activated electrophiles at room temperature. Despite their reactivity and base sensitivity, even α-bromo esters and ketones react only once in high yield (entries 6 and 7). Most reactions are complete within a few hours and products are easily isolated by column chromatography.
| Entry | 2 R2 | Alkylating agent R1–X | Product (methoda) | Yield (%) |
|---|---|---|---|---|
| a Conditions: (a) NaOMe (1.1 equiv.), R1–X (1.2 equiv.), THF; (b) as in (a) with NaI (1 equiv.) added; (c) NaH (1.1 equiv.), R1–X (1.2 equiv.), DMF, 80 °C; (d) NaH (1.1 equiv.), R1–X (1.2 equiv.), DMF; (e) as in (c) with NaI (1 equiv.) added. b Conversion by NMR. | ||||
| 1 | Ph | (E)-PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2Br CHCH2Br |
3e (a) | 99 |
| 2 | Ph | (E)-PhCHCHCH2Cl |
3e (b) | 97 |
| 3 | Ph | (Z)-PhCHCHCH2Br |
3f (a) | 85 |
| 4 | Ph | CH2CHCH2Br |
3g (a) | 74 |
| 5 | Ph | PhCH2Br | 3h (a) | 75 |
| 6 | Ph | PhC(O)CH2Br | 3i (a) | 86 |
| 7 | Ph | BrCH2CO2t-Bu | 3j (a) | 84 |
| 8 | Ph | PhCH2CH2Br | 3k (c) | <17b |
| 9 | Ph | CH3(CH2)11I | 3l (c) | 71 |
| 10 | 2-Furanyl | (E)-PhCHCHCH2Br |
3m (a) | 83 |
| 11 | Me | (E)-PhCHCHCH2Br |
3n (a) | 87 |
| 12 | Me | PhCH2Br | 3o (a) | 72 |
| 13 | Me | CH3(CH2)11I | 3p (d) | 0b |
| 14 | Et | MeO2C(CH2)10Br | 3q (e) | <10b |
| 15 | PhCH2CH2 | (E)-PhCHCHCH2Br |
3r (a) | 95 |
| 16 |

|
CH2CHCH2Br |
3s (a) | 82 |
The alkylation products are generally highly crystalline due to the diphenylphosphinoyl group making it possible to crystallise the product directly from the crude reaction mixture. This is particularly useful when the reactions are performed on a large scale. Owing to the moderate reactivity of β-keto-phosphine oxides towards very reactive electrophiles, we suspected that they would be unreactive towards less reactive alkyl halides. Attempts at alkylating methyl ketone 2a (Table 2, entry 13) and phenyl ketone 2d (entry 9) with an alkyl iodide showed that the nature of the enolate nucleophile has a strong influence on the reaction outcome. Whereas, phenyl ketone 2d produces the desired mono-alkylated product in good yield no reaction occurs with methyl ketone 2a. The same trend is observed with alkyl bromides (Table 2, entries 8 and 14) which give only low conversions even at elevated temperature. The alkylation reactions of methyl ketone 2a and phenyl ketone 2d were investigated further using butyl chloride, bromide and iodide as electrophiles, and NaOMe/THF and NaH/DMF as base (with and without added sodium iodide) at room temperature and at 60 °C (Table 3). All reactions were examined by NMR after 20 h. Butyl chloride was completely inert under all the studied conditions (entry 1) and butyl bromide gave hardly any conversion at room temperature and only poor conversion at elevated temperature (entries 2–6). The addition of sodium iodide did not have any significant effect on conversion rates. Only butyl iodide gave conversions that could be considered useful from a synthetic point of view when sodium hydride in DMF was employed (entries 8 and 10). Both the methyl ketone 2a and phenyl ketone 2d gave good conversions.
|
|
||||
|---|---|---|---|---|
| Entry | Bu–X | Methoda/°C | Yield (%)b | |
| 3t | 3u | |||
| a Conditions: (a) NaOMe (1.1 equiv.), THF, Bu–X (1.1 equiv.), 20 h; (b) as in (a) with NaI (20 mol%) added; (c) NaH (1.1 equiv.), DMF, Bu–X (1.1 equiv.), 20 h; (d) as in (c) with NaI (20 mol%) added. b Conversion by NMR; RT = room temperature. | ||||
| 1 | BuCl | a,b,c,d (RT/60) | 0 | 0 |
| 2 | BuBr | a,b,c,d (RT) | <14 | <4 |
| 3 | BuBr | a (60) | 0 | 4 |
| 4 | BuBr | b (60) | 20 | 35 |
| 5 | BuBr | c (60) | 23 | 42 |
| 6 | BuBr | d (60) | 41 | 49 |
| 7 | BuI | a (RT) | 5 | 4 |
| 8 | BuI | c (RT) | 73 | 39 |
| 9 | BuI | a (60) | 24 | 1 |
| 10 | BuI | c (60) | 67 | 57 |

Introduction of a second substituent at the α-position of mono-alkylated β-keto-phosphine oxides 3 might be difficult due to the complete absence of dialkylated side-products previously (Table 2). As foreseen, the first attempt at alkylating a phenyl ketone failed to give any product (Scheme 4). However, when the same reaction was attempted with methyl ketone 3a, and sodium hydride in DMF, the dialkylated product was obtained in a good yield (Table 4, entry 2) and when sodium methoxide in THF was employed the product was obtained in excellent yield (entry 3). This prompted us to perform a number of experiments with alkyl ketones (entries 4–7) that all produced the dialkylated products 4 in moderate to good yields.
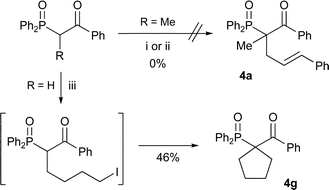 | ||
| Scheme 4 Reagents and conditions: i) NaOMe, THF, (E)-cinnamyl bromide; ii) NaH, DMF, (E)-cinnamyl bromide, 80 °C; iii) NaH, DMF, 1,4-diiodobutane. | ||
| Entry | Starting material 3 | Alkylating agent R3–X | Product (methoda) | Yield (%) | |
|---|---|---|---|---|---|
| R1 | R2 | ||||
| a Conditions: (a) NaOMe, R3–X, THF, 20 °C; (b) NaH, R3–X, DMF, 80 °C; (c) NaH, R3–X, DMF, 20 °C; (d) NaH (2.2 equiv.), R3–X (1.2 equiv.), DMF. | |||||
| 1 | Me | Ph | (E)-PhCHCHCH2Br |
4a (a,b) | 0 |
| 2 | Me | Me | (E)-PhCHCHCH2Br |
4b (c) | 72 |
| 3 | Me | Me | (E)-PhCHCHCH2Br |
4b (a) | 97 |
| 4 | Me | Et | (E)-PhCHCHCH2Br |
4c (a) | 81 |
| 5 | Me | i-Pr | (E)-PhCHCHCH2Br |
4d (a) | 80 |
| 6 | (E)-PhCHCHCH2 |
Me | CH2CHCH2Br |
4e (a) | 54 |
| 7 | (E)-PhCHCHCH2 |
(CH2)2Ph | (CH3)CCHCH2Br |
4f (a) | 87 |
| 8 | H | Ph | ICH2CH2CH2CH2I | 4g (d) | 46 |
To establish if it was possible to do a double alkylation of phenyl ketones, we made the second alkylation intramolecular producing cyclic phenyl ketone 4g (Scheme 4), giving the product in moderate yield.
Using phosphine oxide 3e as a model compound, a number of different dephosphinoylation conditions were examined: (a) sodium hydroxide in refluxing water and ethanol; (b) potassium hydroxide in refluxing methanol; (c) sodium methoxide in refluxing methanol; (d) potassium fluoride in refluxing water and ethanol; (e) potassium carbonate in refluxing water and ethanol. With the exception of potassium fluoride (Table 5, entry 5), all of these dephosphinoylation conditions resulted in conversion to the desired ketone in excellent yield. In many instances, the ‘crude’ reaction product 6 was analytically pure. It is noteworthy that the acetal-protected phosphine oxide 5i (entry 14) was dephosphinoylated in good yield, demonstrating that this method is ideally suited for the synthesis of acid-sensitive ketones. Moreover, (Z)-alkene 5k (entry 16) did not isomerise to the (E)-isomer under the reaction conditions, and furan 5m (entry 18) was stable giving a good yield of ketone.
| Entry | Starting material 3/4 | Product (methoda) | Yield (%) | ||
|---|---|---|---|---|---|
| R1 | R2 | R3 | |||
| a Conditions: (a) 4 M aq. NaOH, EtOH, reflux; (b) KOH (10 equiv.), MeOH, reflux; (c) K2CO3 (10 equiv.), H2O, EtOH, reflux; (d) NaOMe (10 equiv.), MeOH, reflux; (e) KF (10 equiv.), H2O, EtOH, reflux. b Conversion by NMR with Ph2P(O)OMe as the other product. c The carboxylic acid was obtained. | |||||
| 1 | H | Ph | (E)-PhCHCHCH2 |
5a (a) | 99 |
| 2 | H | Ph | (E)-PhCHCHCH2 |
5a (b) | 96 |
| 3 | H | Ph | (E)-PhCHCHCH2 |
5a (c) | 95 |
| 4 | H | Ph | (E)-PhCHCHCH2 |
5a (d) | >95b |
| 5 | H | Ph | (E)-PhCHCHCH2 |
5a (e) | 0 |
| 6 | H | Me | (E)-PhCHCHCH2 |
5b (a) | 92 |
| 7 | H | Me | (E)-PhCHCHCH2 |
5b (c) | 95 |
| 8 | H | Me | PhCH2 | 5c (a) | 86 |
| 9 | H | Ph | PhCH2 | 5d (a) | 96 |
| 10 | H | Ph | CH2CHCH2 |
5e (a) | 78 |
| 11 | H | Ph | CH3(CH2)11 | 5f (a) | 68 |
| 12 | H | Ph | PhC(O)CH2 | 5g (a) | 67 |
| 13 | H | Ph | t-BuOC(O)CH2 | 5h (a) | 77c |
| 14 | H | Ph |
|
5i (b) | 82 |
| 15 | H | PhCH2CH2 | (E)-PhCHCHCH2 |
5j (b) | 81 |
| 16 | H | Ph | (Z)-PhCHCHCH2 |
5k (a) | 93 |
| 17 | H |
|
CH2CHCH2 |
5l (a) | 78 |
| 18 | H | Furan-2-yl | (E)-PhCHCHCH2 |
5m (b) | 72 |
| 19 | Me | Me | (E)-PhCHCHCH2 |
6a (a) | 96 |
| 20 | Me | Et | (E)-PhCHCHCH2 |
6b (a) | 91 |
| 21 | Me | i-Pr | (E)-PhCHCHCH2 |
6c (a) | 97 |
| 22 | CH2CHCH2 |
Me | (E)-PhCHCHCH2 |
6d (b) | 71 |
| 23 | (CH3)2CCHCH2 |
PhCH2CH2 | (E)-PhCHCHCH2 |
6e (a) | 80 |
It was also possible to apply the method to the synthesis of deuterium labelled ketones by performing the dephosphinoylation in deuterated solvents. For example, ketones 5n and 5o (Scheme 5) were synthesised in good yield by treating the respective β-keto-phosphine oxides with sodium deuteroxide in refluxing deuterium oxide and ethanol-d.1
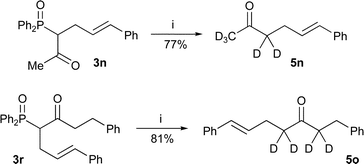 | ||
| Scheme 5 Reagents and conditions: i) NaOD, D2O, EtOD, reflux. | ||
The synthesis and dephosphinoylation of β-keto-phosphine oxides can also be combined into a one-pot reaction with no purification of intermediates. As described above, β-keto-phosphine oxides can be constructed in a number of ways. For example, phosphine oxide 1a (Scheme 6) was acylated followed by dephosphinoylation of the crude product 3v to give a moderate yield of ketone 5p over two steps.
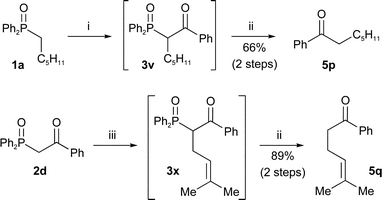 | ||
| Scheme 6
Reagents and conditions: i) n-BuLi, THF, PhCO2Me, −78 °C; ii) NaOH, H2O, EtOH, reflux; iii) NaOMe, THF, (CH3)2CCHCH2Br. | ||
Alternatively, alkylation of β-keto-phosphine oxides can be accomplished first followed by dephosphinoylation. For example, phosphine oxide 2d (Scheme 6) was alkylated with prenyl bromide followed by dephosphinoylation of the crude product 3x to give a high yield of ketone 5q over two steps.
Alkylation of α-,γ-dilithiated β-keto-phosphine oxides can also lead to the synthesis of interesting ketones.45 The treatment of variously substituted β-keto-phosphine oxides with two equivalents of LDA produces a dilithium derivative, which selectively reacted at the less stabilised γ-position. Systematic investigation into the effect that the substitution pattern of the phosphine oxide has on the reaction outcome was conducted using cinnamyl bromide as the electrophile (Table 6). This revealed that increased methylation at either the α- or γ-position reduces the yield of the γ-alkylation. The introduction of one methyl group at either the α- or γ-position reduced the yield by 10–20% (entries 2–4) and the introduction of methyl groups at both the α- and γ-positions reduced the yield by approximately 40% (Entry 5).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Starting material | Product (methoda) | Yield 7a–f (%) | Product (methoda) | Yield 8a–d (%) | ||
| R1 | R2 | R3 | |||||
| a Conditions: (a) LDA (2 equiv.), 9 (1 equiv.), THF, −78 °C; (b) LDA (2 equiv.), 9 (1 equiv.), THF, −78 to 0 °C; (c) LDA (2 equiv.), 9 (1 equiv.), THF, −78 °C to room temperature; (d) 4 M aq. NaOH, EtOH, reflux; (e) KOH (10 equiv.), MeOH, reflux. b Conversion by NMR. | |||||||
| 1 | H | H | H | 7a (a) | 65 | 8a [d (e)] | 91 (86) |
| 2 | Me | H | H | 7b (a) | 55 | 8b (d) | 85 |
| 3 | H | Me | H | 7c (a) | 45 | 8c (e) | 93 |
| 4 | H | Me | H | 7c (b) | 48 | 8c | — |
| 5 | Me | Me | H | 7d (a) | 22 | 8d (d) | 70 |
| 6 | H | Me | Me | 7e (a) | <5b | 8e | — |
| 7 | Me | Me | Me | 7f (a,c) | <5b | 8f | — |

Bis-methylation at the γ-position irrespective of the α-substitution gives only trace amounts of alkylated products 8e and 8f by NMR (entries 6 and 7). Hence, only one of the lithiated centres can carry an alkyl group for the reaction to be synthetically useful (entries 1–3). The synthesised γ-alkylated products were easily dephosphinoylated to give ketones in high yield (Table 6). As a consequence, the same α-,γ-disubstituted β-keto-phosphine oxide 3r can be produced via alkylation in either order. Comparison of the two routes to β-keto-phosphine oxide 3r (Scheme 7) showed that both alkylation yields are higher if the γ-substituent is introduced first.
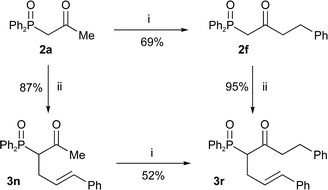 | ||
| Scheme 7
Reagents and conditions: i) LDA, THF, BnBr, −78 °C; ii) NaOMe, THF, PhCHCHCH2Br. | ||
α-,γ-Dilithiated β-keto-phosphine oxides also add to aldehydes to produce β-keto-δ-hydroxy phosphine oxides in high yield (Scheme 8). The double anion equivalent of phosphine oxide 2a and benzaldehyde gave phosphine oxide 7g in high yield. However, this compound gave a complex mixture when dephosphinoylation was attempted.
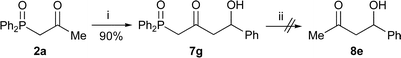
Taking the acylation, α- and γ-alkylation and dephosphinoylation reaction in conjunction, this method has proved to be a general, practical method for the synthesis of branched ketones, which has several advantages over existing methods.
Acknowledgements
D. S. P. would like to thank the Alfred Benzon Foundation, Cambridge European Trust and the Anglo-Danish Society for financial support.References
- B. T. O'Neill, Nucleophilic Addition to Carboxylic Acid Derivatives, Pergamon, Oxford, 1991, pp. 397–458 Search PubMed.
- F. Sato, M. Inoue, K. Oguro and M. Sato, Tetrahedron Lett., 1979, 20, 4303 CrossRef.
- V. Fiandanese, G. Marchese, V. Martina and L. Ronzini, Tetrahedron Lett., 1984, 25, 4805 CrossRef CAS.
- C. Cardellicchio, V. Fiandanese, G. Marchese and L. Ronzini, Tetrahedron Lett., 1987, 28, 2053 CrossRef CAS.
- C. Cardellicchio, V. Viandanese, G. Marchese and L. Ronzini, Tetrahedron Lett., 1985, 26, 3595 CrossRef CAS.
- G. H. Posner, An Introduction to Synthesis Using Organocopper Reagents, Wiley, New York, 1980 Search PubMed.
- M. Onaka, Y. Matsuoka and T. Mukaiyama, Chem. Lett., 1981, 531 CAS.
- R. J. Anderson, C. A. Henrick and L. D. Rosenblu, J. Am. Chem. Soc., 1974, 96, 3654 CrossRef CAS.
- D. Milstein and J. K. Stille, J. Org. Chem., 1979, 44, 1613 CrossRef CAS.
- E. Negishi, V. Bagheri, S. Chatterjee, F. T. Luo, J. A. Miller and A. T. Stoll, Tetrahedron Lett., 1983, 24, 5181 CrossRef CAS.
- H. Tokuyama, S. Yokoshima, T. Yamashita and T. Fukuyama, Tetrahedron Lett., 1998, 39, 3189 CrossRef CAS.
- G. M. Rubottom and C. W. Kim, J. Org. Chem., 1983, 48, 1550 CrossRef CAS.
- M. P. Cooke, J. Org. Chem., 1986, 51, 951 CrossRef CAS.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS.
- T. Mukaiyam, M. Araki and H. Takei, J. Am. Chem. Soc., 1973, 95, 4763 CrossRef CAS.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1345 CrossRef CAS.
- E. J. Corey and T. Durst, J. Am. Chem. Soc., 1966, 66, 5656 CrossRef.
- A. Bell, A. H. Davidson, C. Earnshaw, H. K. Norrish, R. S. Torr, D. B. Trowbridge and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1983, 2879 RSC.
- R. S. Torr and S. Warren, J. Chem. Soc. Pak., 1979, 1, 15 Search PubMed.
- M. Gall and H. O. House, Org. Synth., 1988, 50–59, 121.
- E. I. Negishi and S. Chatterjee, Tetrahedron Lett., 1983, 24, 1341 CrossRef CAS.
- M. Yasuda, T. Ohhata, I. Shibata, A. Baba and H. Matsuda, J. Chem. Soc., Perkin Trans. 1, 1993, 859 RSC.
- M. Yasuda, K. Hayashi, Y. Katoh, I. Shibata and A. Baba, J. Am. Chem. Soc., 1998, 120, 715 CrossRef CAS.
- Y. Morita, M. Suzuki and R. Noyori, J. Org. Chem., 1989, 54, 1785 CrossRef.
- G. Cahiez, K. Chau and B. Blanchot, Org. Synth., 1999, 76, 239 CAS.
- G. Cahiez, B. Figadere and P. Clery, Tetrahedron Lett., 1994, 35, 3065 CrossRef CAS.
- G. Cahiez, K. Chau and P. Clery, Tetrahedron Lett., 1994, 35, 3069 CrossRef CAS.
- T. Cuvigny and H. Normant, Synthesis, 1977, 198 CrossRef CAS.
- E. J. Corey and D. Enders, Tetrahedron Lett., 1976, 11 CrossRef CAS.
- T. Yamamitsu, S. Ohta and T. Suga, J. Chem. Soc., Perkin Trans. 1, 1989, 1811 RSC.
- R. Kluger and M. Brandl, J. Org. Chem., 1986, 51, 3964 CrossRef CAS.
- P. G. Gassman and G. D. Richmond, J. Org. Chem., 1966, 31, 2355 CrossRef CAS.
- H. O. House and J. K. Larson, J. Org. Chem., 1968, 33, 61 CrossRef CAS.
- R. D. Clark, L. G. Kozar and C. H. Heathcock, Synthesis, 1975, 635 CrossRef.
- D. J. Fox, D. S. Pedersen and S. Warren, Chem. Commun., 2004, 2598 RSC.
- S. Y. Lee, C.-W. Lee and D. Y. Oh, J. Org. Chem., 1999, 64, 7017 CrossRef CAS.
- J. Clayden and S. Warren, Angew. Chem., Int. Ed. Engl., 1996, 35, 241 CrossRef CAS.
- T. Boesen, D. J. Fox, W. Galloway, D. S. Pedersen, C. R. Tyzack and S. Warren, Org. Biomol. Chem., 2005, 3, 630 RSC.
- P. Wallace and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1988, 2971 RSC.
- G. Bartoli, M. Bosco, L. Sambri and E. Marcantoni, Tetrahedron Lett., 1996, 37, 7421 CrossRef CAS.
- B. Corbel, L. Medinger, J. P. Haelters and G. Sturtz, Synthesis, 1985, 1048 CrossRef.
- T. Mochizuki, S. Hayakawa and K. Narasaka, Bull. Chem. Soc. Jpn., 1996, 69, 2317 CrossRef CAS.
- C. R. Holmquist and E. J. Roskamp, Tetrahedron Lett., 1992, 33, 1131 CrossRef CAS.
- C. A. Cornish and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1985, 2585 RSC.
- R. S. Torr and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1983, 1173 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: experimental and analytical details for all compounds. See DOI: 10.1039/b606873a. |
| This journal is © The Royal Society of Chemistry 2006 |