Synthesis of the bis-spiroacetal moiety of the shellfish toxins spirolides B and D using an iterative oxidative radical cyclization strategy†
Kai Meilert and Margaret A. Brimble*
Department of Chemistry, University of Auckland, 23 Symonds St., Auckland, New Zealand. E-mail: m.brimble@auckland.ac.nz; Fax: +64 9 3737422
First published on 2nd May 2006
Abstract
The enantioselective synthesis of the bis-spiroacetal fragment of the shellfish toxins, spirolides B 1 and D 2, is reported. The carbon framework was constructed via a Barbier reaction of dihydropyran 10 with aldehyde 11, followed by two oxidative radical cyclizations to construct the bis-spiroacetal ring system. A silyl-modified Prins cyclization and enantioselective crotylation successfully installed the stereocenters in the cyclization precursor 21. The initial unsaturated bis-spiroacetals 9a–d underwent equilibration during epoxidation to trans-epoxide 24 that was converted to tertiary alcohol 7.
Introduction
The spirolides A–D 1–5 (Fig. 1) comprise a novel family of pharmacologically active macrocyclic imines found in the polar lipid fraction obtained from the digestive glands of contaminated mussels (Mytilus edulis), scallops (Placopecten magellanicus) and toxic plankton from the eastern coast of Nova Scotia, Canada. Spirolides A–D 1–5 contain an unusual 5,5,6-bis-spiroacetal moiety together with a rare 6,7-spirocyclic imine.1 Spirolides E and F are keto amine hydrolysis derivatives resulting from the ring opening of the cyclic imine suggesting that this functionality is the pharmacophore responsible for toxicity.2 Recently, isolation and culture of a toxic clone of the dinoflagellate Alexandrium ostenfeldii obtained from the same aquaculture site allowed the structural elucidation of three more congeners, spirolides A 1, C 3 and 13-desmethyl C 5.3 The spirolides A–D (1–5) cause potent and characteristic symptoms in the mouse bioassay (spirolide A: LD50 250 µg kg−1) and are activators of L-type calcium channels. These macrocycles contain a novel 6,5,5-bis-spiroacetal ring system as well as an unusual 7,6-spiroimine moiety and bear close resemblance to pinnatoxin A 6 (LD50 180 µg kg−1) which was isolated from toxic extracts of the clam Pinna muricata and has been linked to several major shellfish poisoning events in Japan and China.4,5 The absolute stereochemistry of the spirolide family of toxins has not been established to date, however, a computer-generated relative assignment of 13-desmethyl spirolide C 5 indicating the same relative stereochemistry as the related toxin pinnatoxin A 65 in the region of their common structure, was later reported.6 Preliminary pharmacological research into the mode of action of the spirolides suggests that they are antagonists of the muscarinic acetylcholine receptor.7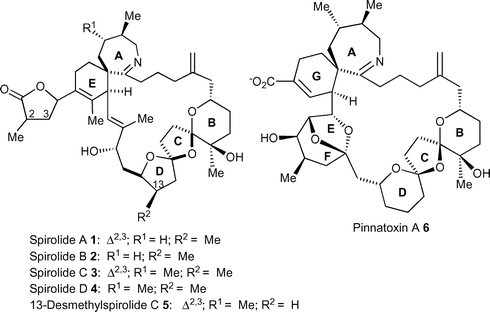 | ||
| Fig. 1 Structure of the spirolides and pinnatoxins. | ||
A total synthesis of the spirolides has not been reported to date, however, an elegant total synthesis of pinnatoxin A 6 has been reported by Kishi et al.8 wherein the BCD bis-spiroacetal ring system was assembled via acid-catalyzed cyclization of a dione precursor. A synthesis of the bis-spiroacetal core of spirolide B 2via acid-catalyzed cyclization of an acyclic triketone has been communicated9 whilst partial syntheses of the bis-spiroacetal moiety of the pinnatoxins are also discussed in a recent review on the synthesis of bis-spiroacetal ring systems.10 Our interest in the synthesis of natural products containing bis-spiroacetal ring systems11 led us to pursue the synthesis of the bis-spiroacetal ring system present in the spirolides using an oxidative radical cyclization to construct the two five-membered rings in the 5,5,6-bis-spiroacetal unit of the spirolides. In addition to our work on the synthesis of model spiroimines12 related to the spirolides, we have previously also reported the synthesis of a C10–C22 bis-spiroacetal fragment lacking the C19 tertiary alcohol group, using a double oxidative radical cyclization.13 However, problems were encountered during the introduction of functionality at C19 and the extension of the carbon framework at C22, thus prompting the adoption of a modified synthetic plan in which disconnection of the C23–C24 bond rather than the C22–C23 bond was a pivotal step. The full details14 of this revised strategy are presented herein providing rapid access to the fully functionalized C10–C23 bis-spiroacetal fragment of spirolides B and D that is homologous to our previous fragment.
Results and discussion
The key disconnection in our proposed retrosynthesis of spirolides B 2 and D 4 (Scheme 1) involves NiII/CrII-mediated Kishi–Nozaki coupling15 between an aldehyde and a vinyl iodide to form the C9–C10 bond of the macrocyclic ring in a similar fashion to that used by Kishi et al.8 in the synthesis of pinnatoxin A 6. Given that our revised synthetic plan relied on the disconnection of the C23–C24 bond rather than the C22–C23 bond, use of a Julia coupling to effect the construction of the C23–C24 bond was envisaged as a key step. Our attention therefore focused on the synthesis of bis-spiroacetal 7, making use of a Julia methylenation16 for the subsequent union with spiroimine sulfone 8. This new approach required access to dihydropyran 9 with the required (S)-configuration at C22 using a silyl-modified Prins cyclization. The two spiroacetal centres in unsaturated spiroacetal 9 are then formed by oxidative radical cyclization of the alcohol resulting from the Barbier coupling of this dihydropyran 10 with aldehyde 11, followed by deprotection of the tert-butyldiphenylsilyl ether and execution of a second oxidative radical cyclization. The syn stereochemistry in aldehyde 11 is available from an enantioselective crotylation. The alkene in bis-spiroacetal 9 provides functionality for subsequent installation of the tertiary alcohol. It is also envisaged that the cis stereochemistry between the terminal rings of the bis-spiroacetal will be established by equilibration after incorporation into the macrocyclic ring. Thus, the initial synthesis of trans-bis-spiroacetals 7 and 9 was required.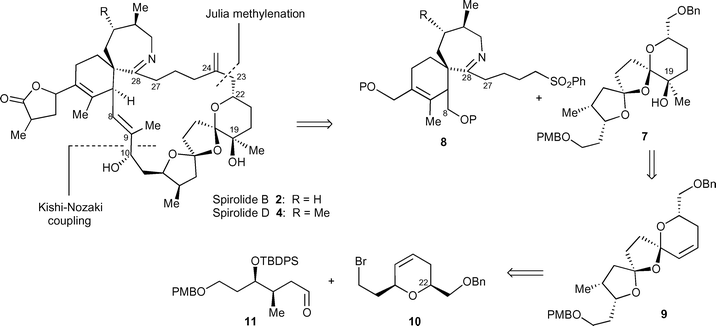 | ||
| Scheme 1 Retrosynthesis of spirolides B 2 and D 4. | ||
The synthesis of the dihydropyran fragment 10 was carried out in 3 steps (51% overall yield), starting from enantiomerically pure O-benzyl protected17 (R)-(+)-glycidol 12 (Scheme 2). Ring opening of epoxide 12 with lithium trimethylsilylacetylide in the presence of a catalytic amount of trimethylaluminium18 afforded homopropargyl alcohol 13 in a higher yield than when using a stoichiometric amount of boron trifluoride diethyl etherate.19 Vinylsilane 14 was initially prepared by semi-hydrogenation of the corresponding acetylene 13 in the presence of a poisoned catalyst. Use of the Rosenmund catalyst (Pd/BaSO4) gave moderate E/Z selectivities and poor yields, while Lindlar's catalyst (Pd/CaCO3/Pb) gave variable selectivities. The optimum solvent using Lindlar’s catalyst was found to be THF affording vinylsilane 14 as a 15 : 85 mixture of E/Z isomers in 69% yield when carried out on a 200 mg scale. Somewhat surprisingly, scaling up of this reaction to a 2 g scale afforded a reversed E/Z selectivity of 100 : 0. Similar selectivity issues when effecting the semi-hydrogenation of alkynes bearing a trimethylsilyl substituent have been reported by others.20 The (Z)-configuration of vinylsilane 14 is crucial for the formation of dihydropyran 10, as elimination of the trimethylsilyl group from the resultant 6-membered ring formed from the (E)-isomer is very slow (<10% conversion after 12 hours).
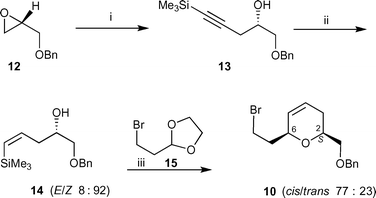 | ||
Scheme 2 Reagents and conditions and yields: (i) Me3SiC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, BuLi, Me3Al (cat.), toluene, −78 °C to room temp., 98%; (ii) DIBALH, Et2O, room temp. then reflux, 24 h, 72%; (iii) InCl3, CH2Cl2, room temp., 48 h, 73%. CH, BuLi, Me3Al (cat.), toluene, −78 °C to room temp., 98%; (ii) DIBALH, Et2O, room temp. then reflux, 24 h, 72%; (iii) InCl3, CH2Cl2, room temp., 48 h, 73%. | ||
Fortunately, after the frustrating attempts to effect the stereoselective semi-hydrogenation of acetylene 13, hydroalumination21 of 13 in ether using DIBALH (1 M in hexane) gave the desired vinylsilane 14 with high (Z)-selectivity (92 : 8). The desired dihydropyran 10 was then prepared in low yield using a silyl-modified Prins cyclization developed by Marko et al.22 by the reaction of vinylsilane 14 with acetal 15 in dichloromethane using trimethylsilyl triflate. The desired dihydropyran 10 was later formed more efficiently using the Lewis acids indium trichloride (72%) or iron trichloride (52%) in dichloromethane at room temperature. For the indium trichloride-catalyzed reaction the 1,3-cis isomer was the major product (cis–trans 77 : 23), however this was of minor importance given that both isomers of the dihydropyran can be used in the radical oxidative cyclization step as an allylic oxocarbenium ion is formed at C6. The relative configuration between C2 and C6 of the major isomer of dihydropyran 10 was assigned as 1,3-cis due to the observation of a strong correlation between H2 and H6 in the NOESY spectrum.
Aldehyde 11 was prepared in five steps from monoprotected 1,3-propanediol 1623,24via reagent-controlled enantioselective crotylation25 of aldehyde 17 using (Z)-2-butene and (−)-β-methoxydiisopinocampheylborane to give (3R,4R)-alcohol 18 in 97% optical purity and dr >95 : 5 (Scheme 3). The absolute configuration of ent-18 was assigned by X-ray diffraction of the derived camphanic ester.26 The enantiomeric excess was measured by 19F NMR after the formation of the Mosher ester using similar methodology to an analogous aldehyde.13 After protection as a tert-butyldiphenylsilyl ether 19, hydroboration with borane-dimethylsulfide followed by oxidative work-up, afforded alcohol 20 that was oxidized with Dess–Martin periodinane to the desired aldehyde 11.
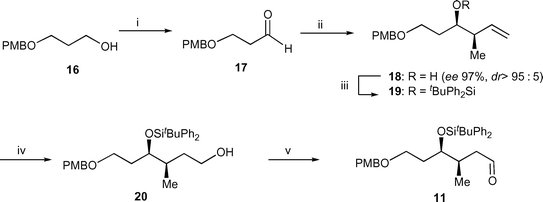 | ||
| Scheme 3 Reagents and conditions and yields: (i) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C, 96%; (ii) (Z)-butene, tBuOK, n-BuLi, (−)-(Ipc)2B(OMe), BF3·Et2O, THF, −78 °C, NaOH, H2O2, 72%; (iii) tBuPh2SiCl, imid, DMF, 100 °C, 99%; (iv) BH3-DMS, THF, room temp., then H2O2, NaOH, 78%; (v) Dess–Martin periodinane, py, CH2Cl2, room temp., 84%. | ||
With aldehyde 11 and bromide 10 in hand, attention next turned to their union via the generation of a Grignard reagent from bromide 10. Previous studies in our group13 have shown that the coupling of similar substrates works best with the use of Barbier's conditions. Magnesium powder was dried under high vacuum with a heat gun and after activation with iodine and 1,2-dibromoethane, a solution of bromide 10 and aldehyde 11 in diethyl ether was added dropwise. After heating for 3 hours under reflux the coupled product 21 was isolated in 88% yield as a ∼1 : 1 mixture of the diastereomers at C3′ (Scheme 4). The two diastereomers could be easily separated by flash chromatography, but usually the mixture was used throughout the synthesis, as equilibration of the bis-spiroacetal ring system was carried out at a later stage. Activation of the magnesium powder was deemed to be a crucial step before the addition of the iodine and 1,2-dibromoethane.
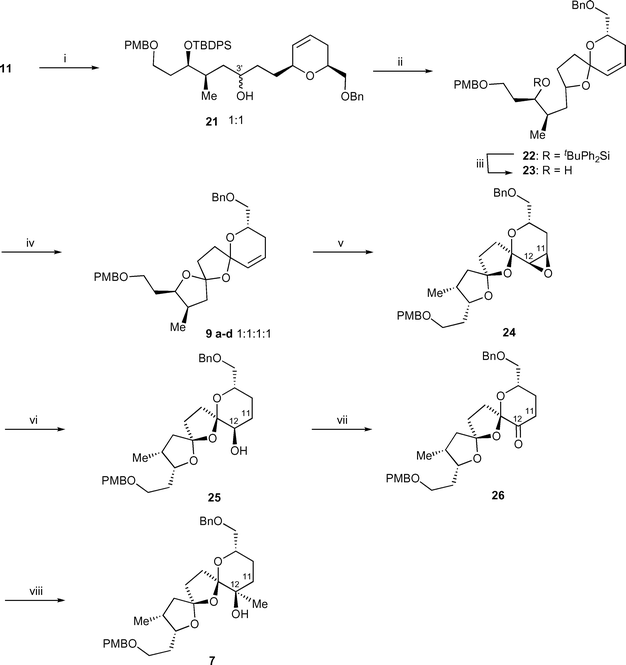
With the basic carbon skeleton fully assembled, use of iterative radical oxidative cyclizations then allows the formation of the bis-spiroacetal. Irradiation of the mixture of alcohols 21 with a 60 W standard desk lamp in the presence of iodobenzene diacetate and iodine27 in cyclohexane afforded spiroacetal 22 as a mixture of diastereomers in 86% yield. Spirocyclization takes place via the formation of an alkoxy radical (generated from a hypoiodite), 1,5-hydrogen transfer to generate a carbon-centred radical, oxidation of the radical to a cation and finally intramolecular trapping of the cation by the hydroxyl group. The tert-butyldiphenyl silyl ether in spiroacetal 22 was removed using tetrabutylammonium fluoride with gentle heating at 80 °C resulting in the formation of alcohol 23. The final bis-spiroacetal ring system was then formed upon execution of a second oxidative radical cyclization providing bis-spiroacetals 9a–d in 81% yield as a 1 : 1 : 1 : 1 mixture of diastereomers.
 | ||
| Scheme 4 Reagents and conditions and yields: (i) 10, Mg, Br(CH2)2Br, I2, Et2O, room temp., 88%; (ii) PhI(OAc)2, I2, hν, cyclohexane, room temp., 86%; (iii) Bu4NF, DMF, 80 °C, 82%; (iv) PhI(OAc)2, I2, hν, cyclohexane, room temp., 81%; (v) m-CPBA, CH2Cl2, 0 °C to room temp., 63%; (vi) DIBALH, hexane, 0 °C, 54%; (viii) Dess–Martin periodinane, CH2Cl2, room temp., 88%; (viii) MeMgBr, Et2O, −78 °C, 86%. | ||
Acid-catalyzed spiroacetalisation of the 1 : 1 : 1 : 1 mixture of 9a–d gave a ∼4 : 1 mixture of the two major isomers (9a and 9b) together with trace quantities (<5%) of two other minor isomers (Table 1). Interestingly, use of indium trichloride gave better results than the commonly used reagents such as HF·py, PPTS, ZnBr2 or ZnCl2 affording a 87 : 13 mixture of the thermodynamically favoured isomers 9a and 9b (entry 5). This is the first example of the use of indium trichloride as a Lewis acid to effect the equilibration of a mixture of spiroacetals allowing convergence to one major diastereomer. The absolute configuration at C5 and C7 in isomer 9a was assigned unambiguously using 2D NMR NOESY experiments, that showed clear correlations between H9 and H4, and between 3-CH3 and H14, respectively (Table 1). Use of a 600 MHz spectrometer was essential in order to see the splitting of the H2 and H9 resonances.
| ||
|---|---|---|
| Entry | Conditions | 9a : 9b (yield) |
| 1 | HF·Pyr, MeCN, room temp., 12 h | 76 : 24 (81%) |
| 2 | PPTS (0.2 equiv), MeCN, room temp., 18 h | ∼81 : 19 (89%) |
| 3 | ZnBr2 (0.2 equiv), CH2Cl2, room temp., 19 h | 76 : 24 (95%) |
| 4 | ZnCl2 (0.2 equiv), CH2Cl2, room temp., 24 h | ∼83 : 17 (88%) |
| 5 | InCl3 (0.2 equiv), MeCN, room temp., 1 h | 87 : 13 (85%) |
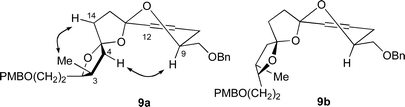
Introduction of the tertiary alcohol at C12 onto the unsaturated bis-spiroacetals 9a–d was initially investigated using a hydroboration–oxidation sequence. Treatment of the unsaturated bis-spiroacetals 9a–d with either BH3·SMe2 or BH3·THF resulted in the complete disappearance of any starting material, but after oxidation of the resultant alcohol using Dess–Martin periodinane only low yields of the undesired C11 ketone could be isolated. Use of different procedures known to be mild for the oxidation of alkylboranes (H2O2, AcONa;28 oxone;29 or NaBO330) did not result in improved yields.
Attempts to direct the formation of a ketone at C12 by the use of a Wacker oxidation31 only afforded recovered starting material (using PdCl2, CuCl and O2) or an intractable complex mixture of products (using PdCl2 and 1,4-benzoquinone) in which oxypalladation appeared to be directed towards the formation of the undesired C11 ketone. Notably, starting with a 1 : 1 : 1 : 1 mixture of unsaturated bis-spiroacetals 9a–d, palladium-catalyzed equilibration of the bis-spiroacetals was also observed to give predominantly bis-spiroacetal 9a.
Finally, treatment of the 1 : 1 : 1 : 1 mixture of bis-spiroacetals 9 with mCPBA afforded β-epoxide 24 as a single diastereomer together with recovered starting material. Remarkably, the presence of meta-chlorobenzoic acid and water in the mCPBA (mCPBA purchased from Fluka contains ∼10% m-chlorobenzoic acid and ∼20% H2O) effected equilibration of the mixture of bis-spiroacetals 9a–d to the most thermodynamically favoured isomer 9a, that then underwent stereoselective epoxidation from the β-face presumably due to the involvement of the neighbouring oxygens in hydrogen bonding to the mCPBA.
Epoxide 24 underwent regioselective reductive opening with DIBALH in hexane and the resultant alcohol 25 was oxidized to ketone 26 upon treatment with Dess–Martin periodinane. Use of hexane as the solvent was crucial for the reductive opening of epoxide 24. Additionally, the reaction was difficult to carry to completion with starting material also being recovered from the reaction. Addition of methylmagnesium bromide to ketone 26 proceeded stereoselectively from the axial direction affording the desired tertiary alcohol 7 with the same stereochemistry as that present in the spirolides. Ishiara and coworkers have similarly reported the use of MeLi in THF to effect the stereoselective axial introduction of the methyl group.9
The stereochemistries of epoxide 24 and tertiary alcohol 7 were assigned unambiguously by 2D NMR NOESY (Fig. 2). For epoxide 24, correlations between H9 and H4, between H12 and H13 and between 3-CH3 and H14 established the stereochemistry as indicated. No correlations were observed between H9 and H11 or H12 as would be expected if epoxidation had taken place from the α-face. For tertiary alcohol 7, clear correlations between H9 and H4 and between H2 and H11 clearly established the trans arrangement of the oxygen atoms about the central ring. The absolute configuration of the CH3 group at C12 was assigned by the observed correlation with H13.
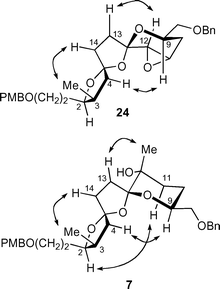 | ||
| Fig. 2 Characteristic NOESY correlations for the assignment of the absolute configuration of epoxide 24 and alcohol 7. | ||
The trans stereochemistry of the bis-spiroacetal ring system adopted by tertiary alcohol 7 represents the thermodynamically favoured isomer and it is hoped that re-equilibration of the bis-spiroacetal to the desired cis stereochemistry as found in the spirolides will take place upon the incorporation of this moiety into the larger macrocyclic system. A comparison of the 1H NMR and 13C NMR chemical shifts recorded for trans bis-spiroacetal 7 with the analogous resonances in the cis bis-spiroacetal moiety of natural product spirolide B 2 is summarized in Table 2.
| |||||
|---|---|---|---|---|---|
| Carbon no. for spirolide B 2 | 13C chemical shift (δC) for spirolide B 2a | Carbon no. for alcohol 7 | 13C chemical shift (δH) for alcohol 7b | 1H chemical shift (δH) for spirolide B 2c | 1H chemical shift (δH) for alcohol 7d |
| a Measured at 125 MHz in CDCl3.b Measured at 100 MHz in CDCl3.c Measured at 500 MHz in CDCl3.d Measured at 600 MHz in CDCl3. | |||||
| 10 | 75.1 | 2′ | 67.7 | 4.25 | 3.58 |
| 11 | 37.1 | 1′ | 30.8 | 1.77, 2.02 | 1.56 |
| 12 | 81.5 | 2 | 78.0 | 4.38 | 4.23 |
| 13 | 35.1 | 3 | 34.8 | 2.46 | 2.42 |
| 14 | 42.5 | 4 | 44.0 | 2.24, 1.93 | 2.37, 1.75 |
| 15 | 116.3 | 5 | 114.9 | — | — |
| 16 | 35.4 | 14 | 35.4 | 2.38, 2.04 | 2.21, 1.94 |
| 17 | 30.8 | 13 | 35.0 | 2.16, 1.81 | 2.40, 1.76 |
| 18 | 111.1 | 7 | 110.4 | — | — |
| 19 | 69.8 | 12 | 68.9 | — | — |
| 20 | 35.8 | 11 | 30.8 | 1.65 (2x) | 1.89, 1.67 |
| 21 | 29.0 | 10 | 26.9 | 1.62, 1.25 | 1.63, 1.54 |
| 22 | 68.3 | 9 | 69.5 | 3.96 | 4.04 |
| 39 | 15.1 | CH3C(3) | 14.6 | 1.18 | 0.92 |
| 40 | 20.9 | CH3C(12) | 21.2 | 1.23 | 1.26 |

The present work demonstrates the efficient construction of the bis-spiroacetal ring system present in the spirolides using an iterative oxidative radical cyclization strategy. Use of InCl3 and m-CPBA to effect the equilibration of the 6,5,5-bis-spiroacetal ring system provides further examples of reagents to effect spiroacetalizations in a stereoselective fashion. Furthermore, the use of a silyl-modified Prins cyclization provides an efficient entry to the dihydropyran unit of the cyclization precursor. Further synthetic work towards the synthesis of spirolides B 2 and D 4 awaits the synthesis of the spiroimine unit of these marine biotoxins.
Experimental
Electronic supplementary material
General experimental details together with full experimental procedures, 1H NMR, 13C NMR and mass spectral data for compounds 10–14 and 17–20 have been deposited as ESI.†Spectroscopic data for less polar diastereomer 21a. υmax (film)/cm−1 3445, 3070, 3030, 2930, 2855, 1613, 1585, 1515, 1465, 1425, 1365, 1305, 1245, 1175, 1110, 935, 820, 740, 700, 615; δH (400 MHz; CDCl3) 7.66–7.70 (m, 4H, PhSi), 7.26–7.45 (m, 11H, PhSi and ArH), 7.11 (d, 2H, J 8.6, ArH), 6.84 (d, 2H, J 8.6, ArH), 5.83 (tdd, 1H, J 1.7, 5.3 and 10.0, 5′-H), 5.62 (tdd, 1H, J 1.2, 2.2 and 10.0, 4′-H), 4.64 (d, 1H, J 12.3, CH2Bn), 4.58 (d, 1H, J 12.3, CH2Bn), 4.19–4.21 (m, 1H, 6′-H), 4.16 (s, 2H, CH2PMBn), 3.81–3.88 (m, 2H, 6-H, 2′-H), 3.81 (s, 3H, CH3O), 3.57–3.62 (m, 1H, 3-H), 3.57 (dd, 1H, J 6.4 and 10.8, 1″-Ha), 3.49 (dd, 1H, J 5.4 and 10.8, 1″-Hb), 3.33 (td, 1H, J 7.5 and 15.0, 8-Ha), 3.20 (td, 1H, J 7.3 and 15.0, 8-Hb), 1.93–2.10 (m, 2H, 3′-H), 1.93–2.09 (m, 2H, 4-H), 1.82–1.96 (m, 2H, 2-H), 1.72–1.82 (m, 2H, 7-H), 1.55–1.70 (m, 2H, 1-H), 1.46–1.55 (m, 1H, 5-H), 1.07 (s, 9H, (CH3)3CSi) and 0.84 (d, 3H, J 6.8, CH3); δC (100 MHz; CDCl3) 158.9 (C, Carom(PMBn)), 138.3 (C, Carom(PMBn)), 136.1, 136.0 (4 × CH, PhSi), 134.5 (C, PhSi), 134.0 (C, PhSi), 130.6 (C, Carom(Bn)), 130.2 (CH, Carom(Bn)), 129.6 (2 × CH, PhSi), 129.5 (CH, C-5′), 129.1 (2 × CH, Carom(PMBn)), 128.3 (2 × CH, Carom(Bn)), 127.7 (CH, Carom(Bn)), 127.6 (CH, Carom(Bn)), 127.5, (CH, PhSi), 127.4 (CH, PhSi), 124.2 (CH, C-4′), 113.6 (2 × CH, Carom(PMBn)), 75.3 (CH, C-3), 74.8 (CH, C-6), 73.4 (CH2, CH2Bn), 73.3 (CH, C-6′), 73.0 (CH2, C-1″), 72.2 (CH2, CH2PMBn), 69.3 (CH, C-2′), 67.3 (CH2, C-8), 55.2 (CH3, CH3O), 40.5 (CH2, C-4), 35.0 (CH, C-5), 34.4 (CH2, C-2), 34.2 (CH2, C-7), 31.8 (CH2, C-3′), 27.6 (CH2, C-1), 27.1 (3 × CH3, (CH3)3CSi), 19.5 (C, (CH3)3CSi) and 14.7 (CH3, CH3); MS (FAB) m/z (%) 737 (0.5, [M + H]+), 481 (0.5), 359 (1), 197 (4), 135 (11), 121 (100), 105 (2) and 91 (19, [Bn]+); HRMS (CI) m/z 737.4237 calcd for C46H61O6Si 737.4235.
Spectroscopic data for more polar diastereomer 21b. [α]20589 −1 (c = 0.15, CHCl3); υmax (film)/cm−1 3435, 3030, 2930, 2855, 1960, 1890, 1830, 1610, 1590, 1515, 1455, 1430, 1365, 1300, 1170, 1110, 1040, 820, 740, 700, 615; δH (400 MHz; CDCl3) 7.65–7.69 (m, 4H, PhSi), 7.26–7.43 (m, 11H, PhSi, ArH), 7.09 (d, 2H, J 8.6, ArH), 6.83 (d, 2H, J 8.6, ArH), 5.83 (m, 1H, 5′-H), 5.58 (bd, 1H, J 10.3, 4′-H), 4.62 (d, 1H, J 12.2, CH2Bn), 4.55 (d, 1H, J 12.2, CH2Bn), 4.15–4.18 (m, 1H, 6′-H), 4.13 (s, 2H, CH2PMBn), 3.81–3.89 (m, 2H, 6-H, 2′-H), 3.80 (s, 3H, CH3O), 3.55 (dd, 1H, J 6.4 and 10.2, 1″-Ha), 3.44 (dd, 1H, J 6.0 and 10.2, 1″-Hb), 3.40–3.47 (m, 1H, 3-H), 3.28 (td, 1H, J 6.8 and 9.3, 8-Ha), 3.17 (td, 1H, J 7.0 and 9.3, 8-Hb), 2.63 (d, 1H, J 3.8, OH), 1.90–1.97 (m, 1H, 3′-Ha), 2.01–2.11 (m, 1H, 3′-Hb), 1.73–1.78 (m, 2H, 7-H), 1.23–1.69 (m, 7H, 1-H, 2-H, 4-H, 5-H), 1.05 (s, 9H, (CH3)3CSi) and 0.90 (d, 3H, J 6.9, CH3); δC (100 MHz; CDCl3) 158.9 (C, Carom(PMBn)), 138.3 (Ct, Carom(PMBn)), 136.0 (CH, PhSi), 135.9 (CH, PhSi), 134.7 (C, PhSi), 133.9 (C, PhSi), 130.5 (C, Carom(Bn)), 130.0 (CH, Carom(Bn)), 129.6 (CH, PhSi), 129.4 (CH, PhSi), 129.1 (2 × CH, Carom(PMBn)), 128.3 (2 × CH, Carom(Bn)), 127.7 (2 × CH, Carom(Bn)), 127.6 (CH, C-4′), 127.5 (2 × CH, PhSi), 127.4 (2 × CH, PhSi), 124.4 (CH, C-5′), 113.6 (2 × CH, Carom(PMBn)), 74.7 (CH, C-3), 73.6 (CH, C-6), 73.4 (CH2, CH2Bn), 73.3 (CH, C-6′), 73.0 (CH2, C-1″), 72.1 (CH2, CH2PMBn), 69.9 (CH, C-2′), 67.3 (CH2, C-8), 55.2 (CH3, CH3O), 40.0 (CH2, C-4), 34.8 (CH, C-5), 33.6 (CH2, C-2), 33.0 (CH2, C-7), 31.7 (CH2, C-3′), 27.6 (CH2, C-1), 27.1 (CH3, (CH3)3CSi), 19.5 (C, (CH3)3CSi) and 14.7 (CH3, CH3); MS (FAB) m/z 737 (0.8, [M + H]+), 481 (0.5), 359 (1), 197 (5), 135 (11), 121 (100, [PMBn]+) and 91 (26). HRMS (CI) m/z 737.4256 (calcd for C46H61O6Si 737.42352).
Spectroscopic data for less polar diastereomer 23a. [α]20589 −32 (c = 0.18, CHCl3); υmax (film)/cm−1 3450, 2860, 1720, 1610, 1510, 1455, 1360, 1300, 1250, 1090, 1030, 990, 870, 820, 740, 700; δH (400 MHz; CDCl3) 7.24–7.34 (m, 5H, ArH), 7.23 (d, 2H, J 8.7, ArH), 6.85 (d, 2H, J 8.7, ArH), 5.95 (ddd, 1H, J 1.9, 5.6 and 9.8, 9-H), 5.62 (ddd, 1H, J 1.3, 2.6 and 9.8, 10-H), 4.60 (d, 1H, J 12.2, CH2Bn), 4.56 (d, 1H, J 12.2, CH2Bn), 4.43 (s, 2H, CH2(PMBn)), 4.28 (dtd, 1H, J 3.5, 7.2 and 10.3, 2-H), 4.12–4.19 (m, 1H, 7-H), 3.77 (s, 3H, CH3O), 3.73 (dt, 1H, J 2.5 and 9.9, 3′-H), 3.57–3.78 (m, 2H, 5′-H), 3.56 (dd, 1H, J 5.7 and 10.5, 1″-Ha), 3.50 (dd, 1H, J 4.3 and 10.5, 1″-Hb), 1.94–2.03 (m, 2H, 4-H), 1.78 (m, 1H, 2′-H), 1.69–2.09 (m, 2H, 8-H), 1.66–1.78 (m, 2H, 4′-H), 1.54–2.22 (m, 2H, 3-H), 1.46–1.69 (m, 2H, 1′-H) and 0.90 (d, 3H, J 6.8, CH3); δC (100 MHz; CDCl3) 159.1 (C, Carom(PMBn)), 138.4 (C, Carom(PMBn)), 130.2 (C, Carom(Bn)), 129.2 (2 × CH, Carom(PMBn)), 128.6 (CH, C-10), 128.2 (2 × CH, Carom(Bn)), 128.0 (CH, C-9), 127.4 (2 × CH, Carom(Bn)), 127.3 (CH, Carom), 113.7 (2 × CH, Carom(PMBn)), 103.6 (C, C-5), 76.6 (CH, C-2), 73.6 (CH, C-3′), 73.0 (CH2, CH2(Bn)), 72.8 (CH2, CH2(PMBn)), 72.5 (CH2, C-1″), 69.1 (CH2, C-5′), 67.9 (CH, C-7), 55.1 (CH3, CH3O), 39.2 (CH2, C-1′), 37.2 (CH2, C-4), 36.4 (CH, C-2′), 33.3 (CH2, C-4′), 31.3 (CH2, C-3), 26.7 (CH2, C-8) and 14.3 (CH3, CH3); MS (EI) m/z (%) 496 (1, [M]+), 478 (1, [M − H2O]+), 425 (2), 384 (5), 320 (12), 307 (9), 266 (4), 199 (3), 157 (6), 121 (100) and 91 (56); HRMS (EI) m/z 496.2819 (calcd for C30H40O6 496.2825).
Spectroscopic data for more polar diastereomer 23b. [α]20589 −34 (c = 0.07, CHCl3); υmax (film)/cm−1 3450, 2860, 1720, 1610, 1510, 1455, 1360, 1300, 1250, 1090, 1030, 990, 870, 820, 740, 700; δH (400 MHz; CDCl3) 7.26–7.34 (m, 5H, ArH), 7.23 (d, 2H, J 8.5, ArH), 6.85 (d, 2H, J 8.5, ArH), 5.90 (ddd, 1H, J 1.8, 4.4 and 9.9, 9-H), 5.60 (ddd, 1H, J 2.0, 3.0 and 9.9, 10-H), 4.56 (s, 2H, CH2Bn), 4.41 (s, 2H, CH2(PMBn)), 4.24–4.30 (m, 1H, 2-H), 4.15–4.21 (m, 1H, 7-H), 3.77 (s, 3H, CH3O), 3.73 (dt, 1H, J 2.6 and 10.1, 3′-H), 3.55–3.68 (m, 2H, 5′-H), 3.54 (dd, 1H, J 5.8 and 10.5, 1″-Ha), 3.48 (dd, 1H, J 5.0 and 10.5, 1″-Hb), 1.96–2.07 (each m, 2H, 8-H), 1.90–2.09 (each m, 2H, 3-H), 1.85–2.09 (each m, 2H, 4-H), 1.70–1.88 (each m, 2H, 1′-H), 1.69 (m, 1H, 2′-H), 1.63–1.77 (each m, 2H, 4′-H) and 0.90 (d, 3H, J 6.5, CH3); δC (100 MHz; CDCl3) 159.1 (C, Carom(PMBn)), 138.4 (C, Carom(PMBn)), 130.1 (C, Carom(Bn)), 129.2 (2 × CH, Carom(PMBn)), 128.5 (CH, C-9), 128.3 (CH, C-10), 128.2 (2 × CH, Carom(Bn)), 127.4 (2 × CH, Carom(Bn)), 127.3 (CH, Carom), 113.7 (2 × CH, Carom(PMBn)), 103.4 (C, C-5), 78.5 (CH, C-2), 73.5 (CH, C-3′), 73.0 (CH2, CH2(Bn)), 72.8 (CH2, CH2(PMBn)), 72.6 (CH2, C-1″), 69.3 (CH2, C-5′), 67.2 (CH, C-7), 55.1 (CH3, CH3O), 41.7 (CH2, C-1′), 38.2 (CH2, C-4), 36.2 (CH, C-2′), 33.7 (CH2, C-4′), 30.9 (CH2, C-3), 26.8 (CH2, C-8) and 13.8 (CH3, CH3); MS (FAB) m/z (%) 497 (12, [M + H]+), 479 (8, [M + H − H2O]+), 357 (2), 121 (100), 91 (39, [Bn]+); HRMS (FAB) m/z 497.2895 (calcd for C30H41O6 497.2903).
Spectroscopic data for major diastereomer 9a. [α]20589 − 23 (c = 0.25, CHCl3); υmax (film)/cm−1 3495, 2930, 2860, 2060, 1880, 1735, 1655, 1610, 1585, 1515, 1500, 1455, 1300, 1245, 1205, 1175, 1095, 1035, 1005, 980, 875, 820, 735, 700; δH (600 MHz; CDCl3) 7.26–7.35 (m, 5H, ArH), 7.26 (d, 2H, J 8.5, ArH), 6.87 (d, 2H, J 8.5, ArH), 5.97 (ddd, 1H, J 2.2, 5.5 and 9.9, 11-H), 5.70 (brd, 1H, J 9.9, 12-H), 4.59 (s, 2H, CH2Ph), 4.35 (s, 3H, CH2(PMBn)), 4.14–4.25 (m, 2H, 2-H, 9-H), 3.80 (s, 3H, CH3O), 3.38–3.64 (m, 4H, 2′-H, 1″-H), 2.50 (dq, 1H, J 14.1 and 7.4, 3-H), 2.28–2.40 (m, 2H, 4-Ha, 13-Ha), 1.89–2.11 (m, 5H, 13-Hb, 14-H, 10-H), 1.62–1.70 (m, 3H, 1′-H, 4-Hb) and 0.90 (d, 3H, J 7.0, CH3); δH (100 MHz; CDCl3) 159.0 (C, Carom(PMBn)), 138.5 (C, Carom(PMBn)), 130.8 (C, Carom(Bn)), 129.3 (2 × CH, Carom(PMBn)), 129.2 (CH, C12), 128.3 (2 × CH, Carom(Bn)), 128.1 (CH, C-11), 127.5 (3 × CH, Carom(Bn)), 114.6 (C, C-5), 113.7 (2 × CH, Carom(PMBn)), 103.4 (C, C-7), 78.6 (CH, C-2), 73.1 (CH2, CH2Ph), 72.8 (CH2, CH2Ar), 72.7 (CH2, C-1″), 68.0 (CH, C-9), 67.8 (CH2, C-2′), 55.3 (CH3, CH3O), 45.5 (CH2, C-4), 35.6, 36.9 (each CH2, C-13, C-14), 34.5 (CH, C-3), 31.1 (CH2, C-1′), 26.8 (CH2, C-10), 14.3 (CH3, CH3); MS (EI) m/z (%) 494 (1, [M]+), 477 (9, [M − OH]+), 476 (25, [M − H2O]+), 403 (1, [M − Bn]+), 485 (1, [M − Bn − H2O]+), 373 (8, [M − PMBn]+), 358 (8), 355 (3, [M − PMBn − H2O]+), 229 (6), 203 (6), 157 (11), 137 (10), 121 (100), 91 (65); HRMS (EI) m/z 494.2668 (calcd for C30H38O6 494.26684).
Spectroscopic data for minor diastereomer 9b. δH (600 MHz; CDCl3) 7.28–7.32 (m, 5H, ArH), 7.22 (d, 2H, J 8.5, ArH), 6.83 (d, 2H, J 8.5, ArH), 5.92–5.96 (m, 1H, 11-H), 5.58 (brd, 1H, J 9.9, 12-H), 4.44 (s, 2H, CH2Ph), 4.30 (s, 2H, CH2(PMBn)), 4.10–4.16 (m, 2H, 2-H, 9-H), 3.79 (s, 3H, CH3O), 3.38–3.42 (m, 4H, 2′-H, 1″-H), 2.50 (m, 1H, 3-H), 2.20–2.33 (m, 2H, 4-Ha, 13-Ha), 1.72–2.01 (m, 5H, 13-Hb, 14-H, 10-H), 1.61–1.69 (m, 3H, 1′-H, 4-Hb) and 0.89 (m, 3H, CH3).
Acknowledgements
The authors would like to thank the Swiss National Science Foundation and the Royal Society of New Zealand Marsden Fund for financial support, Michael Walker for helpful discussions concerning NMR experiments and L. Ravi Sumoreeah for preliminary experimentation on the silyl-modified Prins cyclization.References
- T. Hu, J. M. Curtis, Y. Oshima, J. A. Walter, W. M. Watson-Wright and J. L. Wright, J. Chem. Soc., Chem. Commun., 1995, 2159 RSC.
- T. Hu, J. M. Curtis, J. A. Walter and J. L. C. Wright, Tetrahedron Lett., 1996, 37, 7671–7674 CrossRef CAS.
- T. Hu, I. W. Burton, A. D. Cembella, J. M. Curtis, M. A. Quilliam, J. A. Walter and J. L. C. Wright, J. Nat. Prod., 2001, 64, 308 CrossRef CAS.
- D. Uemura, T. Chuo, T. Haino, A. Nagatsu, S. Fukuzawa, S. Zheng and H. Chen, J. Am. Chem. Soc., 1995, 117, 1155 CrossRef CAS.
- T. Chou, O. Kamo and D. Uemura, Tetrahedron Lett., 1996, 37, 4023–4026 CrossRef CAS.
- M. Falk, I. W. Burton, T. Hu, J. A. Walter and J. L. C. Wright, Tetrahedron, 2001, 57, 8659 CrossRef CAS.
- S. Gill, M. Murphy, J. Clausen, D. Richard, M. Quilliam, S. MacKinnon, P. LaBlanc, R. Mueller and O. Pulido, Neurotoxicology, 2003, 24, 593 CrossRef CAS.
- J. A. McCauley, K. Nagasawa, P. A. Lander, S. G. Mischke, M. A. Semones and Y. Kishi, J. Am. Chem. Soc., 1998, 120, 7647 CrossRef CAS.
- J. Ishihara, T. Ishizaka, T. Suzuki and S. Hatakeyama, Tetrahedron Lett., 2004, 45, 7855 CrossRef CAS.
- For a review on the synthesis of bis-spiroacetal-containing natural products see: M. A. Brimble and D. P. Furkert, Curr. Org. Chem., 2003, 7, 1461 Search PubMed.
- For an example of our previous work in this area see: M. A. Brimble, P. R. Allen and H. Prabaharan, J. Chem. Soc., Perkin Trans. I, 2001, 379 Search PubMed.
- M. A. Brimble and M. Trzoss, Tetrahedron, 2004, 60, 5613 CrossRef CAS.
- M. A. Brimble and D. P. Furkert, Org. Biomol. Chem., 2004, 2, 3573 RSC.
- For an earlier communication on this work see: K. Meilert and M. A. Brimble, Org. Lett., 2005, 7, 3497 Search PubMed.
- (a) K. Takai, K. Kimura, T. Kuroda, T. Hiyama and H. Nozaki, Tetrahedron Lett., 1983, 24, 5281 CrossRef CAS; (b) H. Jin, J. Uenishi, W. J. Christ and Y. Kishi, J. Am. Chem. Soc., 1986, 108, 5644 CrossRef CAS; K. Takai, M. Tagashira, T. Kuroda, K. Oshima, K. Utimoto and H. Nozaki, J. Am. Chem. Soc., 1986, 108, 6048 CrossRef CAS; (c) D. P. Stamos, X. C. Sheng, S. S. Chen and Y. Kishi, Tetrahedron Lett., 1997, 38, 6355 CrossRef CAS.
- C. De Lima, M. Julia and J. N. Vepeaux, Synlett, 1992, 133 CrossRef CAS.
- M. T. Lai, E. Oh, Y. Shih and H.-W. Liu, J. Org. Chem., 1992, 57, 2471 CrossRef CAS.
- T. Ooi, N. Kagoshima, H. Ichikawa and K. Maruoka, J. Am. Chem. Soc., 1999, 121, 3328 CrossRef CAS.
- Y. Ichikawa, M. Isobe, D.-L. Bai and T. Goto, Tetrahedron, 1987, 43, 4737 CrossRef CAS.
- (a) J. A. Soderquist and B. Santiago, Tetrahedron Lett., 1990, 31, 5113 CrossRef CAS; (b) M. C. McIntosh and S. M. Weinreb, J. Org. Chem., 1993, 58, 4823 CrossRef CAS; (c) B. M. Trost and R. Braslau, Tetrahedron Lett., 1989, 30, 4657 CrossRef CAS; (d) A. D. Kini, D. V. Nadkarni and J. L. Fry, Tetrahedron Lett., 1994, 35, 1507 CrossRef CAS.
- I. E. Marko and D. J. Bayston, Tetrahedron, 1994, 50, 7141 CrossRef CAS.
- The silyl-modified Prins cyclization and the silyl-modified Sakurai cyclization are in fact the same reaction and the name silyl Prins is adopted herein. See: (a) I. E. Marko, A. Mekhalfia, D. J. Bayston and H. Adams, J. Org. Chem., 1992, 57, 2211 CrossRef CAS; (b) A. P. Dobbs and S. Martinovic, Tetrahedron Lett., 2002, 43, 7055 CrossRef CAS; (c) A. P. Dobbs, S. J. J. Guesne, S. Martinovic, S. J. Coles and M. B. Hursthouse, J. Org. Chem., 2003, 68, 7880 CrossRef CAS; (d) P. O. Miranda, D. D. Diaz, J. I. Padron, J. Bermejo and V. S. Martin, Org. Lett., 2003, 5, 1979 CrossRef CAS.
- F. Coelho and G. Diaz, Tetrahedron, 2002, 58, 1647 CrossRef CAS.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 889 CrossRef CAS.
- H. C. Brown and K. S. Bhat, J. Am. Chem. Soc., 1986, 108, 293 CrossRef CAS.
- K. T. Meilert, G. R. Clark, T. Groutso and M. A. Brimble, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, E61, O6 CAS.
- P. De Armas, C. G. Francisco and E. Suarez, Angew. Chem., Int. Ed. Engl., 1992, 31, 772 CrossRef.
- H. C. Brown, J. Org. Chem., 1981, 54, 3987.
- D. H. B. Ripin, W. Cai and S. J. Brenek, Tetrahedron Lett., 2000, 41, 5817 CrossRef CAS.
- G. W. Kabalka, T. M. Shoup and N. M. Goudgaon, J. Org. Chem., 1989, 54, 5930 CrossRef CAS.
- For examples of heteroatom directed Wacker oxidations see: (a) J. Tsuji, Synthesis, 1984, 5, 369 CrossRef; (b) S. K. Kang, K. Y. Jung, J. U. Chung, E. Y. Namkoong and T. H. Kim, J. Org. Chem., 1995, 60, 4678 CrossRef CAS; (c) J. Y. Lai, X. X. Shi and L. X. Dai, J. Org. Chem., 1992, 57, 3485 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details together with full experimental procedures, 1H NMR, 13C NMR and mass spectral data for compounds 10–14 and 17–20. See DOI: 10.1039/b604334h |
| This journal is © The Royal Society of Chemistry 2006 |