Raman scattering and FT-IR spectroscopic studies on dithienylethene switches—towards non-destructive optical readout†
Jaap J. D.
de Jong
a,
Wesley R.
Browne
a,
Martin
Walko
b,
Linda N.
Lucas
a,
Lindsay J.
Barrett
c,
John J.
McGarvey
c,
Jan H.
van Esch
a and
Ben L.
Feringa
*a
aOrganic and Molecular Inorganic Chemistry, Stratingh Institute, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: B.L.Feringa@rug.nl; Fax: +31 50 3634296; Tel: +31 50 3634235
bInstitute of Chemistry, Faculty of Science, P. J. Safarik University in Kosice, Moyzesova 11, Kosice, 04001, Slovakia
cSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, Northern Ireland, UK BT5 9AG
First published on 22nd May 2006
Abstract
The non-destructive readout of photochromic memory materials based on the dithienylethene unit both by IR spectroscopy and Raman scattering is explored. A representative series of C5-substituted thienyl hexahydro- and hexafluoro-cyclopentene based photochromes was investigated to explore the effect and potential usefulness of substitution for the development of multicomponent memory materials. The effect of the deposition method on the photochemistry of solid materials containing photochromic dithienylcyclopentene switches was also explored. Photoconversion in the solid state to the closed form was found to be low when starting from the open form, but, in contrast, ring opening to the open state from the closed form was found to be complete. The effect was found to be due to inner filter rather than conformational phenomena. Characteristic vibrational bands for the central dithienyl core are assigned and a comparison made of the vibrational spectroscopic properties of the perhydro- and perfluoro switches. The data enable the determination of the photoconversion achievable in the solid state as well as some assessment of the influence of the deposition method on the photoconversion. The potential of Raman spectroscopy as a method of achieving non-destructive optical readout is demonstrated through the large differences in absolute Raman scattering intensity between the open and closed states, when monitored at wavelengths which do not result in photochemical ring opening.
Introduction
Photo- and electro-chromic switches, and especially dithienylethene switches,1 continue to receive considerable attention in the development of functional addressable molecular materials, which respond to non-destructive external stimuli, e.g. electrochemical potential changes and light, and are of particular interest in areas such as electrochromic displays and memory devices.2 An essential prerequisite to the successful application of organic materials in memory devices is the achievement of non-destructive readout.1 Several approaches based on the use of IR spectroscopy, low intensity optical methods (which minimise rather than avoid ring opening) or the use of materials with low photochemical quantum yields have been explored in an effort to achieve non-destructive readout.3In recent years, dithienylethene based photochromic switches have been shown to be particularly attractive as candidates for application in such devices.1 Dithienylethene based switches exist in two photochemically active states, i.e. the open and closed states (Fig. 1). Although, the open and closed states show large differences in their UV–vis spectra (Fig. 1), the use of visible detection is often unsuitable for non-destructive readout as visible irradiation results in ring opening. In contrast to optical detection in the visible range, IR spectroscopy has proven useful in achieving non-destructive readout,4 as demonstrated with a recent example of a three bit eight state system.5 Despite this recent progress in the application of vibrational spectroscopy as a non-destructive readout method,6 a general examination of the vibrational properties essential in systematically tuning vibrational structure has not been undertaken so far.
 | ||
| Fig. 1 Dithienylcyclopentene switches described in the text: suffix o denotes open form, c denotes closed form, H and F denote hexahydrocyclopentene and hexafluorocyclopentene based compounds respectively. Th denotes thiophene, Ph denotes phenyl group with all substituents on the para position. | ||
Raman spectroscopy offers an alternative to FTIR spectroscopy as a technique for non-destructive readout, due to its quite different technical requirements and its use of visible to near-IR lasers and CCD detectors,7 but its application has so far received relatively little attention.8,9 Since the use of visible excitation may result in photoconversion during the measurement, only excitation at wavelengths longer than the lowest energy absorption bands of the closed form are useful in achieving non-destructive optical readout.8b
Recently we reported the synthesis,10 photo- and electrochemical properties11 of perfluorocyclopentene (1–4F) and the perhydrocyclopentene (1–12H) based switches (Fig. 1). In the present contribution, we report an extensive investigation of the IR and Raman vibrational structure of dithienylcyclopentene based switches. The effect of substitution of the perhydrocyclopentene unit for a perfluorocyclopentene unit and substitution at the C5 position of the thienyl rings on both the IR and Raman spectra in the open and closed states are reported. The relative merits of Raman scattering and IR spectroscopy for non-destructive readout are assessed.
Experimental
Materials
All solvents employed were of UVASOL grade or better and used as received unless stated otherwise. All reagents employed in synthetic procedures were of reagent grade or better and were also used as received unless stated otherwise. Compounds 1–4F and 1–12H were prepared by literature procedures.10b,11b,12 FTIR spectra were recorded (as intimate mixtures in KBr) in reflectance mode using a Nicolet Nexus FTIR spectrometer. Raman spectra were recorded at an excitation wavelength, λexc, of 785 nm on quartz slides or in quartz cuvettes on an Avalon R1-ST Ramanstation. Spectral acquisition and processing was performed using GramsAI software.Computational methods
Density functional theory (DFT) calculations were carried out with the GAUSSIAN 03 W (rev. B.04) program package.13 All the calculations were performed on systems in the gas phase using the Becke's three-parameter hybrid functional14 with the LYP correlation functional15 (B3LYP) and 6–31(g)d basis set. The geometry optimization was followed by the frequency calculation to prove that the energy minimum was found. The Raman frequencies calculated on the optimized geometry were scaled by 0.9614.16Results and discussion
The FTIR spectra of 1–12H and 1–4F were recorded in KBr powder in the range 4000 to 500 cm−1 (see ESI†). The FTIR spectra of 2–4F for both the open and closed forms are in close agreement with spectra reported previously by Zerbi, Uchida and co-workers.17 Comparison of the spectra of 1–4Ho with those of 1–4Fo (Fig. 2 and ESI†) shows a remarkably small influence of the cyclopentene unit on the vibrational structure of the core dithienylethene vibrations and the vibrations of the C5 substituents (1450–1650 cm−1). The C–H stretching region (2700–3200 cm−1) is largely unaffected except for a decrease in aliphatic C–H vibrations of the cyclopentene unit at 2842 cm−1. In the low wavenumber region (650–800 cm−1) for the aromatic C5 substituted compounds (2–4Ho, and 2–4Fo) the aromatic o.o.p. bending vibrations remain clear of interfering cyclopentene vibrations. The most distinctive difference in the spectra between the perhydro- and hexafluoro-cyclopentene compounds is observed in the fingerprint region (800–1400 cm−1). The strong C–F vibrations (see Fig. 2) obscure the vibrations of the central dithienylethene core, rendering them unavailable for use in monitoring ring opening/closing reactions (vide infra).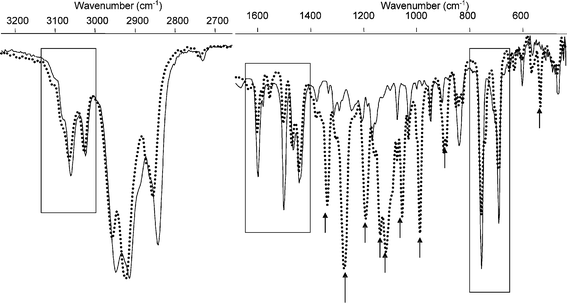 | ||
| Fig. 2 Overlay of IR spectra of 2Ho (solid line) and 2Fo (dotted line), deposited from an acetonitrile solution onto KBr powder. The main vibrations arising from the hexafluoro-cyclopentene ring are noted by black arrows. | ||
Upon irradiation of the open form (1–12Ho and 1–4Fo) in CH3CN solution with UV light (λ = 313 nm) photocyclisation to the closed form (1–4Hc and 1–4Fc) occurs.10,11 The closed forms were prepared by irradiation in solution prior to the deposition onto the KBr powder (Fig. 3). For 2–4F, the spectra obtained closely match those reported in earlier studies (see ESI†).17
 | ||
| Fig. 3 Overlay of IR spectra of 4Hc (dotted lines) with: upper set—4Ho (solid) and lower set—4Fc (solid) deposited from an acetonitrile solution onto KBr powder. | ||
Comparison of the IR spectra of 1–4F with 1–4H indicates that the absorption at ∼1440 cm−1 is common and is associated with the central cis-bis(trans-butadiene) core of the closed dithienylethene unit. For 1H/1F, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations of the aldehyde are red-shifted and become narrower upon ring closure. As for the open forms, the C–F absorptions (1400–800 cm−1, see Fig. 2) in the fingerprint region obscure the absorption of the dithienylethene core, however, the C–F absorptions undergo considerable changes upon ring closure. In particular the changes in the 900 to 1100 cm−1 region are quite pronounced for 1–4F compared with 1–4H.
O vibrations of the aldehyde are red-shifted and become narrower upon ring closure. As for the open forms, the C–F absorptions (1400–800 cm−1, see Fig. 2) in the fingerprint region obscure the absorption of the dithienylethene core, however, the C–F absorptions undergo considerable changes upon ring closure. In particular the changes in the 900 to 1100 cm−1 region are quite pronounced for 1–4F compared with 1–4H.
Comparison of the spectra of the open and closed state of the perhydrocyclopentene based switches (1-12H) reveals several similarities and differences (Fig. 3, upper spectrum). In the C–H stretching region (2700–3200 cm−1) ring closure results in minor changes to the bands observed.18 However, in the 600–1600 cm−1 region large changes are observed, in particular in the aromatic o.o.p. bending bands (∼820 cm−1) and the ring breathing vibrations at 1605 cm−1. The red-shift of the thienyl CC stretching vibration8 at 1515 to 1500 cm−1 reflects the formation of the extended cis-bis(trans-butadiene) system (Fig. 1). The changes observed for the vibrational modes assigned to the peripheral aromatic rings (2–7H), in particular the red-shift of the ring breathing vibration at ∼1600 cm−1, indicates a modest increase in communication between the thiophene and phenyl ring systems upon ring closure. The absorptions at 1437, 1375, 1311, 1291, and 918 cm−1 for 1–12Ho and at 1337, 1194, 1137, 1111 cm−1 for 1–4Fo are characteristic of the CC and C–C stretching vibrations of the thienyl groups and those at 1439, 1363, 1309, 1175, and 1140 cm−1 for 1–12Hc and at 1626, 1440, 1373, 1341, 1225, 1187, 1120, 1077, 1032 cm−1 for 1–4Fc are characteristic of the CC and C–C stretching vibrations of the cis-bis-(trans-butadiene) systems. However, despite these general similarities, it is clear that in the fingerprint region there are significant differences between the variously substituted switches.
Comparison of substituent effects on IR spectra
Although substitution of the perhydro-cyclopentene unit of the dithienylethenes by a hexafluoro-cyclopentene unit results in quite a dramatic difference to the IR spectrum in the 1000–1400 cm−1 fingerprint region due to the appearance of C–F vibrations, the effect of such a substitution on the absorptions associated with the dithienylethene core (in particular in the 1400 to 1650 cm−1 region) appears to be slight (Fig. 2 and 3). In contrast, substitution in the C5 position of the thienyl rings leads to quite marked changes in this latter region. For 1–12Ho 1437, 1375, 1311, 1291, and 918 cm−1 and for 1–12Hc 1439, 1363, 1309, 1175, and 1140 cm−1 are characteristic IR absorption bands, which show minimal sensitivity to substitution. Previous reports17c suggested that the spectra of 2–4F are similar in the 900–1250 cm−1 region (e.g. o.o.p. bending vibrations of the substituted phenyl rings), however, from Fig. 3, 4 and 5 it is evident that there are significant differences, most notably in the region of 950–1000 cm−1. The relative insensitivity of vibrational modes of substituent groups to ring opening/closing (e.g. the CO vibrations of 1F/H, 11Hetc.) may be useful in acting as internal reference points both in single and multicomponent memory systems.
 | ||
| Fig. 4 Overlay of FTIR spectra of 2–6Ho recorded on KBr. | ||
 | ||
| Fig. 5 IR spectra of 11Ho (green) on KBr irradiated at 313 nm to 11Hc (red) followed by >520 nm irradiation (4 complete opening/closing cycles). The CO stretching vibration is convenient for internal referencing of intensity changes. | ||
Photochemical conversion in the solid state; effect of deposition method
Although photoconversion in solution between the open and closed forms is efficient (in terms of the photostationary state {PSS}, which is achievable), photoconversion of solid samples is not straightforward. The IR spectra of the open and closed forms can be obtained by first irradiating a solution of the compound at the appropriate wavelength (vide supra), followed by deposition onto KBr powder. The spectra of the open and closed forms obtained in this manner enable an estimation of the PSS achieved upon irradiation of the solid samples.19 Ring closure of the open form of 11Ho results in poor conversion (<50%),20 however, the reversibility of the conversion is good, with minimal degradation over at least 4 ring opening/closing cycles (Fig. 5). When the same process is performed starting with the closed form, high conversion to the ring open form can be achieved upon visible irradiation. (Fig. 6). However, subsequent ring closure occurred with poor photoconversion.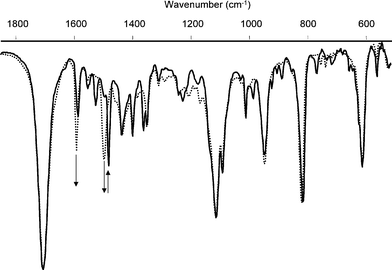 | ||
| Fig. 6 IR spectra of 6Hc (solid line) on KBr irradiated at >520 nm to 6Ho (dotted line). The CO stretching vibration (1705 cm−1) allows for internal referencing of intensity changes. | ||
The origin of the difference in photoconversion being dependent on whether the open or closed form is deposited may arise from the presence of a photochemically inactive conformation of the open form. The open form exists in solution as a dynamic equilibrium between the parallel and anti-parallel states with the parallel state being photochemically inactive.1 In solution, rapid equilibration allows complete photoconversion to be achieved; however, in the solid state where interconversion can be inhibited, only the anti-parallel form may be available to undergo photoconversion. For the closed form, ring opening in the solid state will invariably lead to the anti-parallel conformation of the open form and hence subsequent ring closure would be expected to occur with high photoconversion. However, since complete photoconversion to the ring open form (in the photochemically active conformation only) can be achieved from the closed form, the poor photoconversion observed for ring closure (regardless of whether the open or closed form was deposited initially) may be due to inner filter effects rather than conformational effects in the solid state. That this is the case is confirmed by deposition of either the open or closed forms as a thin layer. Under these conditions full ring opening and closing is achieved, demonstrating that for instances where full photoconversion is not observed, optical rather than conformational effects are at work.
Raman spectroscopy
The solid state Raman spectra of the open forms of the dithienylethene switches were recorded at an excitation wavelength of 785 nm. In contrast to the situation with the IR spectra, substitution of the central cyclopentene unit has little effect on the Raman spectra of the switches, in particular in the 1000–1400 cm−1 region (Fig. 7). The C–F vibrations of the hexafluorocyclopentene unit (e.g. ∼1280, 1050, 750 cm−1) are weak compared with the alkene and aromatic absorptions in the 1400–1650 cm−1 region. Similarly the CC vibrational modes responsible for the absorptions in this latter region are relatively insensitive to the cyclopentene unit, with the notable exception of an increase in the relative intensity of the ∼1620 cm−1 band.
 | ||
| Fig. 7 Solid state Raman spectra of 2Ho, 2Fo, 3Ho and 3Fo (λexc = 785 nm) as neat powders. | ||
Comparison of 2Ho (R = Ph), 2Fo (R = Ph), 9Ho (R = Cl) and 10Ho (R = Me) enables assignment of the major peaks in the 1400–1700 cm−1 region (Fig. 8). The dithienylethene based absorption at ∼1636 cm−1, which is weak in the IR spectrum, is quite strong in the Raman spectra. Its intensity and energy are relatively sensitive to both the substituent in the C5 position (Δ = 7 cm−1) and to substitution of the perhydrocyclopentene unit for a perfluorocyclopentene unit. The sensitivity of the 1636 cm−1 band to the cyclopentene unit is in stark contrast to the 1599–43, 1443–37 and 1466–53 cm−1 bands, which are sensitive to C5 substitution but insensitive to the nature of the cyclopentene unit.
 | ||
| Fig. 8 Solid state Raman spectra of 2Fo, 2Ho, 10Ho and 9Ho (λexc = 785 nm) as neat powders. | ||
For the phenyl substituted compounds 2–7Ho and 2–4Fo, the aromatic ring breathing bands are sensitive to substitution and range from 1607 cm−1 for 4Fo to 1583 cm−1 for 7Ho. By contrast, substitution of the phenyl rings leads to a dramatic difference in the relative intensity of the features in the 1400–1650 cm−1 region (Fig. 7 and 9). Although the effect of variation of substituents in the C5 position can be quite pronounced, the differences are largely confined to the relative band intensities (especially for the C5 X-phenyl substituted compounds, Fig. 9) with the band at ∼1590 cm−1 undergoing a red-shift on increasing the electron withdrawing strength of the substituent.
 | ||
| Fig. 9 Solid state Raman spectra of 2–4Ho, 6Ho and 7Ho (λexc = 785 nm). | ||
For all the dithienylethene switches examined no significant absorption by the closed form is observed at 785 nm, making this wavelength suitable for non-destructive analysis of the open and closed forms (see Fig. 10, Fig. 11 and ESI†). The Raman spectra recorded at 785 nm for 2Ho and 2Hc are presented in Fig. 10. As for the IR spectra, ring closing of the open form results in a dramatic change in the Raman spectra. In addition, a large increase in intensity of bands associated with the dithienylethene core, assigned to the extended conjugated cis-bis(trans-butadiene) unit, is observed (see Fig. 12).21 The very high intensity of this band, relative to the Raman intensity of vibrations associated with the substituents in the C5 position (i.e. aromatic ring breathing at ∼1600 cm−1) and that of the open form, may be attributable to the high polarisability of the extended system.22 Comparison of the Raman spectra obtained in the solid state with those obtained in dilute solution (see ESI Fig. 31†) confirms that other enhancement mechanisms possibly related to the KBr support are not involved.
 | ||
| Fig. 10 Overlay of Raman spectra (λexc 785 nm) of (lower) 2Ho {solid sample} and (upper) 2Hc {deposited from an acetonitrile solution onto KBr powder}. | ||
 | ||
| Fig. 11 Calculated Raman spectra for 2Hc (upper) and 2Ho (lower, ×10). | ||
 | ||
| Fig. 12 Raman spectra of 4Ho (lower spectrum) to 4Hc (upper spectrum)—irradiation at 313 nm, spectra recorded at λexc = 785 nm, 6 × 20 s accumulations. | ||
 | ||
| Fig. 13 Vibrational changes associated with the 1520 cm−1 Raman active mode of 2Hc. | ||
The intense band at 1520 cm−1 is assigned, on the basis of DFT calculations to the extended conjugated polyenic system (Fig. 13). The dramatic increase in scattering intensity is remarkable but is in excellent agreement with the calculated intensity changes.
Thus, Raman spectroscopy offers an alternative approach to IR spectroscopy for the non-destructive readout of photochromic materials. Indeed, in addition to the use of visible/near-infrared laser excitation, the large differences in the Raman scattering cross sections between the open and closed states and the sensitivity of key vibrational modes to peripheral substitution make Raman spectroscopy potentially a more versatile technique than IR spectroscopy. The dramatic changes in the relative intensity of different vibrational features on switching from open to closed forms facilitates global analysis of spectra.
Conclusions
Raman spectroscopy, in particular at excitation wavelengths beyond the lowest energy electronic absorption range, has considerable potential for non-destructive readout studies of photochromic materials, on account of the large intensity differences between the Raman spectra of the open and closed forms. Furthermore, the relatively simple Raman spectra, combined with the sensitivity of core vibrations to substitution, facilitates global analysis procedures. These are distinct advantages in comparison to IR spectroscopy as a non-destructive readout method. Overall however, it is apparent that regardless of the detection method, in the solid state the efficiency of ring opening and closing is critically dependent on the thickness of the deposited layer.Acknowledgements
The authors acknowledge the Materials Science Centre (MSC+) (JJDdJ, MW) and the Dutch Economy, Ecology, Technology (EET) program (WRB) for financial support. We thank Prof. J. C. Hummelen for access to FTIR spectrometry facilities.References
- (a) M. Irie, Chem. Rev., 2000, 100, 1683 CrossRef CAS; (b) Molecular Switches, ed. B. L. Feringa, Wiley VCH, Weinheim, 2001 Search PubMed; (c) H. Tian and S. J. Yang, Chem. Soc. Rev., 2004, 33, 85 RSC.
- Molecular Motors, ed. M. Schliwa, Wiley-VCH, Weinheim, 2003 Search PubMed.
- (a) M. Irie, T. Eriguchi, T. Takada and K. Uchida, Tetrahedron, 1997, 53, 12263 CrossRef CAS; (b) T. Tsuijoka, Y. Hamada and K. Shibata, Appl. Phys. Lett., 2001, 78, 2282 CrossRef CAS.
- (a) K. Uchida, M. Saito, A. Murakami, S. Nakamura and M. Irie, Adv. Mater., 2003, 15, 121 CrossRef CAS; (b) K. Uchida, M. Saito, A. Murakami, S. Nakamura and M. Irie, ChemPhysChem, 2003, 4, 1124 CrossRef CAS.
- K. Uchida, M. Saito, A. Murakami, T. Kobayashi, S. Nakamura and M. Irie, Chem.–Eur. J., 2005, 11, 534 CrossRef CAS.
- D. Majumbar, H. M. Lee, J. Kim and B. J. Mhin, J. Chem. Phys., 1999, 111, 5866 CrossRef.
- S. E. J. Bell, Analyst, 1996, 121, 107R RSC.
- (a) X.-H. Zhou, F. Wei, F.-S. Zhang, F.-Q. Zhao, X.-D. Liu, F.-Y. Xu and C.-H. Tung, J. Photochem. Photobiol., A, 2005, 171, 205 CrossRef CAS; (b) C. Okabe, N. Tanaka, T. Fukaminato, T. Kawai, M. Irie, Y. Nibu, H. Shimada, A. Goldberg, S. Nakamura and H. Sekiya, Chem. Phys. Lett., 2002, 357, 113 CrossRef CAS; (c) C. Bertarelli, M. C. Gallazzi, A. Lucotti and G. Zerbi, Synth. Met., 2003, 139, 933 CrossRef CAS.
- C. Okabe, T. Nakabayashi, N. Nishi, T. Fukaminato, T. Kawai, M. Irie and H. Sekiya, J. Phys. Chem. A, 2003, 107, 5384 CrossRef CAS.
- (a) J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2003, 10, 1887 CrossRef; (b) L. N. Lucas, J. J. D. de Jong, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Eur. J. Org. Chem., 2003, 1, 155 CrossRef.
- (a) W. R. Browne, J. J. D. de Jong, T. Kudernac, M. Walko, L. N. Lucas, K. Uchida, J. H. van Esch and B. L. Feringa, Chem.–Eur. J., 2005, 11, 6414 CrossRef CAS; (b) W. R. Browne, J. J. D. de Jong, T. Kudernac, M. Walko, L. N. Lucas, K. Uchida, J. H. van Esch and B. L. Feringa, Chem.–Eur. J., 2005, 11, 6430 CrossRef CAS.
- L. N. Lucas, J. van Esch, R. M. Kellogg and B. L. Feringa, Chem. Commun., 2001, 759 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.04, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502 CrossRef CAS.
- (a) F. Stellacci, C. Bertarelli, F. Toscano, M. C. Gallazzi and G. Zerbi, Chem. Phys. Lett., 1999, 302, 563 CrossRef CAS; (b) M. Saito, T. Miyata, A. Murakami, S. Nakamura, M. Irie and K. Uchida, Chem. Lett., 2004, 33, 786 CrossRef CAS; (c) A. Bianco, C. Bertarelli, J. F. Rabolt and G. Zerbi, Chem. Mater., 2005, 17, 869 CrossRef CAS.
- The insensitivity of the CH stretching modes to ring closure is in agreement with 1H NMR spectroscopic studies. See ref. 10,11.
- For compounds 9Ho and 10Ho poor photoconversion to the ring closed forms precludes an accurate structural analysis of the closed states.
- The photoconversion during photolysis on KBr powder was determined by comparison with the fully open and closed states deposited directly.
- M. Walko, J. J. D. de Jong, W. R. Browne, F. Hartl, J. J. McGarvey, J. H. van Esch and B. L. Feringa, manuscript in preparation.
- (a) L. C. T. Shoute, M. Blanchard-Desce and A. Myers Kelley, J. Phys. Chem. A, 2005, 109, 10503 CrossRef CAS , and ref. therein; (b) L. D. Ziegler, J. Raman Spectrosc., 1990, 21, 769 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectroscopic data for 1–4F and 1–12H and Raman spectra are provided. See DOI: 10.1039/b603914f |
| This journal is © The Royal Society of Chemistry 2006 |