Anion-templated assembly of [2]rotaxanes
Mark R. Sambrook†, Paul D. Beer*, Michael D. Lankshear, R. Frederick Ludlow and James A. Wisner‡
Department of Chemistry, Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK OX1 3QR. E-mail: paul.beer@chem.ox.ac.uk
First published on 10th March 2006
Abstract
Anion templation is used to develop a general method for rotaxane synthesis. The anion-templated synthesis of three new [2]rotaxanes containing positively charged pyridinium axles and neutral isophthalamide macrocyclic components is described. The incorporation of electron withdrawing substituents, such as the nitro group, into the 5-position of an isophthalamide bis-vinyl acyclic precursor results in a significant improvement in [2]rotaxane assembly yields. Rotaxane anion binding strengths are also enhanced whilst the rotaxane's unique interlocked binding domain ensures selectivity for chloride – the templating anion – is maintained.
Introduction
Over the past couple of decades a number of assembling motifs have been established for the preparation of mechanically bonded supramolecular systems.1 In particular, the use of metal–ligand coordination chemistry,2 hydrogen bonding,3 π–π stacking interactions4 and solvophobic forces5 have become common-place. In spite of their diffuse nature, pH-dependence and relative high solvation energies, the field of anion templation is of intense current interest6 where many serendipitous examples of templated metal-based cages7 and macrocycles8 reveal the anion filling an internal void around which the structure is assembled.9 In sharp contrast, however, the much rarer use of anion templates in assembling interlocked molecules has focused on the use of rigorously designed hydrogen-bond donor anion recognition motifs.10 For example, Vögtle's anion-mediated rotaxane synthesis relies upon the complexation and orientation of a negatively charged phenoxide intermediate by a neutral macrocyclic lactam to form a semi-rotaxane.11 This assembly, shielded on one side by a bulky group, subsequently reacts with a sterically demanding electrophile to form a rotaxane. This imaginative approach has been exploited by Smith's group in the preparation of rotaxanes with ion-pair binding macrocycles and rotaxinated squarine dyes12 and in a related stoppering method of rotaxane formation by Schalley and co-workers.13We have recently reported the use of spherical halide anions as templates for the construction of a number of [2]pseudorotaxane assemblies.14 A chloride anion that is tightly ion-paired to a cationic organic thread, e.g. a pyridinium amide cleft, has been found, in non-competitive solvents, to be coordinatively unsaturated and possess an empty meridian available for further complexation by a second, neutral, anion recognition site.15 Although not possessing the stereochemical preferences inherent in a transition metal centre, sterics force an orthogonal arrangement of the two ligands in a manner analogous to, for example, a [Cu(phen)2]+ complex. Formation of an interpenetrated assembly, or [2]pseudorotaxane, was achieved by incorporation of a neutral amide cleft into a macrocyclic component (Fig. 1). Subsequent chloride anion recognition by this hydrogen-bond donor site results in threading of the organic pyridinium chloride ion-pair and formation of a [2]pseudorotaxane.16
![Anion-templated [2]pseudorotaxane assembly.](/image/article/2006/OB/b518178j/b518178j-f1.gif) | ||
| Fig. 1 Anion-templated [2]pseudorotaxane assembly. | ||
The conversion of these types of pseudorotaxanes to interlocked mechanically-bonded assemblies will produce novel rotaxane and catenane structures that contain unique, three-dimensional, topological binding domains for the specific recognition of anionic guest species. With the goal of using anion templation to develop a general method for rotaxane synthesis and to understand the factors that influence the yields and anion recognition properties of the assembled interlocked species, we report here the synthesis and anion coordination studies of three new [2]rotaxanes.
Results and discussion
Rotaxane construction strategy
We have recently reported the synthesis of a [2]rotaxane using an anion-templated approach, illustrated in Scheme 1.17 The axle component consists of a pyridinium chloride ion-pair motif with two bulky ‘stopper’ groups. In non-competitive solvent media, chloride anion association with a suitably designed neutral acyclic second component that contains an anion recognition site, such as isophthalamide, and is terminally functionalised with allyl groups produces an assembly that after a ring-closing metathesis (RCM) reaction affords the target [2]rotaxane (Scheme 1). RCM has proved an efficient method in the preparation of catenanes18,19 and rotaxanes,20 with the latter having also been accessed by related olefin metathesis strategies.21![Anion-templated synthesis of a [2]rotaxane via ring-closing metathesis (RCM).](/image/article/2006/OB/b518178j/b518178j-s1.gif) | ||
| Scheme 1 Anion-templated synthesis of a [2]rotaxane via ring-closing metathesis (RCM). | ||
In an effort to demonstrate the generality and synthetic versatility of this anion-templated method of rotaxane assembly, an investigation into the effect of incorporating electron withdrawing substituents into the 5-position of an isophthalamide anion binding motif and changing the nature of the aryl spacer motif on the yields and anion binding properties of the resulting rotaxanes was undertaken.
Synthesis of bis-vinyl-functionalised isophthalamide derivatives
Condensation of the appropriate 5-substituted isophthalic acid chloride with two equivalents of the vinyl-functionalised amine derivative (1)19 in the presence of triethylamine in dry tetrahydrofuran solution afforded the corresponding bis-vinyl isophthalamide derivatives 3 and 4 in yields of 40–50% (Scheme 2). The naphthalene-containing isophthalamide derivative was prepared according to the synthetic pathway shown in Scheme 3. | ||
| Scheme 2 Synthesis of bis-vinyl-functionalised isophthalamide dervatives. | ||
 | ||
| Scheme 3 Naphthalene ligand synthesis. Reagents and conditions: i) BrCH2CN, K2CO3, acetone, reflux, 12 h, 69%; ii) LiAlH4, Et2O, 293 K, 1 h, 71%; iii) Boc-anhydride, CH2Cl2, 293 K, 12 h, 53%; iv) Pd/C, 1 atm H2, DMF–MeOH (4 : 1), 293 K, 12 h, 61%; v) 2-allyloxyethanol-p-toluene sulfonate,23 K2CO3, EtOH, reflux, 12 h, 64%; vi) TFA, CH2Cl2, 293 K, 12 h, CHCl3, HCl(g), 20 min, 47%; vii) isophthaloylcarbonylchloride, NEt3, CH2Cl2, 0 °C, 1 h, 44%. | ||
The starting material 1-benzyloxy-5-hydroxynaphthalene was prepared according to a literature procedure.22 Reaction with bromoacetonitrile in acetone using potassium carbonate as base afforded the nitrile compound 5 in moderate yield, which was subsequently reduced using lithium aluminium hydride to give the amine compound 6. Protection of the amine functionality using Boc-anhydride followed by the high yielding removal of the benzyl protecting group via hydrogenation afforded compound 8. The known compound 2-allyloxyethanol-p-toluene sulfonate23 was reacted with compound 8 in the presence of base to give the allyl-appended naphthalene derivative 9. Removal of the Boc protecting group using trifluoroacetic acid (TFA) yielded the deprotected amine as an oil which was more easily handled after protonation to give the hydrochloride salt 10. Condensation of 10 with isophthalic acid chloride gave the naphthalene-containing isophthalmide derivative 11.
The terphenyl-functionalised pyridinium halide and hexafluorophosphate derivatives 12a–d (Fig. 2) was prepared according to literature procedure.17
![Terphenyl-functionalised pyridinium salts 12a–d investigated as potential [2]rotaxane thread components.](/image/article/2006/OB/b518178j/b518178j-f2.gif) | ||
| Fig. 2 Terphenyl-functionalised pyridinium salts 12a–d investigated as potential [2]rotaxane thread components. | ||
1H NMR anion-binding studies of isophthalamide derivatives
In order to ascertain which anion may serve as the potential template in the ensuing rotaxane syntheses, 1H NMR anion binding studies were undertaken with bis-vinyl-appended isophthalamide (2) and 5-nitroisophthalamide (3) compounds. The addition of tetrabutylammonium (TBA) halide, dihydrogen phosphate and hydrogen sulfate salts to dichloromethane-d2 solutions of both derivatives resulted in significant downfield shift perturbations of the respective compounds' aryl b and amide c protons. WinEQNMR![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 analyses of the resulting titration isotherms gave 1 : 1 association constant values as shown in Table 1.
24 analyses of the resulting titration isotherms gave 1 : 1 association constant values as shown in Table 1.
With both derivatives, chloride anions are bound the most strongly of the halides, as a result of possessing the most effective hydrogen-bond acceptor ability and a potential size match with the amide cleft binding site. It is noteworthy that the presence of an electron withdrawing nitro group in the isophthalamide's 5-position substantially enhances the anion binding ability of the ligand for all anionic guests, without altering the selectivity trends. This increased affinity is postulated to be a result of a decrease in the pKa of the amide protons and hence an increase in their hydrogen-bond donor ability. The 5-iodoisophthalamide ligand (4) was found to bind chloride with an association constant of 900 M−1.
Taking into account these findings, chloride was chosen as the templating anion in subsequent rotaxane formation reactions.
Templated macrocyclisation reactions
As a test for the suitability of RCM macrocyclisation reactions using Grubbs' catalyst in the presence of a halide anion template, the isophthalamide derivative 2 was stirred with one equivalent of TBA chloride and 10% Grubbs' catalyst by weight. Macrocycle 13 was isolated by preparative TLC in 30% yield (Scheme 4). | ||
| Scheme 4 Anion-templated macrocyclisation facilitated by ring-closing metathesis (RCM). | ||
A comparison of the 1H NMR spectra of the acyclic and cyclised isophthalamide compound reveals the expected changes in the allyl proton environments as a result of a completed metathesis reaction (Fig. 3).
 | ||
| Fig. 3 Selected region of 1H NMR of ligand 2 (bottom) and the related macrocycle 13 (top) in CDCl3. | ||
The alkene in the cyclised product can exist as either the cis or trans isomer, with the latter expected to dominate as the thermodynamic product of the equilibrium process. In addition, it is expected that no steric strain will be imposed on this functionality due to the large ring size. The trans : cis ratio is calculated from the isomeric olefinic protons in the 1H NMR spectrum (trans = 5.84, cis = 5.77 ppm) to be 6 : 1.
Interestingly, in the absence of a chloride template, or in the presence of the non-coordinating PF6− anion, no cyclisation is observed, indicating the critical nature of the template in the RCM process. These results are in agreement with previously reported observations on related systems in which the lowest energy conformation of 3,5-isophthalamide systems has been determined to be the syn–anti.25 Stabilisation of this conformation is through internal NH⋯O hydrogen bonding. In this conformation the two terminal allyl groups will be at a considerable spatial separation and therefore no cyclisation is observed. Upon addition of a suitable anion template, however, the higher energy syn–syn conformation becomes more favoured. In this conformation the allyl groups are in much closer proximity, thus favouring the RCM reaction (Scheme 5).
 | ||
| Scheme 5 Anion recognition forces a change in ligand conformation from syn–anti to syn–syn, thus facilitating ring-closing metathesis. | ||
The chloride anion-templated RCM reaction was also attempted with the 5-nitroisophthalamide derivative 3 and the cyclic product (14) isolated in 60% yield after purification (Fig. 4). This higher macrocyclisation yield, as compared with the preparation of macrocycle 13, is a result of more effective chloride anion templation and complements the 1H NMR association constant values shown in Table 1.
 | ||
| Fig. 4 Structure of the cyclic product 14. | ||
Anion-templated assembly of [2]rotaxanes
Extension of this cyclisation strategy to the formation of [2]rotaxanes using the 5-nitro-, 5-iodo- and naphthalene-isophthalamide derivatives (3, 4 and 11) involved simply mixing the appropriate component with the pyridinium chloride-stoppered thread 12a in dichloromethane. Of note is the observed solubilisation of the neutral acyclic amide compounds being enhanced in the presence of the pyridinium chloride thread, indicative of association of the two components. Addition of Grubbs' catalyst (10% by weight) facilitates the RCM reaction, and isolation of the [2]rotaxanes was achieved by preparative TLC (Scheme 6).![[2]Rotaxane synthesis.](/image/article/2006/OB/b518178j/b518178j-s6.gif) | ||
| Scheme 6 [2]Rotaxane synthesis. | ||
[2]Rotaxane yields of up to 60% for the 5-nitroisophthalamide derivative 3 were achieved, which compares with the previously reported isophthalamide [2]rotaxane yield of 45%.17 This result is in agreement with the enhanced macrocyclisation yields and the increased strength of chloride anion binding by 3 in comparison to 2, suggesting that the predominant interaction driving the rotaxane assembly process is anion recognition.
Importantly, no [2]rotaxane formation was observed using pyridinium bromide, iodide and hexafluorophosphate salts 12b–d, indicating the crucial nature of the chloride anion template to the assembly process. Over an extended period of time no dissociation of the [2]rotaxane species was observed, indicating that the bulky stopper groups are large enough to prevent a deslipping process.
A comparison of the 1H NMR spectra of 12a, 3 and the [2]rotaxane product 16a is given in Fig. 5. With 5-nitroisophthalamide derivative 3, downfield shift perturbations are observed for the aryl, n, and amide, o, proton environments as a result of chloride anion complexation. Correspondingly, an upfield perturbation in the pyridinium thread's aryl, b, and amide, c, proton environments is observed due to chloride anion polarisation as a result of complexation by the neutral component. Importantly, upfield shift perturbations are observed in the neutral component's hydroquinone proton environments, s and r, as a result of a π–π donor–acceptor interaction between these electron-rich functionalities and the electron-deficient pyridinium ring of the threading component.26 The completed metathesis reaction is again observed by the loss of the characteristic terminal allyl splitting pattern. In addition, high resolution electrospray mass spectrometry and elemental analysis were used to characterise the new rotaxane systems (see the Experimental section).
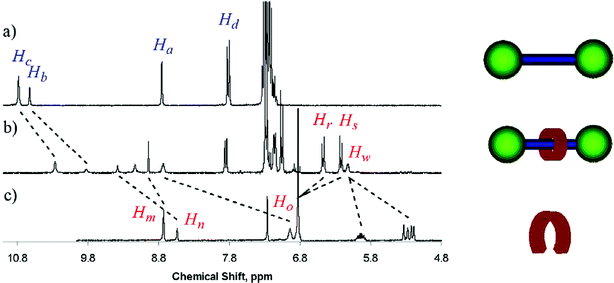 | ||
| Fig. 5 Selected region of the 1H NMR of (a) thread molecule 12a, (b) rotaxane 16a and (c) 5-nitroisophthalamide derivative 3 (CDCl3, 298 K). For proton labelling see Scheme 6. | ||
Despite the much poorer solubility of the naphthalene amide compound (11) solubilisation was observed in dichloromethane in the presence of pyridinium chloride ion-pair 12a. Addition of Grubbs' catalyst once again facilitated RCM and, after purification by preparative TLC, [2]rotaxane 18 (Fig. 6) was obtained in 15% yield. This lower [2]rotaxane product yield can be postulated to arise from both the greater flexibility afforded to the vinyl-terminated oxyethylene chain as a result of the unsymmetrical substitution of the naphthalene linker and the larger resulting macrocycle.
![Structure of the naphthalene-containing [2]rotaxane (18) as assembled though anion templation. Additional structural stabilisation is provided by π–π stacking interactions and N+CH3⋯O hydrogen bonding.](/image/article/2006/OB/b518178j/b518178j-f6.gif) | ||
| Fig. 6 Structure of the naphthalene-containing [2]rotaxane (18) as assembled though anion templation. Additional structural stabilisation is provided by π–π stacking interactions and N+CH3⋯O hydrogen bonding. | ||
Similar 1H NMR shift perturbations to those noted with [2]rotaxanes 16a and 17a were observed for rotaxane 18, namely downfield shifts in environments n and m and upfield shifts in b and c as a result of anion binding (Fig. 7). Although inherently more crowded, the aromatic region of the 1H NMR spectrum also revealed upfield shift perturbations in the electron-rich naphthalene proton environment, t, as a result of π–π donor–acceptor interaction with the electron-deficient pyridinium ring.
 | ||
| Fig. 7 Selected region of the 1H NMR spectrum of rotaxane 18 (293 K, acetone-d6). For proton labelling see Fig. 6. | ||
Rotaxanes as anion hosts
Whilst the anion-templated assembly of [2]rotaxanes presents a novel approach to the preparation of interlocked species, the desire to incorporate a degree of functionality into the final system encouraged us to investigate their ability to function as anion hosts. Removal of the chloride anion template from the cavity of [2]rotaxanes 16a and 17a using silver hexafluorophosphate yielded the corresponding hexafluorophosphate [2]rotaxane salts 16b and 17b. Although perturbations in the 1H NMR chemical shifts of the cleft-sited amide and aryl protons are observed, very few additional perturbations are seen, indicating a retention of the [2]rotaxane structure upon template removal. Titration of chloride, dihydrogen phosphate and acetate anions, as their TBA salts, gave rise to perturbations in the rotaxane amide and aryl protons surrounding the pocket previously occupied by the template. Analysis of the resulting binding curves using the WinEQNMR24 software program allowed determination of 1 : 1 association constants for complexation of the anionic guests by the host [2]rotaxane (Table 2). In order to determine the effect of rotaxanation upon binding phenomena, analogous association constant determinations were carried out with the hexafluorophosphate thread 12d. Owing to a limited supply of material, [2]rotaxane 18 was not investigated with regard to its anion binding properties.
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif)
Despite the possibility of a complementary size match between the amide cleft of the thread and a chloride anion, the pyridinium hexafluorophosphate thread 12d displays a greater selectivity preference for both acetate and dihydrogen phosphate anions. The dominant factor in anion recognition by the free thread is therefore the oxobasicity of the guest anions, hence the much weaker coordination of the chloride anion. In sharp contrast, however, are the binding trends observed for all three hexafluorophosphate rotaxanes, which are completely reversed: Cl− > H2PO4− > OAc−. The selectivity change is postulated to be a result of a unique anion binding site formed by the two orthogonally arranged amide clefts. A chloride anion can be bound tightly in this pocket due to both complementary topography and the formation of four NH⋯Cl− hydrogen bonds. Large anions such as dihydrogen phosphate and acetate bind weakly as a result of either having to penetrate the binding pocket, which would be sterically demanding, or forcing a displacement of the pyridinium thread from the macrocyclic cavity. Both of these binding mechanisms would be highly unfavourable and unlikely to be able to present a full complement of hydrogen-bond donors.
With rotaxanes 16b and 17b the dramatic increase in the magnitude of the association constants can be explained by considering the effect of the nitro and iodo substituents of the isophthalamide ring. As indicated by the 1H NMR binding studies of 3 and 4, these electron withdrawing functionalities appear to increase the acidity and hydrogen-bonding ability of the amide protons and thus result in enhanced anion recognition. It is noteworthy that whilst the anion association constant values are larger for rotaxanes 16b and 17b relative to 15b (Table 2), the unique topology of the interlocked binding domain ensures that selectivity for chloride, the templating anion, is maintained.
Conclusions
Anion templation has been shown to be an effective route to the preparation of macrocycles and [2]rotaxanes. In particular, the synthesis of three new [2]rotaxanes has demonstrated the generality of this anion-templated approach to the assembly of interlocked species. Importantly, rotaxanes templated in this manner have a degree of functionality integral to their structure. The use of unique cavities at the centre of interlocked structures as host molecules has only recently begun to be explored despite the unique properties they possess.12b,17,18,27 Dramatic changes in anion binding selectivities have been demonstrated to occur upon rotaxanation. Simple modification of just one component can improve both rotaxane assembly yields and anion binding strengths significantly. In this instance the incorporation of electron withdrawing substituents, such as the nitro group, into the 5-position of an isophthalamide anion binding motif increases the acidity of the amide protons which leads to impressive yields of up to 60% for [2]rotaxane formation, concomitant with the rotaxane binding domain both augmenting thermodynamic stability and conserving selectivity for chloride, the templating anion. We are currently seeking to exploit this anion templation methodology further in the design of mechanically interlocked species for anion sensing applications.Experimental
The synthesis of compounds 12a–d, 1, 2 and 15a,b have been previously described.17,19 2-Allyloxyethanol-p-toluene sulfonate and 5-iodoisophthalic acid were prepared according to literature procedures.23,28 All starting materials were purchased from Aldrich Chemicals and used as received with the exception of thionyl chloride and triethylamine which were distilled from triethyl phosphite and over potassium hydroxide, respectively, and the latter stored over potassium hydroxide. All solvents were dried using standard laboratory procedures prior to use. 1H and 13C NMR spectra were recorded on Varian 300 MHz VX Works and 500 MHz Unity Plus spectrometers. Mass spectrometry was carried out using a Micromass LCT electrospray mass spectrometer. Elemental analyses were performed by the Inorganic Chemistry Laboratory Microanalysis Service, Department of Chemistry, University of Oxford, and by the Elemental Analysis Service, London Metropolitan University.Preparation of 5-nitroisophthalamide ligand 3
A solution of synthon 1 (0.82 g, 3.0 mmol) and triethylamine (8 ml) in THF (100 ml) was prepared and cooled to 0 °C. 5-Nitroisophthaloyl dichloride was added and the solution stirred for 1 h keeping the temperature below 5 °C. The resulting reaction mixture was filtered and the solvent removed to give an oil. The oil was then dissolved in dichloromethane (100 ml) and washed with aqueous 10% HCl solution (3 × 100 ml) and water (3 × 100 ml). The organic phase was then dried over magnesium sulfate and the solvent removed to give the product (0.49 g, 50%) as a yellow solid (Found: C, 62.9; H, 6.1; N, 6.6. Calc. for C34H39N3O10: C, 62.8; H, 6.05; N, 6.5%); δH(300 MHz, CDCl3, Me4Si) 3.89 (4H, t, J = 4.2 Hz, CH2), 3.89 (4H, m, NHCH2), 4.09 (12H, m, CH2), 5.2–5.3 (2H, m, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.93 (2H, m, CHCH2), 6.83 (8H, m, ArH), 6.94 (2H, br s, NH), 8.54 (1H, s, 4-H) and 8.73 (2H, s, 2-H); δC(300 MHz, CDCl3, Me4Si) 40.1, 68.0, 66.9, 68.6, 72.4, 115.3, 115.6, 117.5, 124.6, 131.2, 134.5, 136.3, 148.2, 152.5, 153.3, 164.6; m/z (ESI) 672.70 (M + H+ requires 672.25).
CH2), 5.93 (2H, m, CHCH2), 6.83 (8H, m, ArH), 6.94 (2H, br s, NH), 8.54 (1H, s, 4-H) and 8.73 (2H, s, 2-H); δC(300 MHz, CDCl3, Me4Si) 40.1, 68.0, 66.9, 68.6, 72.4, 115.3, 115.6, 117.5, 124.6, 131.2, 134.5, 136.3, 148.2, 152.5, 153.3, 164.6; m/z (ESI) 672.70 (M + H+ requires 672.25).Preparation of 5-iodoisophthalamide ligand 4
Method as for compound 3, using synthon 1 (0.3 g, 1.15 × 10−3 mol) and 5-iodoisophthaloyl dichloride (0.10 g, 5.73 × 10−3 mol) to give the product (0.16 g, 40%) as a white solid (Found: C, 55.9; H, 5.4; N, 3.9. Calc. for C34H39IN2O8: C, 55.9; H, 5.4; N, 3.8%); δH(300 MHz, CDCl3, Me4Si) 3.75 (8H, m, CH2), 4.01 (12H, m, CH2), 5.16–5.30 (4H, m, CHCH2), 5.84–5.97 (2H, m, CHCH2), 6.77 (8H, s, ArH), 7.33 (2H, br t, J = 4.4 Hz, NH), 8.11 (1H, s, 4-H) and 8.13 (2H, s, 2-H); δC(300 MHz, CDCl3, Me4Si) 39.8, 66.8, 67.8, 68.4, 72.2, 94.2, 115.3, 115.4, 117.4, 134.3, 136.0, 138.8, 152.5, 153.1 and 165.6; m/z (ESI) 731.1259 (M + H+ requires 731.1829).Preparation of compound 5
1-Benzyloxy-5-hydroxynaphthalene (1.8 g, 7.2 × 10−3 mol), bromoacetonitrile (0.95 g, 7.92 × 10−3 mol) and potassium carbonate (1.09 g, 7.92 × 10−3 mol) were heated under reflux in acetone (100 ml) for 12 h. The resulting reaction mixture was cooled to room temperature and filtered. The solvent was removed to give a brown oil that was redissolved in dichloromethane and filtered through a plug of silica. The solvent was removed to give the product (1.44 g, 69%) as a pale yellow solid; δH(300 MHz, CDCl3, Me4Si) 4.96 (2H, s, OCH2Ph), 5.25 (2H, s, CNCH2), 6.96 (1H, d, J = 7.5 Hz, ArH), 7.51–7.53 (2H, m, ArH), 7.33–7.45 (5H, m, ArH), 7.78 (1H, dt, J 8.7, J 0.8, ArH) and 8.07 (1H, dt, J 8.4, J 0.9, ArH); δC(300 MHz, CDCl3, Me4Si) 53.8, 70.2, 106.1, 106.5, 106.6, 114.0, 114.8, 117.2, 124.8, 125.3, 126.2, 127.5, 128.0, 128.7, 129.1, 137.0, 137.3, 152.2 and 154.4; m/z (ESI) 290.1233 (M + H+ requires 290.1181).Preparation of compound 6
Compound 5 (0.5 g, 4.84 × 10−3 mol) was dissolved in diethyl ether (100 ml) and added carefully to a suspension of lithium aluminium hydride (0.1 g, 2.6 × 10−3 mol) in diethyl ether (150 ml) and stirred at room temperature for one hour. Aqueous 10% sodium hydroxide solution was added slowly (approx. 25 ml) until no further gas production was observed. Saturated aqueous sodium chloride solution was added (50 ml) and the organic layer decanted. Diethyl ether (80 ml) was added to the aqueous phase and stirred for 20 min before being decanted. This procedure was repeated once more. The organic phases were combined and dried over magnesium sulfate. Removal of the solvent gave the product (0.36 g, 71%) as a white solid; δH(300 MHz, CDCl3, Me4Si) 3.12 (2H, t, J = 5.1 Hz, CH2), 4.05 (2H, t, J 5.1, CH2), 5.13 (2H, s, CH2 benzyl), 6.73 (1H, d, J 7.8, ArH), 6.81 (1H, d, J 7.5, ArH), 7.21–7.33 (5H, m, ArH benzyl), 7.41–7.43 (2H, m, ArH), 7.77 (1H, d, J 8.4, ArH) and 7.83 (1H, d, J 8.7, ArH).Preparation of compound 7
Compound 6 (1.35 g, 4.60 × 10−3 mol) and Boc-anhydride (1.80 g) were dissolved in dichloromethane (100 ml) and stirred at room temperature for 12 h. The resulting reaction mixture was filtered through a plug of silica and the solvent was removed to give a pale yellow solid. After stirring in hexane (50 ml) for 20 min and filtering the product (0.96 g, 53%) was obtained as a white solid; δH(300 MHz, CDCl3, Me4Si) 1.40 (9H, s, tBu), 3.61 (2H, q, J = 5.1 Hz, NHCH2), 4.08 (2H, t, J 5.1, OCH2), 5.06 (1H, br s, NH), 6.62 (1H, br s, OH), 6.69 (1H, d, J 7.5, ArH), 6.81 (1H, d, J 7.5, ArH), 7.26 (2H, t, J 8, ArH) and 7.72 (2H, d, J 8.4, ArH); δC(300 MHz, CDCl3, Me4Si) 23.4, 40.2, 67.4, 70.1, 105.2, 109.3, 114.4, 124.9, 125.3, 126.7, 127.3, 127.9, 128.5, 151.8, 154.0 and 156.2; m/z (ESI) 304.1563 (M + H+ requires 304.1549).Preparation of compound 8
Compound 7 (0.96 g, 2.44 × 10−3 mol) was dissolved in a suspension of 10% palladium on activated carbon (0.09 g) in DMF–methanol (100 ml, 4 : 1). The reaction mixture was stirred overnight under an atmosphere of hydrogen. Filtration through Celite followed by solvent removal gave a crude brown oil. Recrystallisation from water gave the product (0.45 g, 61%) as a white powder; δH(300 MHz, CDCl3, Me4Si) 1.40 (9H, s, tBu Boc), 3.61 (2H, q, J = 5.1 Hz, NHCH2), 4.08 (2H, t, J 5.1, OCH2), 5.06 (1H, br s, NH), 6.62 (1H, br s, OH), 6.69 (1H, d, J 7.5, ArH), 6.81 (1H, d, J 7.5, ArH), 7.26 (2H, t, J 8.0, ArH) and 7.72 (2H, d, J 8.4, ArH); δC(300 MHz, CDCl3, Me4Si) 28.4, 40.2, 67.4, 70.1, 105.2, 109.3, 113.9, 114.4, 124.9, 125.3, 126.7, 127.3, 127.9, 128.5, 151.8, 154.0 and 156.2; m/z (ESI) 304.1563 (M + H+ requires 304.1549)Preparation of compound 9
A solution of compound 8 (0.43 g, 1.42 × 10−3 mol), 2-allyloxyethanol-p-toluene sulfonate (0.36 g, 1.42 × 10−3 mol) and potassium carbonate (0.22 g, 1.56 × 10−3 mol) in ethanol (50 ml) was heated under reflux for 12 h. After cooling to room temperature the reaction mixture was filtered and the solvent removed to give a brown oil. Chloroform (80 ml) was added and a fine precipitate observed which was subsequently removed by filtration. Removal of the solvent gave the product (0.35 g, 64%) as a brown oil; δH(300 MHz, CDCl3, Me4Si) 1.48 (9H, s, tBu), 3.7 (2H, q, J = 5.2 Hz, NHCH2), 3.97 (2H, t, J 4.8, CH2), 4.17–4.22 (4H, m, CH2), 4.32 (2H, t, J 4.8, CH2), 5.23–5.40 (2H, m, CHCH2), 5.93–6.06 (1H, m, CHCH2), 6.84 (1H, t, J 7.5, ArH), 6.88 (1H, d, J 7.5, ArH), 7.37 (1H, t, J 8.0, ArH), 7.39 (1H, t, J 8.0, ArH), 7.85 (1H, d, J 8.4, ArH) and 7.91 (1H, d, J 8.4, ArH); δC(300 MHz, CDCl3, Me4Si) 28.4, 40.2, 67.5, 67.6, 68.6, 70.1, 72.4, 105.5, 105.7, 114.2, 114.8, 117.3, 125.2, 127.3, 127.9, 128.5, 134.6 and 154.4; m/z (ESI) 410.1945 (M + H+ requires 410.1943).Preparation of compound 10
Compound 9 (0.33 g, 8.52 × 10−3 mol) was dissolved in dichloromethane (100 ml) and TFA (5 ml) and stirred for 12 h. The resulting reaction mixture was washed with water (2 × 100 ml) and the combined aqueous layers back-extracted with dichloromethane (100 ml). The organic phases were combined, dried over magnseium sulfate and the solvent was removed to give a light brown oil. The oil was dissolved in chloroform (50 ml) and HCl(g) was bubbled through the solution for 20 min or until no more precipitate was formed. Diethyl ether (50 ml) was added and the product (0.11 g, 47%) collected by filtration; δH(300 MHz, CD3OD, Me4Si) 3.63 (2H, m, CH2), 3.95 (4H, m, CH2), 4.17 (4H, br t, CH2), 5.15–5.25 (2H, m, CHCH2), 5.77–5.87 (2H, m, CHCH2), 6.86 (2H, m, ArH), 7.79–7.93 (2H, m, ArH) and 7.33–7.45 (2H, m, ArH); m/z (ESI) 274.1574 (M+
− Cl− requires 274.169).Preparation of naphthalene ligand 11
A solution of compound 10 (0.27 g, 9.75 × 10−3 mol) and triethylamine (3 ml) in dichloromethane (50 ml) was prepared and cooled to 0 °C. Isophthaloylcarbonylchloride was added and the reaction mixture stirred for one hour, keeping the temperature below 5 °C, during which time a precipitate was observed to form. The product (0.15 g, 44%) was isolated by filtration; δH(300 MHz, CDCl3, Me4Si) 3.79–3.83 (8H, m, CH2), 4.08 (4H, m, CH2), 4.23–4.28 (8H, m, CH2), 5.14–5.32 (4H, m, CHCH2), 5.86–5.98 (2H, m, CHCH2), 6.93 (2H, d, J = 7.2 Hz, ArH), 7.02 (2H, d, J 8.1, ArH), 7.27–7.44 (5H, m, ArH), 7.55 (2H, t, J 8.8, ArH), 7.71 (2H, d, J 9.3, ArH), 7.82 (2H, d, J 7.5, ArH), 8.032 (2H, d, J 8.4, ArH), 8.57 (1H, s, ArH), 9.13 (2H, br t, J 5.7, NH); m/z (ESI) 727.3250 (M + H+ requires 727.2995).Preparation of isophthalamide macrocycle 13
Ligand 2 (75 mg, 0.124 mmol) and tetrabutylammonium chloride (56 mg, 0.2 mmol) were dissolved in dichloromethane (50 ml) and stirred for 15 min. Grubbs' catalyst (7.5 mg) was added and the resulting solution stirred for 12 h. The resulting reaction mixture was purified by preparative TLC (SiO2) using dichloromethane–methanol (93 : 7) as eluent to give the product (21 mg, 30%) as a white powder (Found: C, 65.3; H, 6.3; N, 4.5. Calc. for C32H36N2O8·CH3OH: C, 65.1; H, 6.6; N, 4.6%); δH(300 MHz, CDCl3, Me4Si) 3.76 (4H, m, OCH2CH2O), 3.85 (4H, m, NHCH2), 4.08 (12H, m, CH2), 5.84 (2H, s, CHCH), 6.66 (2H, br s, NH), 6.80 (8H, m, ArH hydroquinone), 7.55 (1H, t, J = 7.6 Hz, Ar-5-H) and 7.99 (3H, m, ArH); δC(300 MHz, CDCl3, Me4Si) 40.0, 67.4, 68.4, 68.7, 71.1, 104.8, 115.6, 116.1, 129.5, 131.0, 134.9, 135.3, 152.9, 153.6 and 167.0; m/z (ESI) 577.69 (C32H36N2O8
+ H+ requires 577.65).Preparation of 5-nitroisophthalamide macrocycle 14
Method as for 13 using ligand 3 (100 mg, 1.5 × 10−4 mol), tetrabutylammonium chloride (42.7 mg, 1.5 × 10−4 mol) and Grubbs' catalyst (10 mg). Purification by preparative TLC using ethyl acetate as eluent furnished the product (52 mg, 54%) as a pale yellow powder (Found: C, 61.1; H, 6.1; N, 6.5. Calc. for C32H35N3O10: C, 61.8; H, 5.7; N, 6.8%); δH(300 MHz, CDCl3, Me4Si) 3.76 (4H, t, J = 4.2 Hz, CH2), 3.86 (4H, m, NHCH2), 4.04 (12H, m, CH2), 5.84 (2H, t, J 2.9, CHCH), 6.73 (8H, m, ArH), 6.91 (2H, br s, NH), 8.35 (1H, s, ArH p-NO2) and 8.78 (2H, s, ArH o-NO2); δC(300 MHz, CDCl3, Me4Si) 40.2, 68.7, 67.1, 68.4, 71.0, 115.5–115.8 (four peaks), 125.4, 129.3, 136.3, 153.0 and 153.4; m/z (ESI) 622.2300 (M + H+ requires 622.2400).Preparation of nitro-rotaxane chloride 16a
Ligand 3 (90 mg, 1.39 × 10−4 mol) and pyridinium chloride thread 12a (100 mg, 9.29 × 10−4mol) were dissolved in dichloromethane (50 ml) and stirred for 20 min. Grubbs' catalyst was added (18 mg, 10% by weight) and the reaction mixture stirred overnight. The solvent was removed and the crude reaction mixture seperated by preparative TLC (SiO2) using dichloromethane–methanol (93 : 7) as eluent. The product (90 mg, 57%) was isolated as a yellow powder (Found: C, 75.0; H, 6.8; N, 4.8. Calc. for C106H113ClN6O12: C, 75.0; H, 6.7; N, 4.95%); δH(300 MHz, CDCl3, Me4Si) 1.31 (36H, s, tBu), 3.77 (12H, m, NHCH2CH2OArOCH2), 4.11 (8H, m, CH2OCH2), 4.45 (3H, s, N+CH3), 6.01 (2H, br s, CHCH), 6.18 (4H, d, J = 9.0 Hz, ArH), 6.43 (4H, d, J 9.0, ArH), 7.02 (8H, m, ArH), 7.19 (2H, m, ArH), 7.23 (12H, m, ArH), 7.81 (4H, d, J 9.0, ArH), 8.70 (2H, br s, NH mac), 8.90 (2H, s, ArH o-NO2), 9.10 (2H, s, ArH o-N+), 9.34 (1H, s, ArH p-NO2), 9.78 (1H, br s, ArH p-N+) and 10.23 (2H, br s, NH thread); m/z (ESI) 1662.85 (M+ − Cl− requires 1161.8416).Preparation of iodo-rotaxane chloride 17a
Method as for rotaxane 16a using ligand 4 (25 mg, 1.41 × 10−5 mol), pyridinium chloride thread 12a (69 mg, 6.39 × 10−5mol) and Grubbs' catalyst (7 mg) to furnish the product (43%) as a yellow powder; δH(300 MHz, CDCl3, Me4Si) 1.31 (36H, s, tBu), 3.74–3.82 (12H, m, CH2), 4.06–4.11 (8H, m, CH2), 4.37 (3H, br s, N+CH3), 6.02 (2H, br s, CHCH), 6.17 (4H, d, J = 8.7 Hz, ArH), 6.42 (4H, d, J 8.4, ArH), 7.07 (8H, d, J 7.8, ArH stopper), 7.16 (8H, d, J 7.8, ArH stopper), 7.21–7.26 (14H, m, ArH stopper), 7.81 (4H, d, J 7.5, NHArH), 8.43 (2H, ArH o-N+), 8.50 (2H, br s, ArH o-CI), 8.92 (1H, br s, ArH p-CI), 9.14 (2H, br s, NH mac), 9.71 (1H, br s, ArH p-N+) and 10.26 (2H, br s, NH thread); m/z (ESI) 1742.7482 (M+
− Cl− requires 1742.7532).Preparation of nitro-rotaxane hexafluorophosphate 16b
The nitro-rotaxane chloride (40 mg, 2.35 × 10−5 mol) was dissolved in dichloromethane and stirred with silver hexafluorophosphate (30 mg, 1.18 × 10−4mol) for 12 h in the absence of light. The resulting solution was filtered through Celite and the solvent removed to give the pure product (85%) as a yellow solid (Found: C, 70.3; H, 6.5; N, 4.8. Calc. for C106H113F6N6O12P: C, 70.4; H, 6.3; N, 4.7%); δH(300 MHz, CDCl3, Me4Si) 1.31 (36H, s, tBu), 3.76–3.81 (12H, m, CH2), 4.05–4.10 (8H, m, CH2), 4.31 (3H, br s, N+CH3), 6.01 (2H, br s, CHCH), 6.19 (4H, d, J = 8.7 Hz, ArH), 6.47 (4H, d, J 8.7, ArH), 7.05–7.21 (14H, m, ArH), 7.78 (4H, d, J 7.8, NHArH), 8.57 (2H, br s, ArH o-N+), 8.96 (2H, m, ArH o-CI, p-CI), 9.04 (2H, br s, NH mac), 9.26 (1H, br s, ArH p-N+) and 10.19 (2H, br s, NH thread).Preparation of iodo-rotaxane hexafluorophosphate 17b
Method as for 16b using the iodo-rotaxane chloride 17a (25 mg, 1.41 × 10−5 mol) and silver hexafluorophosphate (14 mg, 5.62 × 10−5mol) to give the product (18 mg, 100%) as a yellow powder (Found: C, 67.4; H, 6.2; N, 3.7. Calc. for C106H113F6IN5O10P: C, 67.4; H, 6.0; N, 3.7%); δH(300 MHz, CDCl3, Me4Si) 1.28 (36H, tBu), 3.73 (8H, m, CH2), 3.80 (4H, m, CH2), 4.01 (8H, m, CH2), 4.24 (3H, s, N+CH3), 5.94 (2H, CHCH), 6.23 (4H, d, J = 8.0 Hz, ArH), 6.45 (4H, d, J 8.5, ArH), 7.10 (8H, d, J 8.0, ArH), 7.21 (14H, m, ArH), 7.72 (4H, br s, ArHNH), 8.48 (2H, s, ArH o-N+), 8.71 (1H, br s, ArH p-CI), 8.98 (2H, br s, NH mac), 9.30 (1H, br s, ArH p-N+) and 10.05 (2H, br s, NH thread).Preparation of naphthalene-rotaxane chloride 18
Method as for 16a using naphthalene ligand 11 (120 mg, 1.70 × 10−4 mol), pyridinium chloride thread 12a (120 mg, 1.12 × 10−4mol) and Grubbs' catalyst to furnish the product (30 mg, 15%) as a yellow solid (Found: C, 76.7; H, 6.7; N, 3.7. Calc. for C114H118ClN5O10: C, 78.1; H, 6.8; N, 4.0%); δH(300 MHz, acetone-d6, Me4Si) 1.37 (36H, s, tBu), 3.67–3.71 (4H, m, NHCH2), 3.94–3.96 (4H, m, CH2), 4.06–4.07 (4H, m, CH2), 4.11 (4H, t, J = 4.5 Hz, CH2), 4.24 (4H, m, CH2CH), 4.66 (3H, s, N+CH3), 6.24 (2H, br s, CHCH), 6.28 (2H, d, J 7.8), ArHt), 6.40 (2H, d, J 7.5, ArHq) 6.64 (2H, t, J 8.0, ArHr), 6.84 (2H, t, J 8.3, ArHu), 7.10 (2H, d, J 9.0, ArHν), 7.05–7.42 (31H, m, ArHe,f,g,h,i,j,k), 7.54 (2H, d, J 8.4, ArHs), 7.90 (4H, d, J 8.7, ArHd), 8.05 (2H, d, J 7.8, ArHl), 8.88 (2H, br s, NHn), 8.95 (3H, ArHa,m), 9.24 (1H, br s, ArHb) and 9.70 (2H, br s, NHc); m/z (ESI) 1716.8728 (M+ − Cl− requires 1716.8878).Acknowledgements
We thank the EPSRC for a studentship (M. R. S.) and for a postdoctoral fellowship (J. A. W.). We also acknowledge the Natural Sciences and Engineering Research Council of Canada for financial support (J. A. W.).References
- Molecular Catenanes, Rotaxanes and Knots, J.-P. Sauvage and C. Dietrich-Buchecker, eds., Wiley-VCH, Germany, 1999 Search PubMed; G. A. Breault, C. A. Hunter and P. C. Meyers, Tetrahedron, 1999, 55, 5265 Search PubMed.
- J.-C. Chambron, J.-P. Collin, V. Heitz, D. Jouvenot, J.-M. Kern, P. Mobian, D. Pomeranc and J.-P. Sauvage, Eur. J. Org. Chem., 2004, 8, 1627 CrossRef; A.-M. L. Fuller, D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, J. Am. Chem. Soc., 2005, 127, 12612 CrossRef CAS; A. L. Vance, N. W. Alcock, J. A. Heppert and D. H. Busch, Inorg. Chem., 1998, 37, 6912 CrossRef CAS; F. Mohr, D. J. Eisler, C. P. McArdle, K. Atieh, M. C. Jennings and R. J. Puddephatt, J. Organomet. Chem., 2003, 670, 27 CrossRef CAS; A. Hori, K. Kumazawa, T. Kusukawa, D. K. Chand, M. Fujita, S. Sakamoto and K. Yamaguchi, Chem.–Eur. J., 2001, 7, 4142 CrossRef CAS; M. E. Padilla-Tosta, O. D. Fox, M. G. B. Drew and P. D. Beer, Angew. Chem., Int. Ed., 2001, 40, 4235 CrossRef CAS; B. A. Blight, K. A. Van Noortwyk, J. A. Wisner and M. C. Jennings, Angew. Chem., Int. Ed., 2005, 44, 1499 CrossRef CAS.
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS; C. A. Hunter, C. M. R. Low, M. J. Packer, S. E. Spey, J. G. Vinter, M. O. Vysotsky and C. Zonta, Angew. Chem., Int. Ed., 2001, 40, 2678 CrossRef CAS; F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619 CrossRef; C. A. Schalley, W. Reckien, S. Peyerimhoff, B. Baytekin and F. Vögtle, Chem.–Eur. J., 2004, 10, 4777 CrossRef CAS; G. Brancato, F. Coutrot, D. A. Leigh, A. Murphy, J. K. Y. Wong and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4967 CrossRef CAS; D. A. Leigh, A. Venturini, A. J. Wilson, J. K. Y. Wong and F. Zerbetto, Chem.–Eur. J., 2004, 10, 4960 CrossRef CAS.
- D. G. Hamilton, M. Montalti, L. Prodi, M. Fontani, P. Zanello and J. K. M. Sanders, Chem.–Eur. J., 2000, 6, 608 CrossRef CAS; T. Iijima, S. A. Vignon, H.-R. Tseng, T. Jarroson, J. K. M. Sanders, M. Venturi, E. Apostoli, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6375 CrossRef; F. Aricó, T. Chang, S.-J. Cantrill, S. I. Kahn and J. F. Stoddart, Chem.–Eur. J., 2005, 11, 4655 CrossRef; N. Georges, S. J. Loeb, J. Tiburcio and J. A. Wisner, Org. Biomol. Chem., 2004, 2, 2751 RSC; A. L. Hubbard, G. J. E. Davidson, R. H. Patel, J. A. Wisner and S. J. Loeb, Chem. Commun., 2004, 138 RSC.
- K. Moon and A. Kaifer, Org. Lett., 2004, 6, 185 CrossRef CAS; S. W. Choi, J. W. Lee, Y. H. Ko and K. Kim, Macromolecules, 2002, 35, 3526 CrossRef CAS; S. Anderson and H. L. Anderson, Angew. Chem., Int. Ed. Engl., 1996, 35, 1956 CrossRef CAS.
- R. Vilar, Angew. Chem., Int. Ed., 2003, 42, 1460 CrossRef CAS.
- R. Vilar, D. M. P. Mingos, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1258 CrossRef CAS; J. S. Fleming, K. L. V. Mann, C.-A. Carraz, E. Psillakis, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 1998, 37, 1279 CrossRef CAS; Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 2002, 41, 2515 CrossRef CAS; R. L. Paul, Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4883 CrossRef CAS.
- S.-T. Cheng, E. Doxiadi, R. Vilar, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2001, 2239 RSC; C. S. Campos-Fernández, R. Clérac and K. R. Dunbar, Angew. Chem., Int. Ed., 1999, 38, 3477 CrossRef CAS; X. Yang, C. B. Knobler and M. F. Hawthorne, Angew. Chem., Int. Ed. Engl., 1991, 30, 1507 CrossRef.
- O. V. Dolomanov, A. J. Blake, N. R. Champness, M. Schröder and C. Wilson, Chem. Commun., 2003, 682 RSC.
- R. Vilar, Struct. Bonding, 2004, 111, 85 CAS.
- C. Seel and F. Vögtle, Chem.–Eur. J., 2000, 6, 21 CrossRef CAS; C. Reuter, W. Wienand, G. M. Hubner, C. Seel and F. Vögtle, Chem.–Eur. J., 1999, 5, 2692 CrossRef CAS; G. M. Hubner, J. Gläser, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 383 CrossRef CAS.
- (a) J. M. Mahoney, R. Shukla, R. A. Marshall, A. M. Beatty, J. Zajicek and B. D. Smith, J. Org. Chem., 2002, 67, 1757; (b) M. J. Deetz, R. Shukla and B. D. Smith, Tetrahedron, 2002, 58, 799 CrossRef CAS; (c) R. Shukla, M. J. Deetz and B. D. Smith, Chem. Commun., 2000, 2397 RSC; (d) E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288 CrossRef CAS.
- P. Ghosh, O. Mermagen and C. A. Schalley, Chem. Commun., 2002, 2628 RSC.
- J. A. Wisner, P. D. Beer, N. G. Berry and B. Tomapatanaget, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4983 CrossRef CAS.
- J. A. Wisner, P. D. Beer and M. G. B. Drew, Angew. Chem., Int. Ed., 2001, 40, 3606 CrossRef CAS.
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul, A. R. Cowley, F. Szemes and M. G. B. Drew, J. Am. Chem. Soc., 2005, 127, 2292 CrossRef CAS; D. Curiel, P. D. Beer, R. L. Paul, A. Cowley, M. R. Sambrook and F. Szemes, Chem. Commun., 2004, 1162 RSC.
- J. A. Wisner, P. D. Beer, M. G. B. Drew and M. R. Sambrook, J. Am. Chem. Soc., 2002, 124, 12469 CrossRef CAS.
- M. Weck, B. Mohr, J. P. Sauvage and R. H. Grubbs, J. Org. Chem., 1999, 64, 5463 CrossRef.
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul and A. R. Cowley, J. Am. Chem. Soc., 2004, 126, 15364 CrossRef CAS.
- A.-M. Fuller, D. A. Leigh, P. J. Lusby, I. D. H. Oswald, S. Parsons and D. B. Walker, Angew. Chem., Int. Ed., 2004, 43, 3914 CrossRef CAS; A. F. M. Kilbinger, S. J. Cantrill, A. W. Waltman, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 3281 CrossRef CAS.
- J. S. Hannam, T. J. Kidd, D. A. Leigh and A. J. Wilson, Org. Lett., 2003, 5, 1907 CrossRef CAS; R. G. E. Coumans, J. A. A. W. Elemans, P. Throdarson, R. J. M. Nolte and A. E. Rowan, Angew. Chem., Int. Ed., 2003, 42, 650 CrossRef CAS; S. A. Vignon, T. Jarroson, T. Iijima, H.-R. Tseng, J. K. M. Sanders and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 9884 CrossRef CAS.
- J. Becher, O. A. Matthews, M. B. Nielsen, F. M. Raymo and J. F. Stoddart, Synlett, 1999, 3, 330 CrossRef.
- H. D. Maynard and R. H. Grubbs, Macromolecules, 1999, 32, 6917 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311 RSC.
- K. Kavallieratos, S. R. de Gala, D. J. Austin and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2325 CrossRef CAS; K. Kavallieratos, C. M. Bertao and R. H. Crabtree, J. Org. Chem., 1999, 64, 1675 CrossRef CAS; S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 5314 CrossRef CAS; C. A. Hunter and D. H. Purvis, Angew. Chem., Int. Ed. Engl., 1992, 31, 792 CrossRef.
- S. J. Loeb and J. A. Wisner, Chem. Commun., 2000, 845 RSC; B. L. Allwood, H. Shahriari-Zaverah, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 1058 RSC; J. E. Kickham, S. J. Loeb and S. L. Murphy, Chem.–Eur. J., 1997, 3, 1203 CrossRef CAS.
- A. Andrievsky, F. Ahuis, J. L. Sessler, F. Vögtle, D. Gudat and M. Moini, J. Am. Chem. Soc., 1998, 120, 9712 CrossRef CAS; P. H. Kwan, M. J. MacLachlan and T. M. Swager, J. Am. Chem. Soc., 2004, 126, 8638 CrossRef CAS; P. Thordarson, R. J. M. Nolte and A. E. Rowan, Aust. J. Chem., 2004, 57, 323 CrossRef CAS; Y. Nagawa, J.-i. Suga, K. Hiratani, E. Koyama and M. Kanesato, Chem. Commun., 2005, 749 RSC.
- A. Kraft, Liebigs Ann. Recl., 1997, 7, 1463 Search PubMed.
Footnotes |
| † Current address: Department of Materials, University of Oxford, Parks Road, Oxford, UK OX1 3PH. |
| ‡ Current address: Department of Chemistry, The University of Western Ontario, Chemistry Building, London, Ontario, Canada N6A 5B7. |
| This journal is © The Royal Society of Chemistry 2006 |