Michael reactions carried out using a bench-top flow system
Frederic
Bonfils
a,
Isabelle
Cazaux
a,
Philip
Hodge
*a and
Claude
Caze
b
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, UK M13 9PL. E-mail: Philip.Hodge@man.ac.uk; Fax: +44 (0)1524 793 252; Tel: +44 (0)161 275 4707
bLaboratoire de Chimie Macromoleculaire URA CNRS 351, Universite des Sciences et Technolgies de Lille, 59685 Cedex, Villeneuve d'Ascq, France
First published on 15th December 2005
Abstract
The Michael reaction between methyl 1-oxoindan-2-carboxylate and methyl vinyl ketone was achieved successfully by pumping solutions of the reactants in toluene through a fluid bed of Amberlyst A21 at 50 °C. The use of a fluid bed reactor is attractive as it allows gel-type beads, i.e. the type of bead used in most studies of polymer-supported (PS) organic reactions, to be used satisfactorily in a flow system. When polymer-supported cinchonidine was used in place of Amberlyst A21, the Michael product was obtained in high yield with an enantiomeric excess (ee) of 51%. This % ee is comparable to that achieved when the reaction was catalysed by cinchonidine itself.
Introduction
Polymer-supported (PS) organic reactions, i.e. reactions where at least one of the reactants is attached to a polymer, have been of great interest in recent years because the use of a polymer support greatly simplifies the isolation of the desired product.1,2 In some instances reaction systems have been automated.3,4 PS reagents and catalysts are of particular interest because the organic substrates and the desired products are in solution and, as a consequence, reactions using them are easily monitored by traditional methods such as thin layer chromatography and 1H NMR spectroscopy.1,2,5 There is now interest in developing PS reagents and catalysts further and using them in bench-top flow systems. Progress in this area has been reviewed recently.6–12 This article describes some Michael reactions carried out in a bench-top flow system. Whilst anion exchange resins have previously been used in the batch mode to catalyse Michael reactions,13 this appears to be the first time such a Michael reaction has been studied in a flow system.Flow systems have several advantages over batch reactions.6–8 For example, (i) the PS reactants suffer less physical damage than in a stirred reaction; (ii) often they allow reactions to be carried out with little or no work up; (iii) they allow reaction conditions to be reproduced relatively easily; (iv) flow systems have the potential for easy automation including feedback on the progress of the reaction; (v) they have the potential to be adapted for the continuous production of product and an easier scale-up from laboratory to plant; and (vi) they have the potential to allow reactions to be carried out in a greener manner.
Results and discussion
This article is concerned with carrying out Michael reactions between methyl 1-oxoindan-2-carboxylate (1) and methyl vinyl ketone (2) to give adduct 3 [Reaction (1)] in a flow system. This well-known reaction was chosen as a model for the present study because it is catalysed by tertiary amines and it has the possibility to be adapted for asymmetric synthesis. The reaction was studied in considerable detail by Wynberg et al.14,15 They showed that this indanone derivative is more reactive than some closely related cyclohexanone derivatives and they studied asymmetric syntheses using cinchona alkaloids as catalysts.14 Other research groups have since studied the reaction using cinchona alkaloids or derivatives.16–23 A wide variety of other compounds24 have also been used as catalysts.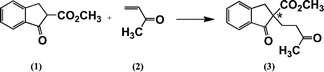 | (1) |
Flow apparatus
In the present study Reactions (1) were achieved by passing solutions of reactants 1 and 2 through a column of a PS tertiary amine. The type of flow system used had a fluid bed of beads. This is attractive for using with gel-type beads, or other types that swell in the reaction solvent, as it allows for the bed volume to change easily and it avoids the clogging that could occur with soft beads in a fixed bed reactor, especially if pressure were to be applied. As the beads can move round in the fluid bed, individual beads do not permanently reside in a part of the reactor where flows may be poor, and so all the beads have an equal opportunity to play a part in the catalysis.The flow apparatus, which is similar to one used previously,25 is shown schematically in Fig. 1. It consists of a glass tube 36 cm long and 14 mm wide (volume 55 ml) with one end closed. The other end is sealed with a septum cap. The PS amine is placed in the tube and the tube immersed in a constant temperature bath to a sufficient depth to cover the column of beads. Using peristaltic pumps solutions of the reactants are pumped, via separate long syringe needles, to the bottom of the tube. The soluble reactants pass up through the fluid bed of beads and the product solution is taken from the top of the bed by a third syringe needle. The placement of this needle allows the liquid level in the column to be controlled easily such that the bed of beads extends from the very bottom of the column up to the take off needle. Thus, under these conditions the volumes of solution above and below the bed are minimized. The eluate was collected as fractions (10 ml). These were evaporated to dryness, dissolved in ether and extracted with aqueous base to remove unreacted compound 1. The solution was dried and the ether evaporated. The residue was analysed by thin layer chromatography and by 1H NMR spectroscopy.
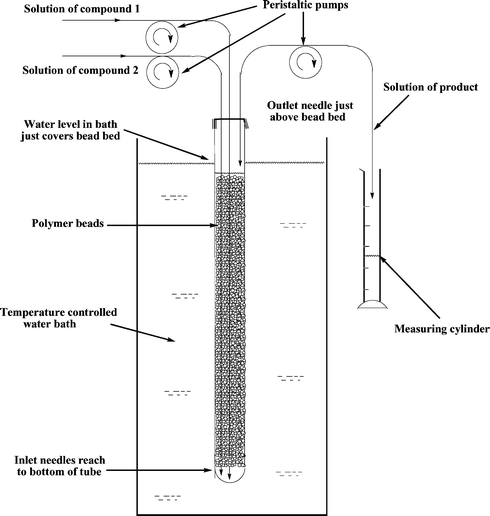 | ||
| Fig. 1 General arrangement of the flow system. | ||
Catalysis of Reaction (1) by Amberlyst A21
Initially, to gain experience of carrying out Michael Reaction (1) in a flow system, reactions were carried out using Amberlyst A21, a commercially available ion exchange resin. This resin contains residues 4 (ca. 5.00 mmol g−1) and is in the form of 490–690 µm diameter macroreticular beads. The flow tube was packed with 15.5 g (79 mmol) of dried beads. These occupied 42 ml. When toluene was added the beads swelled and the bed volume increased to 49 ml. Solutions of the β-ketoester 1 in toluene (0.50 mmol ml−1) and methyl vinyl ketone (2) in toluene (0.53 mmol ml−1) were separately pumped at the same rate into the column. The ketone 2 was used in excess because it is volatile and so an excess can be removed easily from the eluate.Different flow rates and different reaction temperatures were used to optimize the conversion to compound 3: see Table 1. Throughout the volume of the bed was essentially constant at 49 ml. Initially reactions were carried out at 20 °C but it is evident (see Table 1 entries 1 and 2) that the reaction is very slow at this temperature with a residence time (i.e. [volume of solution in the bead bed] divided by [the flow rate]) of >24 h required to obtain significant yields of compound 3. This was considered to be unsatisfactory for a flow system so the reaction temperature was raised to 50 °C and further reactions were carried out: see entries 3–6. At a flow rate of 7.0 ml h−1, corresponding to a residence time of ca. 6 h, very high yields of 3 were obtained: see entry 5. The column was then used continuously under these conditions for 72 h without any drop in the yield of 3; see entry 6. Approximately 10 g of compound 3 could be produced in 24 h.
| Entry | Catalystb | Reaction conditions | Approximate residence time/hc | Average yieldd,e (%) | Duration of experiment/h | % eee,f | Enantiomer in excess | |
|---|---|---|---|---|---|---|---|---|
| Temperature/°C | Total flow rate/ml h−1 | |||||||
| a Solutions of compound 1 (0.5 mol L−1) and compound 2 (0.53 mol L−1) in toluene were pumped at the same rate through the catalyst bed under the indicated conditions. In the reactions using Amberlyst A21 the swollen bead bed (15.5 g of beads) occupied 49 ml. In the reactions using PS-CD the swollen bead bed (15.0 g of beads) occupied 41 ml. b A21 = Amberlyst A21; PS-CD = polymer-supported cinchonidine. c Volume of solution in the bed divided by the flow rate. In the experiments using Amberlyst A21 the volume of solution in the bed was 42 ml. In the experiments using PS-CD the volume of solution in the bed was 28.9 ml. d Yield determined by analysis of 1H NMR spectra of eluate after removal of volatile products. e Figure quoted is the average yield (±4% of the values quoted) for 5 fractions. f Determined by polarimetry. | ||||||||
| 1 | A21 | 20 | 5.0 | 8 | 14 | 12 | — | — |
| 2 | A21 | 20 | 1.0 | 40 | 55 | 48 | — | — |
| 3 | A21 | 50 | 21.0 | 2 | 77 | 18 | — | — |
| 4 | A21 | 50 | 10.5 | 4 | 86 | 18 | — | — |
| 5 | A21 | 50 | 7.0 | 6 | 99 | 24 | — | — |
| 6 | A21 | 50 | 7.0 | 6 | 98 | 72 | — | — |
| 7 | PS-CD | 50 | 7.2 | 4 | 92 | 18 | 52 | (S) |
| 8 | PS-CD | 50 | 5.0 | 6 | 97 | 24 | 51 | (S) |
| 9 | PS-CD | 50 | 5.0 | 6 | 96 | 72 | 52 | (S) |
Catalysis of Reaction (1) by cinchona alkaloids
Since compound 3 contains a chiral centre, asymmetric synthesis can potentially be achieved by using a chiral tertiary amine as the catalyst. Cinchona alkaloids have been used for this purpose.14,19 Quinine (5) and cinchonidine (6) give an excess of the (−)-(S)-enantiomer whilst quinidine (7) and cinchonine (8) give an excess of the (+)-(R)-enantiomer.14,19 In the present work batch reactions were carried out by treating mixtures of compounds 1 and 2 in toluene with 5 mol% of cinchonidine (6) and with 5 mol% of cinchonine (8) at 25 °C. The results are summarized in Table 2, entries 1 and 2. It is evident that the reactions are very slow at this temperature and that cinchonidine (6) affords the higher % ee. In an attempt to shorten the reaction time the reactions were repeated at 50 °C. In many asymmetric syntheses this would result in very much lower % ees, but in the present case there were only modest falls, see Table 2 entries 3 and 4, and reaction times for high yields could now be reduced to a few hours. Previous studies indicate that when catalyzed by cinchona alkaloids, the % ees obtained in Reaction (1) are only moderately sensitive to temperature in the −21 °C to +50 °C range.14,19 The % ees obtained in the present work are comparable with those obtained previously under slightly different reaction conditions.14,19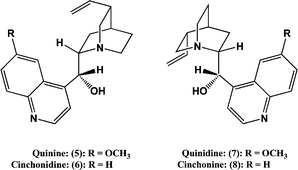
| Entry | Catalystb | Reaction conditions | % Yieldc | % eed | Enantiomer | |
|---|---|---|---|---|---|---|
| Temperature/°C | Time/h | |||||
| a Compound 1 (2.73 mmol) and compound 2 (4.23 mmol) in toluene (10 ml) were stirred with 5 mol% of catalyst under the indicated conditions, b CD = cinchonidine; C = cinchonine; PS-CD = polymer-supported cinchonidine, c Determined by 1H NMR spectra of products after removal of volatile products. d Determined by polarimetry. | ||||||
| 1 | CD | 25 | 48 | 91 | 58 | (S) |
| 2 | C | 25 | 48 | 96 | 47 | (R) |
| 3 | CD | 50 | 3 | 91 | 53 | (S) |
| 4 | C | 50 | 6 | 87 | 47 | (R) |
| 5 | PS–CD | 50 | 24 | 98 | 47 | (S) |
| 6 | PS–CD | 50 | 3 | 96 | 48 | (S) |
Following these preliminary experiments cinchonidine (6) was attached via the vinyl group to gel-type polymer beads using a method first reported by the author: see Reaction (2).26 Polystyrene beads (1% crosslinked; 80–150 µm in diameter) were chloromethylated27 and the product treated with thiourea then aqueous sodium hydroxide to give beads containing thiol groups 9 (1.76 mmol g−1).28 Reaction of these beads with cinchonidine (6) in the presence of azobisisobutyronitrile at 60 °C gave beads with residues 10: unreacted thiol groups were capped by reaction with cyclohex-2-en-3-one.26 By elemental analysis for nitrogen the loading of cinchonidine residues 10 was 0.93 mmol g−1.
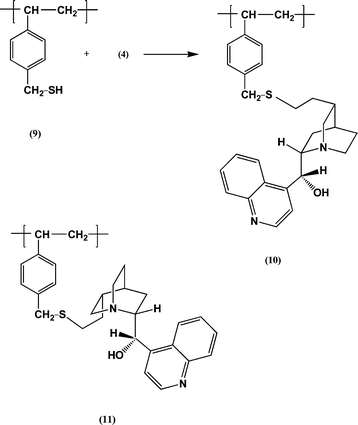 | (2) |
The PS cinchonidine 10 was used in batch reactions to catalyse Reaction (1). The results are summarized in Table 2, entries 5 and 6. Interestingly the % ees obtained were only slightly lower than those obtained with the free alkaloid. Initially this was surprising because in the asymmetric addition of thiols to α,β-enones (another type of Michael addition) the % ees are substantially lower with the PS cinchonidine catalyst 10 and cinchonine catalyst 11 than with the free alkaloids.26 With the PS cinchonine catalyst 11 the difference was shown to be due to the change from vinyl groups to ArCH2SCH2CH2 groups rather than any “polymer effect”.26 It should be noted that in cinchonidine (6) the vinyl and aromatic groups are on opposite sides of the azabicyclcooctane unit, whereas in cinchonine (8) they are on the same side. Also that the arrangement of the reactants in the transition states of the two types of Michael additions may be quite different.21
The PS cinchonidine 10 beads (15.0 g, 14 mmol) were placed in the flow tube and reactions were carried out at 50 °C using the same procedure as with Amberlyst A21. The bed volume was essentially constant at 41 ml. The results are summarized in Table 1, entries 7–9. At an optimum flow rate of 5.0 ml h−1, see entry 8, the chiral product 3 was obtained in high yield and an ee of 51%. The residence time was ca. 6 h. The flow system functioned satisfactorily for at least 72 h: see entry 6. The % ees obtained using the flow system are essentially the same as those obtained in the batch reactions (Table 2, entries 5 and 6). This has been found to be the case in other asymmetric syntheses,6,8 though for the reaction of benzaldehyde with diethylzinc the % ees were found to be substantially higher in flow systems.25,29
Conclusions
Michael Reaction (1) can be carried out successfully by pumping solutions of compounds 1 and 2 through the simple flow system outlined in Fig. 1. This uses a fluid bed of beads and is an attractive type of system in that it allows gel-type beads, i.e. the type of bead used in most studies of PS organic reactions,1–3 to be used satisfactorily in a flow system. In the present studies under optimum conditions chemical yields were high and, when the PS cinchonidine 10 was used as the catalyst the % ees of the (S)-enantiomer of adduct 3 were essentially the same as in the batch reactions. Future studies will be aimed at developing PS scavengers to remove the unreacted starting materials.5,30Experimental
Experimental details are as given previously.25 Beads were dried in a vacuum oven (2 Torr) at 40 °C. Cinchonidine (6) and cinchonine (8) were purchased from Aldrich and used as received.Amberlyst A21
Commercially supplied beads (106.5 g) were washed successively with water (3 × 200 ml), acetone (2 × 100 ml), tetrahydrofuran (2 × 100 ml) and ether (2 × 100 ml) The beads were then dried to constant weight (45.4 g). By elemental analysis they contained 7.11% nitrogen, corresponding to 5.08 mmol g−1 of moieties 4.A portion of the beads (6.60 g) was placed in a measuring cylinder (100 ml). The dry beads occupied 18.0 ml. Toluene (60 ml) was added and the beads allowed to swell and settle over 2 days. The bed of beads then occupied 21.0 ml and 42 ml of toluene remained on top. Thus the bead bed of 21.0 ml contained 18 ml of solvent.
Preparation of polymer-supported cinchonidine 1026
1% Crosslinked polystyrene beads (80–150 µm diameter) were chloromethylated.27 The product (27.0 g; 51.7 mmol of Cl) was treated with thiourea and the intermediate hydrolysed with sodium hydroxide to give beads (28.2 g) containing thiol groups 9.28 By elemental analysis they contained S = 5.63% (1.76 mmol g−1) corresponding to a conversion of 96%. The thiol containing beads (25.0 g) in toluene at 65 °C were reacted with cinchonidine 6 (22.0 g) in the presence of AIBN (3 × 100 mg) to give the PS cinchonidine 10 (35.9 g). Unreacted thiol groups were capped by treating the beads with cyclohex-2-en-1-one in the presence of pyridine. By elemental analysis the final product contained S = 3.94% (1.23 mmol g−1), N = 2.62% (1.87 mmol g−1) corresponding to loading of cinchonidine residues 10 of 0.94 mmol g−1 and a conversion of 76%.A portion of the final beads (3.70 g) was placed in a measuring cylinder (25 ml). The dry beads occupied 6.3 ml. Toluene (15 ml) was added and the beads allowed to swell and settle over 2 days. The bed of beads then occupied 10.2 ml and 7.8 ml of toluene remained on top. Thus the bead bed of 10.2 ml contained 7.2 ml of solvent.
Flow apparatus
The general arrangement of the apparatus used is shown schematically in Fig. 1. The reaction tube was fabricated in house using a commercial Quickfit™ B14 joint and Pyrex glass tubing. It was 36 cm long and had an internal diameter of 14 mm. The appropriate amount of PS catalyst was transferred into the tube, toluene was added and the tube sealed with a standard “B14” rubber septum. The tube was set aside for 2 h to allow the beads to swell. The tube was then immersed (ca. 34 cm) into a water bath whose temperature was controlled by a Techne TU16A Tempunit™ thermoregulator. Two “inlet” hypodermic needles (19G, 24 in) (supplied by Aldrich) were inserted through the septum so as to reach to the bottom of the tube and one “outlet” needle (18G, 10 in) was inserted to just above (ca. 0.5 cm) the top of the bead bed. To help the bed settle, toluene was pumped through it for 2 h using two Watson-Marlow 503U pumps, one equipped with two 501R1 pumpheads (both “inlets” were driven from the same axle) and one equipped with a 303D/A pumphead (for the “outlet”), using Viton™ (0.8 mm ID) tubing. The “inlet” needles were then attached to reservoirs containing 1 in toluene (0.50 mmol ml−1) and compound 2 in toluene (0.53 mml ml−1). The “outlet” needle was passed into a measuring cylinder (25 ml). The pumps were started and fractions (10 ml) collected in the cylinder.Methyl 1-oxoindan-2-carboxylate (1)
This compound was synthesised by the method described by House.31Reactions (1) using Amberlyst A21 in the flow apparatus
The following experiments are typical of those summarized in Table 1, entries 1–6. The tube was charged with Amberlyst A21 beads (15.5 g) and they were allowed to swell in toluene.Michael reactions summarized in Table 2
The following procedures are typical.Reactions (1) using PS cinchonidine 10 in the flow apparatus
The following experiment is typical of those summarized in Table 1, entries 7–9. The tube was charged with PS cinchonidine 10 beads (15.0 g) and they were allowed to swell in toluene.Acknowledgements
We thank the British Council and the French CNRS for financial support (FB, IC) and Ian Goodbody for carrying out several reactions.References
- See, for example, S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Talyor, J. Chem. Soc., Perkin Trans. 1, 2000, 3815 Search PubMed.
- See, for example, Polymeric Materials in Organic Synthesis and Catalysis, ed. M. R. Buchmeiser, Wiley Interscience, Weinheim, 2003 Search PubMed.
- E. Vickerstaffe, B. H. Warrington and M. Ladlow, J. Comb. Chem., 2005, 7, 385 CrossRef CAS.
- E. Vickerstaffe, B. H. Warrington, M. Ladlow and S. V. Ley, J. Comb. Chem., 2004, 6, 332 CrossRef CAS.
- A. Kirschning, H. Monenschein and R. Wittenberg, Angew. Chem., Int. Ed., 2001, 40, 650 CrossRef CAS.
- P. Hodge, Curr. Opin. Chem. Biol., 2003, 1, 2419.
- G. Jas and A. Kirschning, Chem. Eur. J., 2003, 9, 5708 CrossRef CAS.
- P. Hodge, Ind. Eng. Chem. Res., 2005, 44, 8542 CrossRef CAS.
- U. Kunz, H. Schonfeld, W. Solodenko, G. Has and A. Kirschning, Ind. Eng. Chem. Res., 2005, 44, 8548.
- S. France, D. Bernstein, A. Weatherwax and T. Lectka, Org. Lett., 2005, 7, 3009 CrossRef CAS.
- S. Saaby, K. R. Knudsen, M. Ladlow and S. V. Ley, Chem. Commun., 2005, 2909 RSC.
- C. K. Y. Lee, A. B. Holmes, S. V. Ley, I. F. McConvey, B. Al-Duri, G. A. Leeke, R. C. D. Santos and J. P. K. Seville, Chem. Commun., 2005, 2175 RSC.
- See, for example, (a) C. Simon, J.-F. Peyronel, F. Clerc and J. Rodriguez, Eur. J. Org. Chem., 2002, 3359 CrossRef CAS; (b) K. N. Trivedi, J. Sci. Ind. Res., 1959, 18B, 397 Search PubMed.
- K. Hermann and H. Wynberg, J. Org. Chem., 1979, 44, 2238 CrossRef CAS.
- H. Wynberg and R. Helder, Tetrahedron Lett., 1975, 4057 CrossRef.
- P. Hodge, E. Khoshdel and J. Waterhouse, J. Chem. Soc., Perkin Trans. 1, 1983, 2205 RSC.
- E. J. Park, M. H. Kim and D. A. Kim, J. Org. Chem., 2004, 69, 6897 CrossRef CAS.
- K. Onimura, K. Matsuzaki, Y.-K. Lee, H. Tsutsumi and T. Oishi, Polym. J. (Tokyo), 2004, 36, 190 Search PubMed.
- G. Szollosi and M. Bartok, Chirality, 2001, 13, 614 CrossRef CAS.
- R. Alvarez, M.-A. Hourdin, C. Cave, J. d'Angelo and P. Chaminade, Tetrahedron Lett., 1999, 40, 7091 CrossRef CAS.
- A. Sera, K. Takagi, H. Katayama and H. Yamada, J. Org. Chem., 1988, 53, 1157 CrossRef CAS.
- M. Inagaki, J. Hiratake, Y. Yamamoto and J. Oda, Bull. Chem. Soc. Jpn., 1987, 60, 4121 CAS.
- N. Kobayashi and K. Iwai, J. Am. Chem. Soc., 1978, 100, 7071 CrossRef CAS.
- See, for example, (a) M. Nakajima, S. Yamamoto, Y. Yamaguchi, S. Nakamura and S. Hashimoto, Tetrahedron, 2003, 59, 7307 CrossRef CAS; (b) T. Suzuki and T. Torii, Tetrahedron: Asymmetry, 2001, 12, 1077 CrossRef CAS , and references cited therein.
- P. Hodge, D. W. L. Sung and P. W. Stratford, J. Chem. Soc., Perkin Trans. 1, 1999, 2335 RSC.
- P. Hodge, E. Khoshdel, J. Waterhouse and J. M. J. Frechet, J. Chem. Soc., Perkin Trans. 1, 1985, 2327 RSC.
- C. R. Harrison, P. Hodge, J. Kemp and G. M. Perry, Makromol. Chem., 1975, 176, 267 CrossRef CAS.
- J. M. J. Frechet, M. D. deSmet and M. J. Farrall, Polymer, 1979, 20, 675 CrossRef CAS.
- M. I. Burguete, E. Garcia-Verdugo, M. J. Vicent, S. V. Luis, H. Pennemann, N. Graf von Kerserling and J. Martens, Org. Lett., 2002, 4, 3947 CrossRef CAS.
- A. M. Hafez, A. E. Taggi, T. Dudding and T. Lectka, J. Am. Chem. Soc., 2001, 123, 10853 CrossRef CAS.
- H. O. House and C. B. Hudson, J. Org. Chem., 1970, 35, 647 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |