DOI:
10.1039/B514748D
(Paper)
Org. Biomol. Chem., 2006,
4, 1966-1976
Synthetic peptides with selective affinity for apoptotic cells
Received
(in Pittsburgh, PA)
18th October 2005
, Accepted 6th March 2006
First published on 30th March 2006
Abstract
The appearance of anionic phosphatidylserine (PS) in the outer monolayer of the plasma membrane is a universal indicator of the early/intermediate stages of cell apoptosis. The most common method of detecting PS on a cell surface is to use the protein annexin V; however, in certain applications there is a need for alternative reagents. Recent research indicates that rationally designed zinc 2,2′-dipicolylamine (Zn2+–DPA) coordination complexes can mimic the apoptosis sensing function of annexin V. Here, a series of fluorescently-labelled, tri-and pentapeptides with side chains containing Zn2+–DPA are prepared and shown to selectively bind to anionic vesicle membranes. Fluorescein-labelled versions of the peptides are used to detect apoptotic cells by fluorescence microscopy and flow cytometry.
Introduction
Assays for programmed cell death, or apoptosis, are used often in cell biology research and also in the drug discovery process. Various intracellular methods for detecting apoptosis have been reported such as caspase action and nucleic acid fragmentation.1 An attractive extracellular strategy is to monitor the change in phospholipid distribution across the cell plasma membrane. In particular, the appearance of anionic phosphatidylserine (PS)† in the membrane outer monolayer is a universal indicator of the early/intermediate stages of cell apoptosis and can be detected before morphological changes can be observed.2–4 The most common method of detecting PS on a cell surface is to use the Ca2+-dependent, PS-binding protein annexin V.5–7 For in vitro assays, the 35 kDa protein is typically labelled with a fluorescent dye, whereas radioactive and MRI contrast agents are employed for in vivo imaging.8,9 Although it is utilized extensively, the labelled protein is expensive and moderately unstable. Thus, it is not convenient for high throughput screening assays in drug discovery. An additional concern is that approximately 2.5 mM of extracellular Ca2+ is needed for complete binding. This can lead to false positive results because most animal cells have a Ca2+ dependent scramblase that can move PS to the cell surface.10 Furthermore, complete annexin binding requires incubation times of up to one hour, which is problematic for kinetic assays.11,12 In short, dye labelled annexin V is a useful apoptosis sensor, but it has a number of limitations and there is a need for replacement reagents that are cheap, robust, low-molecular weight, rapidly-binding, membrane-impermeable, and Ca2+-independent.
We have discovered that rationally designed zinc 2,2′-dipicolylamine (Zn2+–DPA) coordination complexes can mimic the apoptosis sensing function of annexin V. Our designs were conceived after inspection of the X-ray crystal structure of an annexin V–glycerophosphoserine complex. The X-ray structure shows that one phosphoserine head group is coordinated to two bridging Ca2+ ions that are in turn coordinated to one of the four canonical binding sites at the protein surface.13 This three-component assembly picture suggested to us that synthetic metal coordination complexes with appropriate charge, geometry, and spatial orientation may bind to the head group of anionic phospholipids preferentially over zwitterionic phospholipids (Scheme 1).‡
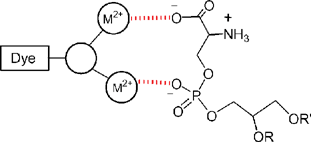 |
| | Scheme 1 Dye-labelled, metal coordination complexes can mimic the protein annexin V and coordinate to a PS head group via the phosphate and/or carboxylate anion. | |
The breakthrough compound was PSS-380 (phosphatidylserine sensor 380 absorbance, see Scheme 2) which was successfully used to detect apoptosis in Jurkat cells.14,15 The utility of PSS-380 as a fluorescent stain for PS has been confirmed independently in publications from other research groups.16,17 Although PSS-380 is a useful fluorescent sensor, its excitation wavelength (380 nm) is not compatible with the lasers in most flow cytometers. Thus, we pursued a more flexible second generation design that allowed conjugation to a range of fluorescent reporter groups.18 This work led to several new apoptosis sensors such as the fluorescein-linked PSS-480.19,20 Here, we describe a third generation design that is based on a synthetically flexible, peptide architecture. The peptide design was pursued for a number of reasons. Previous structural studies suggested that two closely spaced Zn2+–DPA units were required for selective binding to an anionic membrane surface, however, this requirement had not been thoroughly tested.18–20 Indeed, we find that highly selective binding of anionic membranes can be achieved using peptides containing two widely spaced Zn2+–DPA units. Moreover, reasonably selective binding is observed with a peptide containing just a single Zn2+–DPA unit. To facilitate the peptide synthesis we have developed a simple method of converting the amine terminus of an ornithine side chain into a DPA unit. The preparation of dye-labelled, zinc coordinated tripeptides 1–NBD and 1–Fl is described, as well as the analogous pentapeptides 2–NBD and 2–Fl (Scheme 3). The NBD-labelled peptides are employed as probes to measure affinity to vesicle membranes, and the Fl-labelled peptides as fluorescent stains for detecting cell apoptosis by fluorescence microscopy and flow cytometry.
 |
| | Scheme 2 PS sensors PSS-380 and PSS-480. | |
Results and discussion
Synthesis
The pentapeptides 2–NBD and 2–Fl were designed with glycine residues between the two amino acids bearing the DPA ligands, which allows the two coordinated zinc(II) ions to simultaneously contact a membrane surface without causing molecular strain. The central building block is the ornithine derivative 4, with a DPA side chain. Attempts to prepare this compound by direct alkylation of the amino terminus of N-Boc-ornithine, 3,21 were only partially successful due to difficulties with product purification. Therefore, we turned to a reductive amination procedure,22 treating 3 with two equivalents of picolylaldehyde in the presence of sodium triacetoxyborohydride to give 4 in good yield (Scheme 4). Material prepared via this method was readily purified by column chromatography on silica gel. The acid 4 is readily incorporated into peptides using standard solution phase peptide coupling techniques. Coupling to glycine methyl ester using EDC–HOBt as the activating agents gave the dipeptide 5 in good yield. Treatment of 5 with a 4 M solution of HCl in dioxane gave 6 as the hydrochloride salt, which was then coupled with N-Boc-glycine to give the tripeptide 8 in good yield (Scheme 5). Boc-deprotection using the conditions described above gave the amine 9 as the hydrochloride salt, which was treated with FITC in the presence of triethylamine to give the fluorescein labelled tripeptide 10. Attempts to react 9 directly with 7-chloro-4-nitrobenzofurazan (NBD-Cl) to give the analogous NBD-labelled tripeptide were unsuccessful. We considered the probable reason for this to be steric congestion and so redesigned the compound to contain a six-carbon alkyl chain between the NBD fluorophore and the peptide. Therefore, amine 6 was coupled with the known NBD-5-aminohexanoic acid 1123 to give the NBD-labelled tripeptide 12 in moderate yield.
 |
| | Scheme 4 Reagents and conditions: (a) 2-pyridinecarboxaldehyde (3 equiv.), NaHB(OAc)3 (9 equiv.), CH2Cl2, 85%; (b) EDC·HCl (1.2 equiv.), HOBt·H2O (1.2 equiv.), NMM (3 equiv.), CH2Cl2–DMF, 95%; (c) 4 M HCl in dioxane, quant.; (d) NaOH (3 equiv.), MeOH–H2O, 90%. | |
 |
| | Scheme 5 Reagents and conditions: (a) N-Boc-Gly-OH, EDC·HCl, HOBt·H2O, NMM (3 equiv.), CH3CN, 89%; (b) 4 M HCl in dioxane, quant.; (c) FITC (2 equiv.), NEt3 (4 equiv.), MeOH, 84%; (d) EDC·HCl (1.8 equiv.), HOBt·H2O (1.8 equiv.), NMM (5 equiv.), CH3CN, 58%. | |
A fragment coupling approach was employed to convert 5 into a pentapeptide. That is, the acid fragment 7 was coupled to amine 6 to yield the tetrapeptide 13 (Scheme 6). Boc-deprotection of 13 using standard conditions gave the amine 14 as the hydrochloride salt, which was coupled with 11 to give the NBD-labelled pentapeptide 15 in high yield. Similarly, coupling of amine 14 with N-Boc-protected hexanoic acid 16 under the standard conditions described above gave the pentapeptide 17 in good yield. Deprotection of 17 followed by treatment with FITC, gave the fluorescein labelled pentapeptide 18 in good yield. The fluorescently labelled peptides 10, 12, 15 and 18 were then treated with the appropriate molar equivalents of zinc(II) nitrate to give the zinc-coordinated peptides 1–Fl, 1–NBD, 2–NBD and 2–Fl, respectively.
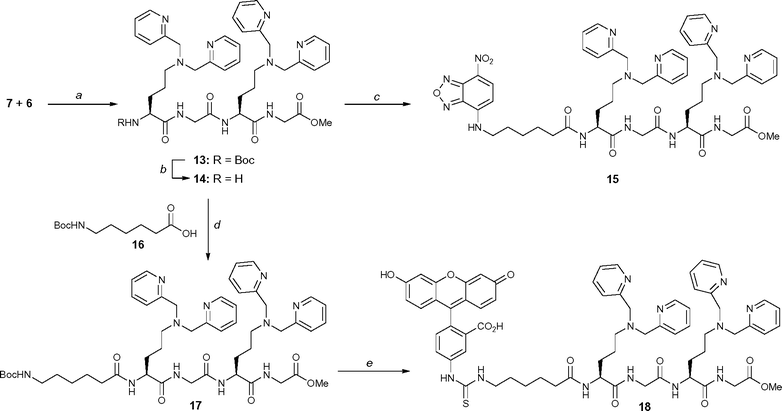 |
| | Scheme 6 Reagents and conditions: (a) EDC·HCl (1.1 equiv.), HOBt·H2O (1.1 equiv.), NMM (5 equiv.), CH2Cl2–DMF, 80%; (b) 4 M HCl in dioxane, quant.; (c) 11 (1.5 equiv.), EDC·HCl (2.5 equiv.), HOBt·H2O (2.5 equiv.), NMM (10 equiv.), CH3CN, 82%; (d) EDC·HCl (1.1 equiv.), HOBt·H2O (1.1 equiv.), NMM (6 equiv.), CH3CN, 78%; (e) Step 1: 4 M HCl in dioxane; Step 2: FITC (1.3 equiv.), NEt3 (10 equiv.), MeOH, 86%. | |
Vesicle studies
The fluorescent derivatives 1–NBD and 2–NBD were prepared because previous work had shown how the NBD fluorophore can be used to measure binding and partitioning into vesicle membranes.18,24 The emission intensity of an NBD-labelled Zn2+–DPA conjugate increases when it associates with a vesicle membrane, which allows titration experiments to be conducted. Thus, separate samples of 1–NBD and 2–NBD were titrated with either zwitterionic vesicles composed only of POPC, or anionic vesicles composed of 1 : 1 mixtures of POPG–POPC, POPA–POPC, or POPS–POPC (see Scheme 7 for phospholipid structures). The unilamellar vesicles (100 nm diameter, 25 µM total phospholipid, pH 7.4) were prepared by standard extrusion methods. The titration curves in Fig. 1 show that both peptides have very weak affinities for the zwitterionic vesicles, but moderately strong affinities for the anionic vesicles. The apparent binding constants are all around 5 × 104 M−1 (Table 1), which are very similar to those obtained previously with other Zn2+–DPA complexes.18 In the case of 1–NBD there is some variation in the change in fluorescence intensity upon vesicle binding, which is suggestive of differences in the non-covalent interactions of the monozinc peptide with the vesicle membrane. However, in the case of 2–NBD the changes in fluorescence intensity upon binding to anionic vesicles are very similar for all three vesicle compositions. This suggests that the attractive interaction between the strongly hydrophilic bis(zinc) peptide and the anionic vesicle membrane is dominated by electrostatic effects, and is independent of the specific structure of the phospholipid head-groups. The peptides 1–NBD and 2–NBD only respond to membrane-bound phospholipids because no changes in emission are observed when the titrations are repeated with short acyl chain phospholipids that do not form a bilayer membrane. For example, addition of a 1 : 1 mixture (100 µM) of dihexanoylphosphatidylcholine (DHPC) and dihexanoylphosphatidylserine (DHPS) to a 1 µM solution of 2–NBD results in no detectable increase in NBD fluorescence intensity.
| Peptide |
K
a
× 104/M−1b |
| 100% POPC |
1 : 1 POPA–POPC |
1 : 1 POPS–POPC |
1 : 1 POPG–POPC |
|
5 mM TES, 100 mM NaCl, pH 7.4.
Average of 3 separate experiments.
|
|
1–NBD |
<1 |
3.9 ± 0.5 |
2.4 ± 1.5 |
1.8 ± 1.2 |
|
2–NBD |
<1 |
4.7 ± 0.5 |
4.9 ± 0.6 |
4.4 ± 1.2 |
 |
| | Fig. 1 Fluorescence intensity change of: (a) 1–NBD, or (b) 2–NBD; (1 µM) in the presence of 100 mol% POPC (✦), 1 : 1 POPG–POPC (×), 1 : 1 POPS–POPC (■) and POPS–POPC (△) vesicles in buffer (5 mM TES, 100 mM NaCl, pH 7.4) at 25 °C. | |
 |
| | Scheme 7 Phospholipid structures. | |
The vesicle partitioning assay is based on the ability of dithionite to chemically reduce the NBD fluorophore and quench its fluorescence. Sodium dithionite cannot cross vesicle membranes, so addition of the reagent to vesicle dispersions can only quench fluorophores that are in the external solution or exposed on the membrane surface. Any NBD-labelled compound that partitions into the vesicle membrane is protected from immediate quenching by added sodium dithionite. The results of our NBD protection assays are shown in Fig. 2. The peptides 1–NBD and 2–NBD were added to various zwitterionic and anionic vesicle dispersions, and after standing for 2 h, an aliquot from each sample was treated with excess sodium dithionite. In the case of 2–NBD, the fluorescence of all samples was completely and immediately quenched. In other words, the bis(zinc) 2–NBD does not partition into any of the vesicle compositions. In the case of 1–NBD, however, there is clear evidence that anionic vesicles composed of 1 : 1 POPG–POPC can protect about 40% of the NBD-conjugate from immediate reaction with the dithionite. The 1–NBD quenching curve in Fig. 2b shows biphasic kinetics. Upon dithionite addition there is a sudden drop in fluorescence emission due to rapid reaction with the exposed NBD groups, followed by a slower rate of quenching attributed to either transport of the dithionite into the vesicles or re-exposure of internalized NBD on the membrane surface. In other words, the mono zinc 1–NBD not only associates with the anionic POPG–POPC vesicle membranes but it subsequently diffuses to the inner membrane surface. In summary, both the monozinc 1–NBD and the bis(zinc) 2–NBD appear to associate equally well with anionic vesicle membranes. However, the less charged 1–NBD appears to interact with the phospholipid head-groups and in one case (1 : 1 POPG–POPC) there is clear evidence that it can partition into the vesicle membrane. The more highly charged 2–NBD stays only at the anionic membrane surface and its binding interaction is essentially independent of the specific structure of the phospholipid head-groups.
 |
| | Fig. 2 NBD protection assay. An aliquot (1.25 µM) of 1–NBD (■) or 2–NBD (×) was added to a 25 µM solution of vesicles in TES buffer, at 25 °C. After 2 h, a sample was taken and assayed. Sodium dithionite was added at assay time of 50 s and Triton-X 100 was added at 150 s. (a) 1 : 1 POPA–POPC vesicles, (b) 1 : 1 POPG–POPC vesicles, (c) 1 : 1 POPS–POPC vesicles, (d) 100 mol% POPC vesicles. | |
Cell studies
Fluorescence microscopy was used to determine if the fluorescein-labelled peptides 1–Fl and 2–Fl could selectively stain apoptotic cells. Separate samples of HeLa and Jurkat cells were treated first with the anticancer drug camptothecin to induce apoptosis, and then simultaneously with 1–Fl (or 2–Fl) and the nuclear stain 7AAD (7-aminoactinomycin D) (Fig. 3–6). Necrotic cells, as well as those cells in the advanced stages of apoptosis, have permeabilized membranes and allow 7AAD to stain the cell nucleus. Healthy cells and those cells in the early to intermediate stages of apoptosis retain their membrane integrity and exclude 7AAD. This allows cells in early apoptosis to be identified by selective staining with 1–Fl (or 2–Fl) and exclusion of 7AAD. The images in Fig. 3–6 indicate that both peptides are quite effective at selectively staining apoptotic cells. A close comparison of the staining in Fig. 5–6 suggests that the bis(zinc) 2–Fl is perhaps slightly better at discriminating between vital and apoptotic cells. However, in both cases the staining selectivity is very similar to that obtained with annexin V–FITC. Furthermore, the staining rate is essentially instantaneous which is an improvement over the sluggish kinetics of annexin V.11,12
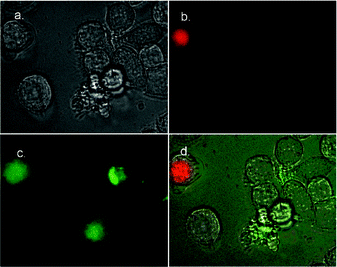 |
| | Fig. 3 Fluorescence micrographs (600× magnification) of HeLa cells treated with camptothecin (10 µM) for 3.5 h to induce apoptosis, then simultaneously stained with 1–Fl (10 µM) and 7AAD (500 ng mL−1). Phase contrast image of treated cells is shown in (a). Fluorescence emission due to 7AAD is shown in (b), fluorescence emission due to 1–Fl is shown in (c). Overlay of both (b) and (c) onto phase contrast image in (d). | |
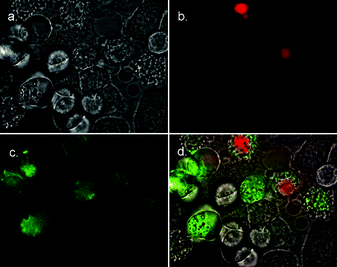 |
| | Fig. 4 Fluorescence micrographs (600× magnification) of HeLa cells treated with camptothecin (10 µM) for 3.5 h to induce apoptosis, then simultaneously stained with 2–Fl (10 µM) and 7AAD (500 ng mL−1). Phase contrast image of treated cells is shown in (a). Fluorescence emission due to 7AAD is shown in (b), fluorescence emission due to 2–Fl is shown in (c). Overlay of both (b) and (c) onto phase contrast image in (d). | |
 |
| | Fig. 5 Fluorescence micrographs (600× magnification) of Jurkat cells treated with camptothecin (10 µM) for 3.5 h to induce apoptosis, then simultaneously stained with 1–Fl (10 µM) and 7AAD (500 ng mL−1). Phase contrast image of treated cells is shown in (a). Fluorescence emission due to 7AAD is shown in (b), fluorescence emission due to 1–Fl is shown in (c). Overlay of both (b) and (c) onto phase contrast image in (d). | |
 |
| | Fig. 6 Fluorescence micrographs (600× magnification) of Jurkat cells treated with camptothecin (10 µM) for 3.5 h to induce apoptosis, then simultaneously stained with 2–Fl (10 µM) and 7AAD (500 ng mL−1). Phase contrast image of treated cells is shown in (a). Fluorescence emission due to 7AAD is shown in (b), fluorescence emission due to 2–Fl is shown in (c). Overlay of both (b) and (c) onto phase contrast image in (d). | |
The utility of 2–Fl in flow cytometry was demonstrated using a population of Jurkat cells that were treated with different concentrations of camptothecin (0 µM, 10 µM, or 12.5 µM for 16 h) to induce apoptosis. The histograms in Fig. 7 indicate that the fraction of apoptotic cells increases with the dose of camptothecin. Nearly identical percentages of apoptotic cells were identified in each group using annexin V–FITC (data not shown).
 |
| | Fig. 7 Flow cytometry histograms illustrating staining of Jurkat cells by 2–Fl. Cells treated with camptothecin (II, 10 µM for 16 h and III, 12.5 µM for 16 h) exhibit significantly more staining by 2–Fl than do control cells (I, no camptothecin). About 30% of treated cells were identified as apoptotic using 2–Fl, while less than 4% of the untreated cells were stained with 2–Fl. a.u., arbitrary units. | |
Conclusions
The fluorescent Zn2+–DPA peptides, 1–Fl and 2–Fl, are able to identify apoptotic cells using fluorescence microscopy. Furthermore, 2–Fl is an effective reagent for determining the fraction of apoptotic cells in a cultured population using flow cytometry. The cationic peptide complexes selectively stain apoptotic cells because the cell plasma membrane becomes increasingly anionic due to the appearance of phosphatidylserine. The peptides are practical alternatives to the 35 KD protein annexin V currently used in apoptosis assays. The results reported here demonstrate that the molecular design of synthetic phosphatidylserine sensors does not necessarily require two closely spaced Zn2+–DPA units. Indeed, it is reasonable to expect that a wide range of Zn2+–DPA coordination complexes will have selective affinity for apoptotic cells. An attractive feature with the peptides reported here is they have two orthogonal conjugation points. That is, their N-terminus is labelled with a dye but their C-terminus remains available for attachment to groups that can fine-tune the recognition properties of the peptide. Furthermore, it should possible to attach these peptides to the surfaces of nanoparticles which should enhance affinity due to multivalent interactions. This may a useful way to construct novel contrast agents for in vivo imaging of apoptotic tissue.8,25,26 Another potential application is tumor targeting, since it is known that PS exposure is increased on the surface of tumor blood vessels.27
Experimental
General
Unless specified otherwise, all reactions were performed under an inert atmosphere of nitrogen with dry, freshly distilled solvents under anhydrous conditions and monitored by TLC using aluminium backed plates coated with Merck Silica Gel 60 F254 and visualised using either UV light (254 or 366 nm) or a molybdenum staining reagent [a solution of (NH4)6Mo7O24·4H2O (20 g) and Ce(SO4)2 (0.4 g) in 10% sulfuric acid (400 mL)]. Flash chromatography was performed on silica gel (Merck silica gel 60, 40–63 µm) at a pressure of 0.3–0.4 bar and the yields given refer to chromatographically and spectroscopically (1H NMR) homogenous material. Ratios of solvent systems for TLC and column chromatography are expressed in v/v as specified. Reverse phase HPLC (RP-HPLC) was performed using a Waters apparatus with a tunable absorbance detector. Analytical RP-HPLC was performed using an Alltech-Altima C18 column (5 µm, 4.6 mm ID, 250 mm) with a flow rate of 0.8 mL min−1. Preparative RP-HPLC was performed using an Alltech-Altima C18 column (10 µm, 22 mm ID, 300 mm) with a flow rate of 7.0 mL min−1. Retention times given are for the analytical column. Melting points were determined using a Gallenkamp melting point apparatus and are reported in degrees Celsius (uncorrected). Infrared absorption spectra were obtained using a Shimadzu FTIR-8400S spectrometer as a thin film between sodium chloride plates. NMR spectra were recorded on a Bruker Avance DPX 200 or a Bruker Avance DPX 300 spectrometer. The solvent 1H and 13C signals, δH 7.26 for residual CHCl3 and δC 77.0 for CDCl3, δH 3.31 and δC 49.0 for d4-MeOH, δH 2.50 and δC 39.5 for d6-DMSO, were used as internal references. J values are given in Hz. Low resolution mass spectra were recorded on a Finnigan LCQ ion trap mass spectrometer (ESI). High resolution mass spectra were recorded on a BioApex Fourier Transform Ion Cyclotron Resonance Mass Spectrometer (ESI). Optical rotations were measured on a Polaar 2001 dual wavelength polarimeter monitor in a 2.5 dm cell at 22 °C using the indicated spectroscopic grade solvents. Elemental analyses were performed by Campbell Microanalytical Laboratories. Most reagents were commercially available from Aldrich, Fluka or Novabiochem and used as supplied. NBD-Hex-OH was prepared according to the method of Jürss and Maelicke.23 All polar lipids were purchased from Avanti Polar Lipids, Inc. annexin V and 7AAD were obtained from BD Biosciences.
Synthesis
Boc-Orn(DPA)-OH (4).
2-Pyridinecarboxaldehyde (13.8 g, 129 mmol) was added to a fine suspension of 3 (10.5 g, 45 mmol) in dichloromethane (400 mL) and the resulting mixture was stirred for 15 min. Sodium triacetoxyborohydride (40.2 g, 430 mmol) was added and the resulting cloudy yellow solution was stirred for 18 h at ambient temperature under a nitrogen atmosphere. The reaction was quenched by the addition of aqueous sodium hydroxide (2 M, 150 mL) then the mixture was acidified to pH 6 by addition of 1 M HCl. The organic phase was separated and the aqueous phase was extracted with chloroform–isopropanol (3 : 1, v/v) (2 × 100 mL). The organic phases were combined and washed with brine (200 mL) then dried with MgSO4. The solvent was removed under reduced pressure to give a yellow oil which was purified by column chromatography on silica gel (9 : 1 v/v chloroform–methanol elution). Concentration of the appropriate fractions gave 4 as a colourless solid (15.9 g, 85%); mp 128–131 °C; [α]22D +14.9 (c 1.5 in CHCl3); νmax (NaCl)/cm−1 3417, 3311, 2975, 1660, 1479, 1249, 983, 763; δH (300 MHz, d6-DMSO) 8.45 (d, J 4.3, 2H), 7.74 (dd, J 7.7 and 7.3, 2H), 7.50 (d, J 7.7, 2H), 7.22 (m 2H) 6.31 (br. s, 1H), 3.77 (m, 1H), 3.67 (s, 4H), 2.41 (m, 2H), 1.85–1.49 (m, 4H), 1.35 (s, 9H), OH not observed; δC (75 MHz, d6-DMSO) 176.2, 159.5, 155.0, 148.7, 136.4, 122.4, 122.0, 77.5, 59.5, 54.4, 53.5, 30.1, 28.2, 22.7; m/z (ESI) 415.2352 (C22H31N4O4 requires 415.2346), 415 [M + H]+ (100%), 437 [M + Na]+ (30%), 315 (38%).
Boc-Orn(DPA)-Gly-OMe (5).
Acid 4 (6.80 g, 16 mmol) and HCl.NH2-Gly-OMe (2.40 g, 19 mmol) were dissolved in a mixture of dichloromethane (40 mL) and dimethylformamide (12 mL). The solution was cooled to 0 °C under an atmosphere of nitrogen, then HOBt·H2O (2.76 g, 20 mmol) and EDC·HCl (3.45 g, 18 mmol) were added, followed by N-methyl morpholine (5.4 mL, 49 mmol). The mixture was warmed to room temperature and stirred for 18 h, then partitioned between saturated aqueous sodium bicarbonate (100 mL) and ethyl acetate (100 mL). The aqueous phase was extracted with ethyl acetate (3 × 100 mL) and the combined organic phases were washed with water (100 mL) and brine (2 × 100 mL) then dried (MgSO4). The solvent was removed under reduced pressure to give a yellow oil which was purified by column chromatography on silica gel (190 : 9 : 1, v/v chloroform–methanol–aqueous ammonia elution). Concentration of the appropriate fractions gave 5 as a yellow oil (7.54 g, 95%); [α]22D +1.08 (c 13.7, CHCl3); νmax (CHCl3)/cm−1: 3304, 2976, 2951, 2934, 2818, 1750, 1700, 1675, 1591, 1526, 1435, 1366, 1207, 1175; δH (300 MHz, d6-DMSO) 8.47 (m, 2H), 8.24 (t, J 5.7, 1H), 7.75 (ddd, J 7.7, 7.7 and 1.8, 2H), 7.51 (d, J 7.7 Hz, 2H), 7.23 (m, 2H), 6.90 (d, J 8.0, 1H), 3.93–3.79 (m, 3H), 3.70 (s, 4H), 3.60 (s, 3H), 2.44 (m, 2H), 1.56–1.54 (m, 4H), 1.35 (s, 9H); δC (75 MHz, d6-DMSO) 172.7, 170.2, 159.4, 155.2, 148.7, 136.4, 122.5, 122.0, 79.2, 59.5, 53.9, 53.3, 51.6, 40.6, 29.9, 28.2, 22.9; m/z (ESI) 486.2703 [(M + H)+; C25H37N5O5 requires 486.2716], 508 [M + Na]+ (75%).
HCl·NH2-Orn(DPA)-Gly-OMe (6).
A solution of HCl in dioxane (4 M, 2 mL) was added to dipeptide 5 (1.0 g, 2.1 mmol) and the resulting mixture stirred at ambient temperature under a nitrogen atmosphere for 8 h. The solvent was removed under reduced pressure and the residue azeotroped with toluene (3 × 10 mL) to give 6 as a colourless solid (2.1 mmol, quant.) which was used without further purification; δH (300 MHz, d6-DMSO) 9.21 (m, 1H), 8.76 (d, J 4.5, 2H), 8.47 (m, 2H), 8.34 (m, 2H), 7.96 (m, 2H), 7.74 (m, 2H), 4.45 (s, 4H), 4.00–3.75 (m, 3H), 3.62 (s, 3H), 2.90 (m, 2H), 1.8 (m, 4H); m/z (ESI) 386 [M + H]+ (100%).
Boc-Orn(DPA)-Gly-OH (7).
A solution of NaOH (0.35 g, 8.8 mmol) in H2O (2 mL) was added to a solution of 5 (1.4 g, 2.9 mmol) in methanol (7 mL). The solution was stirred at ambient temperature for 4 h, then adjusted to pH 7 by the addition of 1 M HCl. The resulting mixture was extracted with chloroform–isopropanol (3 : 1 v/v, 4 × 50 mL) and the extracts combined, then the solvent removed under reduced pressure to give the carboxylic acid 7 as a pale yellow solid (0.46 g, 90%) which was used without further purification; δH (300 MHz, d6-DMSO) 8.49 (d, J 4.3, 2H), 8.12 (m, 1H), 7.76 (m, 2H), 7.51 (d, J 7.8, 2H), 7.26 (m, 2H), 6.89 (d, J 8.2, 1H), 4.31 (m, 1H), 3.9–3.45 (m, 6H), 2.49 (partially obscured, 2H), 1.65–1.40 (m, 4H), 1.36 (s, 9H), OH not observed; m/z (ESI) 494 [M + Na]+ (40%), 472 [M + H]+ (100%).
Boc-Gly-Orn(DPA)-Gly-OMe (8).
Boc-Gly-OH (0.12 g, 0.68 mmol) and 6 (0.28 g, 0.68 mmol) were dissolved in acetonitrile (8 mL) and the solution cooled to 0 °C under an atmosphere of nitrogen. HOBt·H2O (0.10 g, 0.66 mmol) and EDC·HCl (0.13 g, 0.66 mmol) were then added followed by N-methylmorpholine (0.18 g, 1.8 mmol) and the mixture was warmed to room temperature and stirred for 23 h. Saturated aqueous sodium bicarbonate (50 mL) was then added and the resulting mixture was extracted with ethyl acetate (3 × 50 mL). The combined organic phases were washed with water (50 mL) and brine (70 mL) then dried (MgSO4). The solvent was removed under reduced pressure to give a yellow solid which was purified by column chromatography on silica gel (190 : 9 : 1, v/v chloroform–methanol–aqueous ammonia elution). Concentration of the appropriate fractions gave the tripeptide 8 as a pale yellow solid (0.33 g, 89%); mp 36–42 °C (hygroscopic); [α]22D +0.72 (c 5.0 in CHCl3); found: C, 56.1; H, 7.0; N, 14.6. C27H38N6O6.2H2O requires C, 56.0; H, 7.3; N, 14.5; νmax (CHCl3)/cm−1: 3300, 3295, 3055, 3007, 2977, 2950, 2935, 2816, 1755, 1660, 1435, 1367, 1281, 1248, 1207, 1173, 1049, 864, 754; δH (300 MHz, d6-DMSO) 8.47 (dd, J 4.7 and 0.8, 2H), 8.40 (t, J 5.7, 1H), 7.86 (d, J 8.0, 1H), 7.75 (ddd, J 7.8, 7.7 and 1.8, 2H), 7.50 (d, J 7.8, 2H), 7.23 (m, 2H), 6.95 (t, J 5.8, 1H), 4.28 (m, 1H), 3.82 (t, J 5.5, 2H), 3.70 (s, 4H), 3.60 (s, 3H), 3.55 (d, J 5.9, 2H), 2.43 (m, 2H), 1.70–1.40 (m, 4H), 1.36 (s, 9H); δC (75 MHz, d6-DMSO) 172.0, 170.1, 169.5, 159,4, 155.8, 148.7, 136.4, 122.5, 122.0, 78.0, 59.4, 53.2, 52.0, 51.6, 43.2, 40.4, 30.1, 28.1, 22.5; m/z (ESI) 543.2940 [(M + H)+; C27H39N6O6 requires 543.2931], 565 [M + Na]+ (100%).
HCl·H2N-Gly-Orn(DPA)-Gly-OMe (9).
A solution of HCl in dioxane (4 M, 2 mL) was added to tripeptide 8 (80 mg, 0.15 mmol) and the resulting mixture stirred at ambient temperature under a nitrogen atmosphere for 8 h. The solvent was removed under reduced pressure and the residue azeotroped with toluene (3 × 10 mL) to give 9 as a colourless solid (0.15 mmol, quant.) which was used without further purification; δH (200 MHz, CD3OD) 8.77 (dd, J 5.5 and 1.0, 2H), 8.36 (ddd, J 7.8, 7.7 and 1.6, 2H), 7.95 (d, J 7.8, 2H), 7.83 (m, 2H), 4.36 (s, 4H), 4.05 (m, 2H), 4.00–3.68 (m, 3H), 3.68 (s, 3H), 2.94 (m, 2H), 1.82 (m, 4H), 2 × NH and NH3+ not observed; m/z (ESI) 465 [M + Na]+ (15%), 443 [M + H]+ (100%).
Fl–Gly-Orn(DPA)-Gly-OMe (10).
FITC (0.30 g, 0.77 mmol) was added to a solution of tripeptide 9 (0.16 g, 0.36 mmol) in MeOH (2.5 mL) then NEt3 (0.19 mL, 1.4 mmol) was added. The mixture was stirred at room temperature under an atmosphere of nitrogen for 2.5 h in the dark then cooled in an ice bath to give 10 as a dark red precipitate which was collected by filtration (0.25 g, 84%). A portion of this precipitate (30 mg) was purified by preparative HPLC (gradient of 30 to 100% MeOH (0.05% TFA) in H2O (0.05% TFA) over 30 min; tR 24.6 min) for characterization and use in assays; δH (200 MHz, CD3OD) 8.55 (2H, d, J 4.2), 8.23 (1H, d, J 1.7), 7.82–7.68 (3H, m), 7.42–7.30 (4H, m), 7.08 (1H, d, J 8.2), 6.67–6.47 (6H, m), 4.49 (4H, s), 4.45 (1H, m), 4.03 (2H, ABq, J 16.7), 3.87 (2H, m), 3.61 (3H, s), 3.20 (2H, m), 2.2–1.6 (4H, m), 2 × OH and 4 × NH not observed; m/z
(ESI) 830.2649 [(M − H+)−; C43H40N7O9S requires 830.2617].
Fl–Gly-Orn(DPA)-Gly-OMe·[Zn(NO3)2] (1–Fl).
A solution of 10 (0.024 mmol) in MeOH (2 mL) and an aqueous solution of Zn(NO3)2·6H2O (0.024 mmol) were mixed and stirred for 0.5 h. The solvent was removed and the residue lyophilized to afford the complex 1–Fl in quantitative yield.
NBD–Hex-Orn(DPA)-Gly-OMe (12).
NBD–5-aminohexanoic acid 11 (66 mg, 0.22 mmol) was added to a solution of 6 (74 mg, 0.15 mmol) in CH3CN (2.5 mL) and N-methylmorpholine (0.12 mL, 1.1 mmol), then EDC·HCl (52 mg, 0.27 mmol) and HOBt·H2O (36 mg, 0.27 mmol) were added. The resulting solution was stirred at room temperature for 24 h, then partitioned between CHCl3–i-PrOH (3 : 1, 20 mL) and sat. aq. NaHCO3 (20 mL). The aqueous layer was extracted with CHCl3–i-PrOH (3 : 1, 2 × 20 mL), then the organic fractions were combined and washed with H2O (2 × 20 mL) and brine (20 mL), dried (MgSO4) and the solvent was removed under reduced pressure to give a dark brown oil. Purification by preparative thin layer chromatography (CHCl3–MeOH–aq. NH3; 140 : 18 : 2) gave 12 as an orange oil (58 mg, 58%). δH (300 MHz, CD3OD) 8.39 (m, 3H), 7.75 (dt, J 1.7 and 7.7, 2H), 7.57 (d, J 7.7, 2H), 7.22 (m, 2H), 6.24 (d, J 8.9, 1H), 4.33 (m, 1H), 3.92 (ABq, J 17.6, 2H), 3.74 (s, 4H), 3.66 (s, 3H), 3.47 (br. s, 2H), 2.52 (t, J 6.5, 2H), 2.26 (t, J 7.3, 2H), 1.84–1.59 (br. m, 8H), 1.50–1.42 (m, 2H), 3 × NH not observed; δC (75 MHz, CD3OD) 178.8, 175.9, 174.9, 160.5, 149.3, 146.5, 145.7, 145.4, 138.6, 124.9, 123.7, 122.8, 99.6, 60.9, 55.1, 54.3, 52.6, 44.7, 41.8, 36.5, 31.0, 29.0, 27.5, 26.4, 24.1, 1 signal obscured or overlapping; m/z (ESI) 662.3038 [(M + H)+; C32H40N9O7 requires 662.3045], 684 [M + Na]+ (21%).
NBD–Hex-Orn(DPA)-Gly-OMe·[Zn(NO3)2] (1–NBD).
A solution of 12 (0.030 mmol) in MeOH (2 mL) and an aqueous solution of Zn(NO3)2·6H2O (0.030 mmol) were mixed and stirred for 0.5 h. The solvent was removed and the residue lyophilized to afford the complex 1–NBD in quantitative yield.
Boc-Orn(DPA)-Gly-Orn(DPA)-Gly-OMe (13).
Acid 7 (0.28 g, 0.59 mmol) and amine 6 (0.29 g, 0.59 mmol) were dissolved in a solution of dichloromethane–dimethylformamide (3 : 1, v/v, 8 mL) which was cooled to 0 °C under an atmosphere of nitrogen. HOBt·H2O (0.1 g, 0.68 mmol) and EDC·HCl (0.13 g, 0.68 mmol) were then added followed by N-methylmorpholine (0.32 mL, 2.95 mmol) before the mixture was warmed to room temperature and stirred for 23 h. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate (50 mL) and ethyl acetate (40 mL). The aqueous phase was extracted with ethyl acetate (3 × 50 mL) then the organic phases were combined and washed with water (4 × 50 mL), followed by brine (50 mL) then dried with MgSO4. The solvent was removed under reduced pressure to give a yellow oil. Subjection of the crude material to column chromatography on silica gel (190 : 9 : 1, v/v chloroform–methanol–aqueous ammonia elution) and concentration of the appropriate fractions gave the tetrapeptide 13 as a pale yellow solid (0.40 g, 80%); mp 58–62 °C; [α]22D +0.27 (c 5.7 in CHCl3); found: C, 59.2; H, 6.8; N, 15.6. C44H58N10O7.3H2O requires C, 59.2; H, 7.2; N, 15.7; νmax (CHCl3)/cm−1: 3295, 3053, 2949, 2934, 2818, 1751, 1684, 1647, 1433, 1366, 1248, 1207, 1171; δH (300 MHz, CD3OD) 8.43 (m, 4H), 7.77 (m, 4H), 7.61 (m, 4H), 7.26 (m, 4H), 4.33 (m, 1H), 4.00–3.83 (m, 5H), 3.77 (s, 8H), 3.67 (s, 3H), 2.53 (m, 4H), 1.95–1.50 (m, 8H), 1.39 (s, 9H), 4 × NH not observed; δC (75 MHz, CD3OD) 175.9, 174.7, 171.6, 160.7, 160.6, 149.5, 138.8, 125.1, 123.8, 80.7, 61.0, 56.3, 55.1(9), 55.1(3), 54.5, 52.6, 43.7, 41.9, 30.9, 28.8, 24.3, 24.2; m/z (ESI) 839.4542 [(M + H)+; C44H59N10O7 requires 839.4568], 861 [M + Na]+ (66%).
HCl·H2N-Orn(DPA)-Gly-Orn(DPA)-Gly-OMe (14).
A solution of HCl in dioxane (4 M, 2 mL) was added to tetrapeptide 13 (0.13 g, 0.16 mmol) and the resulting mixture stirred at ambient temperature under a nitrogen atmosphere for 8 h. The solvent was removed under reduced pressure and the residue azeotroped with toluene (3 × 10 mL) to give 14 as a colourless solid (0.16 mmol, quant.) which was used without further purification; δH (200 MHz, CD3OD) 8.89 (br. s, 4H), 8.61 (br. s, 4H), 8.19 (br. s, 4H), 8.03 (br. s, 4H), 4.38 (s, 8H), 4.40–3.75 (m, 6H), 3.66 (s, 3H), 2.71 (br. s, 4H), 1.87–1.65 (m, 8H), 3 × NH and NH3+ not observed; m/z (ESI) 762 [M + Na]+ (100%), 740 [M + H]+ (55%).
NBD–Hex-Orn(DPA)-Gly-Orn(DPA)-Gly-OMe (15).
Acid 11 (66 mg, 0.22 mmol) was added to a solution of 14 (110 mg, 0.1 mmol) in CH3CN (2.5 mL) and N-methylmorpholine (0.12 mL, 1.1 mmol), then EDC·HCl (52 mg, 0.27 mmol) and HOBt·H2O (41 mg, 0.27 mmol) were added. The resulting solution was stirred at room temperature for 24 h, then partitioned between CHCl3–i-PrOH (3 : 1, 20 mL) and sat. aq. NaHCO3 (20 mL). The aqueous layer was extracted with CHCl3–i-PrOH (3 : 1, 2 × 20 mL), then the organic fractions were combined and washed with H2O (2 × 20 mL) and brine (20 mL), dried (MgSO4) and the solvent was removed under reduced pressure to give a dark brown oil. Purification by preparative thin layer chromatography (CHCl3–MeOH–aq. NH3; 170 : 27 : 3) gave 15 as an orange oil (83 mg, 82%). δH (300 MHz, CD3OD) 8.40 (m, 5H), 7.75 (m, 4H), 7.57 (m, 4H), 7.23 (m, 4H), 6.26 (d, J 8.9, 1H), 4.34 (m, 1H), 4.19 (m, 1H), 3.98–3.82 (m, 4H), 3.73 (s, 8H), 3.64 (s, 3H), 3.48 (br. s, 2H), 2.52 (m, 4H), 2.23 (t, J 7.2, 2H), 1.89–1.40 (br. m, 14 H), 5 × NH not observed; δC (75 MHz, CD3OD) 176.0, 175.1, 174.7, 171.5, 171.4, 160.6, 160.5, 149.4, 149.3, 146.5, 145.7, 138.6, 124.9, 123.7, 122.7, 99.6, 60.9, 55.1, 55.0, 54.5, 52.6, 44.6, 43.6, 41.8, 36.4, 30.8, 30.4, 29.0, 28.8, 27.5, 26.3, 24.2, 24.0, 6 signals obscured or overlapping; m/z (ESI) 1037.4690 [(M + Na)+; C51H62N14O9Na requires 1037.4717], 1016 [M + H]+ (100%).
NBD–Hex-Orn(DPA)-Gly-Orn(DPA)-Gly-OMe·2[Zn(NO3)2] (2–NBD).
A solution of 15 (0.020 mmol) in MeOH (2 mL) and an aqueous solution of Zn(NO3)2.6H2O (0.040 mmol) were mixed and stirred for 0.5 h. The solvent was removed and the residue lyophilized to afford the complex 2–NBD in quantitative yield.
Boc-Hex-Orn(DPA)-Gly-Orn(DPA)-Gly-OMe (17).
Boc-Hex-OH 16 (0.035 g, 0.15 mmol) and 14 (0.16 g, 0.16 mmol) were dissolved in anhydrous acetonitrile, and the resulting solution was cooled to 0 °C under an atmosphere of nitrogen. HOBt·H2O (0.026 g, 0.17 mmol) and EDC·HCl (0.033 g, 0.17 mmol) were then added followed by N-methyl morpholine (0.11 mL, 1.0 mmol) then the mixture was warmed to room temperature and stirred for 23 h. The reaction mixture was partitioned between saturated aqueous sodium bicarbonate (50 mL) and ethyl acetate (40 mL). The aqueous phase was extracted with ethyl acetate (3 × 50 mL) then the organic phases were combined and washed with water (4 × 50 mL), followed by brine (50 mL) then dried with MgSO4. The solvent was removed under reduced pressure to give a yellow oil. Subjection of the crude material to column chromatography on silica gel (190 : 9 : 1, v/v chloroform–methanol–aqueous ammonia elution) and concentration of the appropriate fractions gave the pentapeptide 17 as a pale yellow foam (0.11 g, 78%); δH (300 MHz, CD3OD) 8.42 (4H, m), 7.78 (4H, m), 7.60 (4H, dd, J 6.4 and 7.5), 7.26 (4H, m), 4.33 (1H, m), 4.14 (1H, m), 3.93– 3.84 (4H, m), 3.81 (8H, s), 3.67 (3H, s), 2.99 (2H, t, J 6.8), 2.54 (4H, m), 2.18 (2H, t, J 7.3), 1.87–1.19 (14H, br. m), 1.45 (9H, s), 5 × NH not observed; δC (75 MHz, CD3OD) 176.3, 175.2, 174.8, 171.5, 171.4, 160.7, 160.6, 158.5, 149.4(4), 149.4(2), 138.7, 125.0, 123.8(7), 123.8(6), 61.0, 60.9, 55.1(3), 55.1(1), 54.6, 52.6, 43.6, 41.8, 41.2, 36.6, 30.8, 30.7, 30.4, 28.2, 27.5, 26.5, 24.3, 24.1, 3 signals obscured or overlapping.
Fl–Hex-Orn(DPA)-Gly-Orn(DPA)-Gly-OMe (18).
A solution of HCl in dioxane (4 M, 2 mL) was added to pentapeptide 17 (0.10 g, 0.11 mmol) and the resulting mixture stirred at ambient temperature under a nitrogen atmosphere for 8 h. The solvent was removed under reduced pressure and the residue azeotroped with toluene (3 × 10 mL) to give the corresponding amine as a colourless solid (0.11 mmol, quant.) which was used without further purification.
FITC (28 mg, 0.073 mmol) was added to a solution of this amine (50 mg, 0.056 mmol) in MeOH (2.5 mL) then NEt3 (0.08 mL, 0.58 mmol) was added. The mixture was stirred at room temperature under an atmosphere of nitrogen for 2.5 h in the dark then cooled in an ice bath to give 18 as a dark red precipitate which was collected by filtration (60 mg, 86%). A portion of this precipitate (20 mg) was purified by preparative HPLC (gradient of 10 to 60% MeOH (0.05% TFA) in H2O (0.05% TFA) over 30 min; tR 26.1 min) for characterization and use in assays. δH (200 MHz, CD3OD) 8.65 (4H, d, J 4.6), 8.25 (1H, d, J 1.6), 7.94 (4H, ddd, J 7.8, 7.7 and 1.5), 7.77 (1H, d, J 6.3), 7.55–7.45 (8H, m), 7.21 (1H, d, J 8.2), 6.88– 6.64 (6H, m), 4.55 (8H, s), 4.48–4.26 (2H, m), 3.89 (4H, m), 3.67 (3H, s), 3.66 (2H, m), 3.24 (4H, m), 2.29 (2H, t, J 7.0), 2.01–1.22 (14 H, br. m), 2 × OH and 6 × NH not observed; m/z (ESI) 1241 [(M + H)+ (10%)].
Fl–Hex-Orn(DPA)-Gly-Orn(DPA)-Gly-OMe·2[Zn(NO3)2] (2–Fl).
A solution of 18 (0.012 mmol) in MeOH (2 mL) and an aqueous solution of Zn(NO3)2·6H2O (0.024 mmol) were mixed and stirred for 0.5 h. The solvent was removed and the residue lyophilized to afford the complex 2–Fl in quantitative yield. m/z (ESI) 683.1705 [(M–2H+ + 2Zn2+)2+; C66H70N12O11SZn22+ requires 683.1800].
Vesicle preparation
Stock solutions of 10 mM vesicles (either 100 mol% POPC, 1 : 1 POPA–POPC, 1 : 1 POPG–POPC or 1 : 1 POPS–POPC) were made by rehydration of thin films at room temperature with TES buffer (5 mM TES, 100 mM NaCl, pH 7.4). The resulting multilamellar vesicles were extruded to form unilamellar vesicles with a Basic LiposoFast device purchased from Avestin, Inc. The samples were extruded 29 times through a 19-mm polycarbonate Nucleopore filter with 100-nm diameter pores.
NBD protection assay
The excitation was set at 460 nm while the emission was measured at 530 nm using a 515 nm cut off filter on a Perkin Elmer Luminescence Spectrometer LS50B. An aliquot of 1–NBD or 2–NBD (4.5 µL of a 5 mM solution to give a final concentration 1.25 µM) in 1 : 1 methanol–water was added to unilamellar vesicles (25 µM final concentration) in TES buffer (22.5 mL) at 25 °C. After 2 h, a 3 mL sample was withdrawn and treated at assay time t = 50 s, with sodium hydrosulfate (180 µL of 60 mM in 1 M Tris, pH ca. 10); at t = 150 s, Triton X-100 (20 µL of 20% v/v) was added to lyse the vesicles. The curves shown in Fig. 2 are typical results from three separate trials.
Vesicle titrations
The excitation was set at 460 nm while the emission was measured at 530 nm using a 515 nm cut off filter on a Perkin Elmer Luminescence Spectrometer LS50B. To a solution of 1–NBD or 2–NBD (1 µM in 3 mL TES buffer, 10 mM) was added aliquots of vesicles in TES buffer. The change in fluorescence emission that was induced by the vesicle addition was rapid (complete within 10 s). The resulting fluorescence intensity was plotted against effective phospholipid concentration (60% of total concentration) and then fitted to a 1 : 1 binding model using an iterative curve-fitting method.18,24 Each binding constant is the average of three independent measurements.
Jurkat cells were cultured in RPMI 1640, 10% FCS and incubated at 37 °C, 5% CO2. Aliquots of cells were treated with camptothecin (10 µM final concentration) in growth media for 3.5 h at 37 °C, 5% CO2. The cells were spun down and re-suspended in annexin binding buffer (10 mM HEPES–sodium salt, 2.5 mM CaCl2, 140 mM NaCl, pH 7.4) for experiments in which annexin V was used, or in growth media for experiments in which annexin V was not used. Cells were then treated with the appropriate staining reagents at the indicated concentrations. annexin V–FITC was used according to the manufacturer's protocol. All reagents were added simultaneously. The cell suspensions were mixed thoroughly by repeated inversion and then incubated 15 min at 37 °C. Cells were then centrifuged at 2500 rpm for 2 min, re-suspended and washed three times in phenol-free RPMI 1640, 10% FCS growth media. At this point, 200 µL of each cell suspension was transferred to a 8-well chamber slide for microscopy. The HeLa cells were cultured in 75 cm2 tissue culture flasks using Eagle's minimum essential medium, 10% FCS, at 37 °C, 5% CO2 and passed according to ATCC protocols. For imaging experiments, the cells were transferred to 8-well chamber slides and allowed to grow to ca. 70% confluence. Cells were treated with camptothecin to induce apoptosis (10 µM final concentration) in growth media for 3.5 h at 37 °C, 5% CO2. The rest of the staining procedure is described above. Fluorescence microscopy was performed immediately after cell staining on an Axiovert S100 TV microscope (Carl Zeiss) equipped with filter sets DAPI/Hoechst/AMCA, FITC/RSGFP/Bodipy/Fluo3/DiO, Cy3 (Chroma). Pictures were taken using a black and white digital camera and coloured upon acquisition using Metamorph software version 6.2.
Flow cytometry
Jurkat cells were cultured as described for fluorescence microscopy. A 10.0 mL volume of cells was treated with camptothecin (10 µM, or 12.5 µM final concentration) in growth media for 16.5 h at 37 °C, 5% CO2. Cells were spun down and resuspended in annexin binding buffer for experiments in which annexin V was used, or in a buffer of 5 mM TES, 145 mM NaCl, pH 7.4 for experiments in which annexin V was not used. Cell aliquots (1.0 mL) were stained with 7AAD (500 ng mL−1) and either 1–Fl, 2–Fl or annexin V-FITC (5 µL mL−1). All reagents were added simultaneously. The cell suspensions were mixed thoroughly by repeated inversion and then incubated 15 min at 37 °C. Immediately after staining, flow cytometry was performed on a Beckman Coulter Cytomics FC 500 MPL (Fullerton, CA). Software colour compensation was used and data analysis was performed using CXP Software (Fullerton CA).
Acknowledgements
This work was supported by the NIH, and Philip Morris USA Inc. and Philip Morris International. The Australian group thank the ARC for funding and for a QEII Fellowship to KAJ.
References
- R. G. Hanshaw and B. D. Smith, Bioorg. Med. Chem., 2005, 13, 5035 CrossRef CAS.
- S. J. Martin, C. P. Reutelingsperger, A. J. McGahon, J. A. Rader, R. C. van Schie, D. M. LaFace and D. R. Green, J. Exp. Med., 1995, 182, 1545 CrossRef CAS.
- R. A. Schlegel and P. Williamson, Cell Death Differ., 2001, 8, 551–563 CrossRef CAS.
- W. L. van Heerde, P. G. de Groot and C. P. Reutelingsperger, Thromb. Haemostasis, 1995, 73, 172 Search PubMed.
- B. Plasier, D. R. Lloyd, G. C. Paul, C. R. Thomas and M. Al Rubeai, J. Immunol. Methods, 1999, 229, 81 CrossRef.
- M. van Engeland, L. J. Nieland, F. C. Ramaekers, B. Schutte and C. P. Reutelingsperger, Cytometry, 1998, 31, 1 CrossRef CAS.
- P. Williamson, S. van den Eijnde and R. A. Schlegel, Methods Cell Biol., 2001, 66, 339 Search PubMed.
- C. M. M. Lahorte, J. L. Vanderheyden, N. Steinmetz, C. Van de Wiele, R. A. Dierckx and G. Slegers, Eur. J. Nucl. Med. Mol. Imaging, 2004, 31, 887 CrossRef CAS.
- T. Z. Belhocine, J. F. Tait, J. L. Vanderheyden, C. Li and F. G. Blankenberg, J. Proteome Res., 2004, 3, 345 CrossRef CAS.
- D. Kamp, T. Sieberg and C. W. M. Haest, Biochemistry, 2001, 40, 9438 CrossRef CAS.
- A. Zweifach, Biochem. J., 2000, 349, 255 CrossRef CAS.
- J. Dachary-Prigent, J. M. Pasquet, J. M. Freyssinet and A. T. Nurden, Biochemistry, 1995, 34, 11625 CrossRef CAS.
- M. A. Swairjo, N. O. Concha, M. A. Kaetzel, J. R. Dedman and B. A. Seaton, Nat. Struct. Biol., 1995, 2, 968 CrossRef CAS.
- A. V. Koulov, K. A. Stucker, C. Lakshmi, J. P. Robinson and B. D. Smith, Cell Death Differ., 2003, 10, 1357 CrossRef CAS; A. V. Koulov, R. G. Hanshaw, K. A. Stucker, C. Lakshmi and B. D. Smith, Isr. J. Chem., 2005, 45, 373 CrossRef CAS.
- A. Ojida, Y. Mito-Oka, M. A. Inoue and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 6256 CrossRef CAS.
- J. Manaka, T. Kuraishi, A. Shiratsuchi, Y. Nakai, H. Higashida, P. Henson and Y. Nakanishi, J. Biol. Chem., 2004, 279, 48466 CrossRef CAS.
- R. A. Fratti, Y. Jun, A. J. Merz, N. Margolis and W. Wickner, J. Cell Biol., 2004, 167, 1087 CrossRef CAS.
- C. Lakshmi, R. G. Hanshaw and B. D. Smith, Tetrahedron, 2004, 60, 11307 CAS.
- R. G. Hanshaw, E. J. O'Neil, M. Foley, R. T. Carpenter and B. D. Smith, J. Mater. Chem., 2005, 15, 2707 RSC.
- R. G. Hanshaw, C. Lakshmi, T. N. Lambert, J. R. Johnson and B. D. Smith, ChemBioChem, 2005, 12, 2214 CrossRef.
- S. R. Banerjee, M. K. Levadala, N. Lazarova, L. H. Wei, J. F. Valliant, K. A. Stephenson, J. W. Babich, K. P. Maresca and J. Zubieta, Inorg. Chem., 2002, 41, 6417 CrossRef CAS.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849 CrossRef CAS.
- R. Jürss and A. Maelicke, J. Biol. Chem., 1983, 258, 10272 CAS.
- H. Jiang, E. J. O'Neil, K. M. DiVittorio and B. D. Smith, Org. Lett., 2005, 7, 3013 CrossRef CAS.
- F. A. Jaffer and R. Weissleder, JAMA, J. Am. Med. Assoc., 2005, 293, 855 Search PubMed.
- F. A. Jaffer and R. Weissleder, Circ. Res., 2004, 94, 433 Search PubMed.
- S. Ran, A. Downes and P. E. Thorpe, Cancer Res., 2002, 62, 6132 CAS.
Footnotes |
| † Abbreviations: 7AAD: 7-aminoactinomycin D, DHPC: 1,2-dihexanoyl-sn-glycero-3-phosphocholine, DHPS: 1,2-dihexanoyl-sn-glycero-3-[phospho-L-serine], DPA: 2,2′-dipicolylamine, EDC: N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide, FITC: fluorescein isothiocyanate, HOBt: 1-hydroxybenzotriazole, NBD: 7-nitro-1,3-benz-2-oxa-1,3-diaza-4-yl, NMM: N-methylmorpholine, POPA: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate, POPC: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, POPG: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol, POPS: 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-L-serine], PS: phosphatidylserine, TES: N-tris[hydroxymethyl]methyl-2-aminoethanesulfonic acid, Zn2+–DPA: zinc 2,2′-dipicolylamine. |
| ‡ The membrane recognition ability of annexin V is due primarily to its ability to bind Ca2+ cations in a geometrical way that allows simultaneous metal coordination by a PS head group. The DPA ligand acts in a functionally similar manner. It can bind Zn2+ cations with high affinity while leaving a vacant metal coordination site for a PS head group. The three-component assembly does not work with Ca2+ because Ca2+ association with the DPA ligand is too weak. |
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here. 