Synthesis of carbosilane dendritic wedges and their use for the construction of dendritic receptors
Rieko
van Heerbeek
,
Paul C. J.
Kamer
,
Piet N. M. W.
van Leeuwen
and
Joost N. H.
Reek
*
Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Nieuwe Achtergracht 166, 1018 WV Amsterdam, The Netherlands. E-mail: reek@science.uva.nl; Fax: (+31)20-5256422; Tel: (+31)20-5256437
First published on 14th December 2005
Abstract
A divergent route for the synthesis of carbosilane wedges that contain either a bromine or amine as focal point has been developed. These new building blocks enable the construction of various core-functionalized carbosilane dendrimers. As a typical example carbosilane dendrimers up to the third generation containing a N,N′,N″-1,3,5-benzenetricarboxamide core (G1–G3) have been synthesized. This new class of molecules has been studied as host molecules and they have been found to bind protected amino acids as guest molecules via hydrogen bonding interactions. A decrease in the association constants was observed for the higher generation dendritic hosts, which is attributed to the increased steric hindrance around the core where the binding site is located. The binding properties of the dendritic host molecules can be tuned by modifying the binding motif at the core of the carbosilane dendrimers. A higher association constant for N-CBZ-protected glutamic acid 1-methyl ester (5) was observed when the third generation N,N′,N″-1,3,5-tris(L-alaninyl)benzenetricarboxamide core-functionalized carbosilane dendrimer (G3′) was used as the host molecule compared to G3. Different association constants for the formation of the diastereomeric G3′·L-5 (K = 295 M−1) and G3′·D-5 (K = 236 M−1) host–guest complexes were observed, pointing to a small enantioselective recognition effect. The difference between the association constants for the formation of the G3′·(L-5)2 and G3′·(D-5)2 host–guest complexes was much more pronounced, K = 37 M−1versusK = 10 M−1, respectively.
Introduction
Dendrimers are interesting macromolecules owing to their well-defined, highly branched structures and physical properties, and they have drawn the attention of chemical, biological and materials scientists for more than two decades.1 The propensity of dendrimers to enclose guest molecules and their potential application in drug delivery and gene therapy2 has been studied since the early developments in dendrimer chemistry. Dendrimers specifically designed for the molecular recognition of substrates,3 including chiral ones,4 are generally synthesized by connecting wedges (dendrons) to receptors.Within the different dendrimer families known, the carbosilane dendrimers form a special class. The physical properties of the carbosilane dendrimers, such as their viscosity, core-accessibility, the size of the internal voids and the generation at which surface congestion occurs, can be tuned easily by choosing the appropriate building blocks for the dendrimer growth sequence.5,6 Furthermore, carbosilane dendrimers are robust, highly apolar macromolecules with low glass transition temperatures. The synthesis of carbosilane dendrimers consists of a repetitive sequence of ω-alkenylations, using either Grignard or lithium reagents, and platinum-catalyzed hydrosilylations with chlorosilanes.7 The degree of branching can be varied between 1–3 by performing the hydrosilylation with either mono-, di- or trichlorosilanes. The distance between the branching points depends on the choice of the ω-alkenylation reagent. In principle, it is possible to alter the degree of branching as well as the branch-length at every generation. The presence of either silicon-chloride or allyl end-groups can be used for the introduction of a functionality at the periphery of the dendrimer. This versatility offered by carbosilane dendrimers, has led to the extensive use of carbosilane dendrimers as scaffolds with various functional groups. Functional materials such as liquid crystals,8 catalysts,9 sensors10 and unimolecular micelles11 have been synthesized by functionalization of carbosilane dendrimers with mesogens, catalysts, metal-complexes12 and water-soluble groups, respectively.
Surprisingly, little attention has been devoted to core-functionalized carbosilane dendrimers. The reaction conditions applied during the carbosilane synthesis impose strong limitations to the functional groups that can be present in the core during the synthesis. Polar groups such as amides and esters are neither compatible with the Grignard nor with the hydrosilylation reactions. Therefore the number of core-functionalized carbosilane dendrimers directly obtained through divergent carbosilane dendrimer synthesis starting from a core-functionality is scarce.13
Convergent dendrimer synthesis, pioneered by Hawker and Fréchet,14 follows a strategy in which dendritic wedges are first prepared, which subsequently can be connected to functional building blocks leading to core-functionalized dendrimers. This synthetic route has led to the synthesis of a large number of core-functionalized dendrimers.15 The availability of carbosilane wedges would greatly facilitate the synthesis of core-functionalized carbosilane dendrimers. A first example of a core-functionalized hyper-branched carbosilane polymer—obtained through a convergent approach—has been reported by Lach et al.16 They synthesized an oxazoline core-functionalized hyper-branched carbosilane polymer, which subsequently was condensed to 1,3,5-benzenetricarboxylic acid yielding a N,N′,N″-1,3,5-benzenetricarboxamide core-functionalized hyper-branched carbosilane polymer. Gossage et al. reported the divergent synthesis of phenol core-functionalized carbosilane dendritic wedges up to the third generation.17 The first generation wedge was condensed successfully with 1,3,5-benzenetricarbonyl trichloride. Oosterom et al. synthesized p-bromophenyl core-functionalized wedges starting from p-bromostyrene. These dendritic building blocks were used for the synthesis of core-functionalized dendritic phosphines.18 Müller et al. synthesized an o-diphenylphosphinophenol core-functionalized carbosilane wedge and applied this wedge as a ligand in the nickel-catalyzed Shell Higher Olefin Process.19
This article describes the synthesis of three generations of 3-bromopropyl core-functionalized carbosilane dendritic wedges (1–3) as useful building blocks for the synthesis of core-functionalized dendrimers. Bromine was selected as a focal point of the carbosilane wedges because it is inert towards the conditions applied in the dendrimer growth sequence and can readily be modified via simple substitution reactions.20 These wedges have been used previously to synthesize a number of core-functionalized dendrimers in collaboration with the groups of Nolte,21 Jerôme22 and van Maarseveen.23 In this article we illustrate the versatility of the building blocks by the facile synthesis of three generation N,N′,N″-1,3,5-benzenetricarboxamide core-functionalized carbosilane dendrimers (G1–G3). The dendrimers G1–G3 have been used as hosts for the binding of (amino) acid guests. The effect of the introduction of an L-alanine spacer, between the core and the wedge of G3, on the binding of chiral guest molecules is evaluated.
Results and discussion
Synthesis of carbosilane wedges and core-functionalized dendrimers
(3-Bromopropyl)triallylsilane 1 was prepared as the smallest wedge and starting compound for the synthesis of the higher generations (Scheme 1). A platinum-catalyzed hydrosilylation of allyl bromide with trichlorosilane yielded (3-bromopropyl)trichlorosilane in 70% yield after vacuum distillation, which gave 1 after a Grignard reaction with allylmagnesium bromide. The catalyst used in the hydrosilylation reaction was bis(tetrabutylammonium)platinum hexachloride (Lukevics catalyst, 0.002 mol%).24 The side-product of the hydrosilylation of allyl bromide is propyltrichlorosilane, which is also observed in the synthesis of (3-chloropropyl)trichlorosilane,25 and originates from the hydrosilylation of propene that is generated via a platinum-catalyzed H/Br exchange of allyl bromide (yielding propene and SiBrCl3). Wedge 1 could be grown in the same way as described for the synthesis of carbosilane dendrimers by Van der Made and Van Leeuwen,7i.e. via a repetitive sequence of hydrosilylation with trichlorosilane followed by a Grignard reaction with allylmagnesium bromide. No cross-coupling of the bromine functionalized wedges with the Grignard reagent was observed within the duration of the experiment (5–6 hours). Wedges up to the third generation were prepared in quantitative conversion, 73–89% isolated yield. | ||
| Scheme 1 Synthesis of higher generation carbosilane wedges from 1 (third generation wedge 3 is shown). Reaction conditions: (i) HSiCl3, (Bu4N)2PtCl6, rt; (ii) (allyl)MgBr, Et2O, rt, 5–6 h. | ||
The use of the Lukevics catalyst in the hydrosilylation reactions gave 100% selectivity for the linear hydrosilylated products. It is well-established that hydrosilylation reactions might suffer from induction periods of variable length and the need for small amounts of oxygen to activate the catalyst, depending on the alkene and hydrosilane reagents.26 In the current reaction the activity of the hydrosilylation catalyst was rather unpredictable, but it was proven that it required the presence of oxygen in the reaction mixture.27 Although the selectivity in all cases remained the same (100% linear product), the reaction could take over one week to run to completion and sometimes required additional catalyst. The poor reproducibility of the activity is most likely related to the oxygen sensitivity of the catalyst. Stein et al. showed that, for poorly coordinating olefins (1-hexene), oxygen is required to prevent the formation of inactive, multi-nuclear Pt species.26 Furthermore Kleyer et al. showed that the hydrosilylation rate can be controlled by the amount of oxygen present in the reaction mixture (introduced as a 1–5 weight percent mixture with an inert gas such as nitrogen or argon).28 The addition of either an insufficient amount or an excess of oxygen can lead to lower reaction rates or the formation of inactive species. The hydrosilylation reactions were performed as follows: the wedges were dissolved together with 0.01 mol% Lukevics catalyst (with respect to the allyl groups) under an inert atmosphere of nitrogen in a mixture of dichloromethane (in order to dissolve the catalyst) and diethyl ether (good solvent for the dendritic wedges), after which trichlorosilane was added to the reaction mixture. Oxygen was allowed to diffuse into the reaction mixture via a calcium chloride tube on the reaction vessel after closing the nitrogen inlet.29 By performing the hydrosilylation in this fashion sufficient time is offered for the platinum complex to be reduced, after which diffusion of air from the head space into the reaction mixture slowly increases the oxygen content, thereby passing through the optimal oxygen concentration.
Substitution of the bromine focal point by an amine could be achieved by stirring the different generations of bromine functionalized wedges in a large excess of liquid ammonia at 70 °C under 15 bar of pressure, resulting in a new series of functionalized wedges. Importantly, the reactions should be performed at sufficiently high dilution (≤0.11 M) since otherwise secondary and tertiary amines are formed as a result of the higher nucleophilicity of the product compared to ammonia. The dendritic wedges 2 and 3 are immiscible with liquid ammonia and require the addition of diethyl ether as a co-solvent. In the absence of co-solvent the reaction with liquid ammonia gave a mixture of the starting compound and the primary, secondary and tertiary amine products. For example, the reaction of 2.50 g of 2 in 50 mL liquid ammonia (0.07 M) at 70 °C yielded a mixture of 20% 2, 50% of the desired primary amine product, 30% of the secondary and traces of tertiary amine products after 20 hours, indicating that the reaction is taking place in a dispersion rather than in a homogeneous solution. When performed under the optimized conditions, the amine wedges could be isolated as colorless oils in 90–96% yield.
The amine functionalized wedges were condensed with 1,3,5-benzenetricarbonyl trichloride in dry CH2Cl2 in the presence of triethylamine yielding carbosilane dendrimers containing an N,N′,N″-1,3,5-benzenetricarboxamide core (G1–G3) in 85–94% yield after column chromatography (Scheme 2). N,N′,N″-1,3,5-Tributylbenzenetricarboxamide G0 was prepared using similar conditions. The quality of the silica gel used for the column chromatography is of great influence on the isolated yields obtained. The silica gel should not be too acidic since this leads to lower yields as a result of the de-allylation of the carbosilane dendrimer. The Si–OH groups present on the silica gel surface lead to immobilization of the dendrimer via Si–O–Si bond formation. In fact Shimada et al. used this hydrolytic procedure with functionalized allylsilanes for the covalent linking of functional groups to silica gel.30
 | ||
| Scheme 2 Synthesis of different generations of carbosilane dendrimers containing an N,N′,N″-1,3,5-benzenetricarboxamide core. | ||
Both G0 and G1 are white solids, whereas G2 and G3 are colorless oils. All dendrimers were characterized by IR spectroscopy, 1H and 13C NMR spectroscopy and elemental analysis. GPC analysis (Fig. 1) revealed that the dendrimers obtained are monodisperse. The plot of the logarithm of the calculated molecular weight of the dendrimers as function of their GPC retention times shows a linear correlation, as is commonly observed for molecules of different molecular weights that have similar shapes.
 | ||
Fig. 1 GPC traces of carbosilane dendrimers G1 (—), G2 (⋯) and G3 (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ). Inset: calculated logMw of G1–G3 as function of the GPC retention times of G1–G3. ). Inset: calculated logMw of G1–G3 as function of the GPC retention times of G1–G3. | ||
The carbosilane dendrimers were subjected to MALDI-TOF MS analysis and satisfactory mass spectra were obtained after Ag+ labeling of the dendrimers with silver trifluoroacetate31 and using dithranol (1,8,9-anthracenetriol) as the matrix (Fig. 2a–c). In all cases the peak corresponding to the [M + Ag]+ ion was the most abundant one and arises from the cation–π interaction between Ag+ and the allylic end groups or the aromatic core. The mass spectrum of G1 (Fig. 2a) displays a peak at m/z = 890 with the isotope pattern corresponding to [M + Ag]+ and a peak at m/z = 769 corresponding to [M − (3 × allyl) + Ag]+. In the case of G2 the mass spectrum (Fig. 2b) displays peaks at m/z = 2259 with an isotope pattern corresponding to [M + Ag]+, and at m/z = 2368 corresponding to the [M + 2Ag]+ ion. For G3 three peaks are observed in the mass spectrum (Fig. 2c), corresponding to [M]+ (m/z = 6267), [M + Ag]+ (m/z = 6375) and [M + 2Ag]+ (m/z = 6484) respectively. The resolution of the mass spectrum of G3 is insufficient for identification of a clear isotope pattern, which indicates that at higher generations the ionization becomes increasingly more difficult. This effect has been observed previously for carbosilane dendrimers.32
 | ||
| Fig. 2 MALDI-TOF MS spectra recorded after Ag+ labeling with silver trifluoroacetate using dithranol as the matrix: (a) G1; (b) G2; (c) G3. Insets: calculated and observed isotope patterns. | ||
Synthesis of a carbosilane dendrimer with an extended core
Focal point manipulation of the amine core-functionalized dendritic wedges can be used to introduce different binding motifs in the core of the carbosilane dendrimers, for example by introducing amino acid spacers using standard peptide chemistry. This provides easy access to new dendritic receptors, which are expected to have different affinities for guest molecules since the introduction of an amino acid spacer increases the number of H-bond donors/acceptors present in the core and enlarges the volume of the void inside the dendrimer. Furthermore, the introduction of a chiral amino acid generates a chiral binding site that might be able to discriminate between two enantiomeric guest molecules. Therefore, an analogue of carbosilane dendrimer G3 containing an L-alanine spacer between the aromatic core and the carbosilane wedges (G3′, Scheme 3) was synthesized, applying the convergent strategy.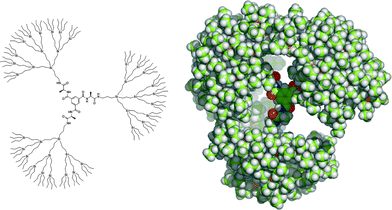 | ||
| Scheme 3 Schematic representation and CPK-representation of a computed structure of N,N′,N″-tris(L-alaninyl)-1,3,5-benzenetricarboxamide core-functionalized carbosilane dendrimer G3′. | ||
To this end a third generation carbosilane wedge, with saturated peripheral groups and an amine focal point was functionalized with L-alanine and a subsequent condensation reaction with 1,3,5-benzenetricarboxylic acid yielded G3′ (Scheme 4). The Gabriel synthesis33 was used to introduce the primary amine as the focal point. The bromine focal point was substituted by a phthaloyl group by reacting dendritic wedge 3 with potassium phthalimide in DMF at 80 °C. The substituted product (7) was obtained in 56% yield after column chromatography. The allylic peripheral groups were fully hydrogenated using palladium on carbon under an atmospheric pressure of hydrogen. Hydrazinolysis of the saturated wedge 8 and an alkaline work-up afforded the amine focal point functionalized wedge 9 in quantitative conversion, 94% isolated yield. Next BOC-protected L-alanine was coupled to the wedge via a DCC coupling, affording wedge 10 in 82% yield. The BOC-group was removed by stirring the wedge in a 50% (v/v) TFA solution in dichloromethane, affording wedge 11 in quantitative yield. Condensation of the amine functionalized wedge 11 with 1,3,5-benzenetricarboxylic acid (trimesic acid) using benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) as the carboxylic acid activating reagent yielded dendrimer G3′ in 93% yield after column chromatography. NMR and IR dilution experiments show that G3′ does not self-associate in apolar solvents, in contrast to its lower generation analogues.34
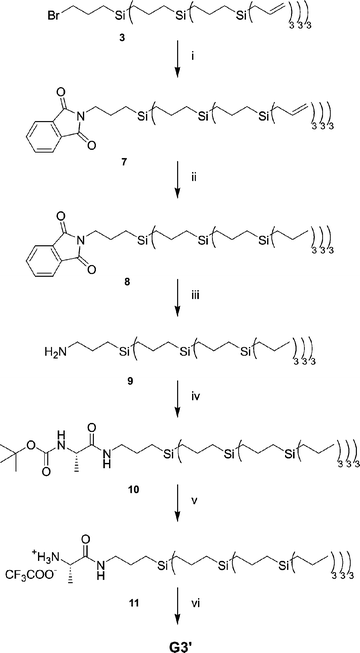 | ||
| Scheme 4 Synthesis of N,N′,N″-1,3,5-tris(L-alaninyl)benzenetricarboxamide core-functionalized dendrimer G3′. Reagents: (i) potassium phthalimide, DMF, 80 °C; (ii) H2, Pd/C (10%), EtOAc; (iii) H2NNH2·H2O, EtOH, reflux followed by HCl (aq), reflux, alkaline work-up; (iv) BOC-L-alanine, DCC, DMAP, CH2Cl2; (v) TFA–CH2Cl2 (1∶1); (vi) trimesic acid, PyBOP, DiPEA, THF. | ||
Binding studies
The N,N′,N″-1,3,5-benzenetricarboxamide core can be used as a binding site for guest molecules.35 CPK models of the dendrimers G1–G3 show that the cores in the higher generation dendrimers are more shielded from the environment by the dendritic carbosilane wedges (Fig. 3). In order to study the ability of these dendrimers to accommodate guest molecules, the binding of three different guest molecules was studied by IR and 1H NMR spectroscopy. | ||
| Fig. 3 CPK-representations of computed structures of the carbosilane dendrimers G1–G3. | ||
Binding studies were performed in dry chloroform-d with G0–G3 at a concentration of 5 mM using FMOC-glycine 4, N-CBZ-L-glutamic acid 1-methyl ester (L-5) and propionic acid (6) as the guest molecules. The IR spectra of 5 mM solutions of G0 and G1 in chloroform-d showed N–H stretching vibrations at 3455 and 3451 cm−1, respectively, characteristic of non-hydrogen bonded amides, indicating that these molecules do not self-associate in this solvent at this concentration.36 The IR spectra of the guest molecules 4 (13 mM)37 and 5 (25 mM) in chloroform-d also showed only non-hydrogen bonded amide stretching vibrations at 3454 and 3430 cm−1, respectively, indicating that these molecules do not aggregate under the conditions applied in the NMR titration experiments. 1H NMR titration experiments were employed to determine the association constants Ka (M−1) and complexation induced shifts ΔδCIS (Hz), defined as the chemical shift difference of a proton of the host–guest complex and that of the free host. The shifts of the aromatic and amide proton resonances of the dendrimers were monitored during the titration experiments (Table 1). The titration curves were fitted assuming the formation of 1∶1 complexes, using a non-linear least-squares fitting procedure, and satisfactory fits were obtained.

| Dendrimer | Guest | K a (M−1)a | ΔδCIS,ArH (ppm) | ΔδCIS,NH (ppm) |
|---|---|---|---|---|
| a Estimated error 5%. b Estimated K values. The small shifts prohibited accurate determination of the association constant. | ||||
| G0 | 4 | 87 | 0.31 | 1.17 |
| G1 | 4 | 69 | 0.26 | 0.99 |
| G2 | 4 | 65 | 0.19 | 0.63 |
| G3 | 4 | 5b | ||
| G0 | L-5 | 40 | 0.33 | 1.05 |
| G1 | L-5 | 38 | 0.29 | 0.94 |
| G2 | L-5 | 10 | 0.32 | 1.14 |
| G3 | L-5 | 5b | ||
| G2 | 6 | 24 | 0.47 | 1.39 |
The observed shifts of the NMR resonances belonging to the amide protons present in the dendrimers clearly indicate that binding of the guest molecules is based on H-bonding. This is substantiated by IR measurements on G2 (νNH = 3448 cm−1) showing a new amide N–H stretching vibration at 3385 cm−1 upon complexation with 6. The low association constant found for 6 suggests that the amide bonds in 4 and L-5 contribute to the binding with the dendrimer core.
The association constants of G0 to G2 reveal that the binding of guests 4 and L-5 is slightly weaker in higher generation dendrimers, while for G3 hardly any binding is observed. These observations reflect the increase in steric hindrance imposed by the dendritic wedges in the higher generation dendrimers, shielding the binding site located at the core of the dendrimer. In G3 the carbosilane wedges almost fully encapsulate the binding site, as can be seen from the CPK model (Fig. 3), hampering the binding of the guest molecules.
The calculated changes in the free energy of formation of the host–guest complexes (ΔG°) range from −2.6 (G0) to −0.95 kcal mol−1 (G3), indicating that the binding is based on one H-bonding interaction (the energy of a hydrogen bond typically varies between 2–5 kcal mol−1 depending on the distance between the donor and acceptor atom). Amino acid L-5 is bound weaker than 4, which might be explained by a difference in the binding geometry. Complexation induced shift values provide some structural information about the binding geometry and a different trend in ΔδCIS within the dendrimer series G0–G2 is observed for 4 compared to L-5. A drop in the ΔδCIS value (upon complexation with 4) is observed for the higher generation hosts, indicating that the geometry of the host–guest complex of G2 differs from that of G0 and G1. Most likely this change in binding geometry is induced by steric hindrance of the dendritic wedges. The different binding geometry, however, reduces the association constant within the dendrimer series to a smaller extent compared to L-5. No drop in ΔδCIS is observed for L-5 within the dendrimer series G0–G2, suggesting that the host–guest complexes have a similar geometry. The inability to adopt a different binding geometry might explain the relatively large drop in association constant observed for L-5 within the dendrimer series compared to 4 and might be attributed to the steric bulk around the amide bond in L-5. The increased steric hindrance in the higher generation dendrimer might hamper, for example, the formation of the hydrogen bond between the dendritic host and the amide bond of L-5 leading to a lower association constant.
Binding studies on a carbosilane dendrimer with an extended core
The binding behavior of dendrimer G3′ with both L- and D-5 was studied using 1H NMR spectroscopy. D-5 was synthesized starting from commercially available N-CBZ-D-glutamic acid γ-tert-butyl ester by subsequent esterification with methanol, via a DCC coupling, and de-esterification of the tert-butyl ester using TFA (Scheme 5). Partial racemization was observed, which is probably the result of direct abstraction of the αH by 4-dimethylaminopyridine (DMAP).38 Optically enriched D-5 was obtained in 87% overall yield and 61% optical purity (determined from the specific optical rotation by comparison with optically pure L-5). | ||
| Scheme 5 Synthesis of optically enriched D-5. Reaction conditions: (i) DCC, DMAP (11%), MeOH, CH2Cl2; (ii) TFA, CH2Cl2. | ||
A Job plot analysis of G3′ with L-5 was performed to determine the binding stoichiometry of the host–guest complex formed (Fig. 4).39 The shift of the resonance of the αH of L-5 was monitored as a probe in solutions containing different [G3′]∶[L-5] ratios while keeping [G3′] + [L-5] constant. The maximum of the Job plot is found at a mole fraction L-5 of 0.50, indicating the formation of a 1∶1 host–guest complex.
 | ||
| Fig. 4 Job plot analysis of a titration of G3′ with L-5. | ||
1H NMR titration experiments (300 MHz) were employed to determine the association constants of G3′ with both enantiomers of 5 (Table 2). The shifts of the resonances belonging to the aromatic protons, the αH of the alaninyl spacer and the amide proton adjacent to the wedge of G3′ were monitored during the titration experiment. Although the Job plot analysis revealed a 1∶1 binding stoichiometry, the best results in the non-linear least-squares fitting procedure were obtained when the formation of a 1∶2 host–guest complex was assumed. This discrepancy can be explained by the fact that the binding of the second guest is relatively weak, and consequently the shift is too small to be detected in the Job plot titration experiment.
| Guest | K 1 (M−1)a | K 2 (M−1)a | ΔδArH (ppm) | ΔδNH (ppm)b | ΔδCH (ppm)c | |||
|---|---|---|---|---|---|---|---|---|
| CIS1 | CIS2 | CIS1 | CIS2 | CIS1 | CIS2 | |||
| a Estimated error 5%. b Complexation induced shifts of the amide protons adjacent to the aromatic core. c Complexation induced shifts of the αH of the alaninyl spacer. d Value corrected for optically pure D-5. | ||||||||
| L-5 | 295 | 37 | 0.045 | 0.47 | 0.27 | 3.5 | −0.076 | −0.95 |
| D-5 | 236 (221)d | 10 (3.3)d | 0.038 | 1.2 | 0.21 | 4.9 | −0.048 | −1.4 |
The association constant for the formation of the G3′·L-5 host–guest complex (K1 = 295 M−1) is considerable higher than the formation of the G3·L-5 host–guest complex (estimated Ka = 5 M−1), which clearly indicates that extending the core leads to a better accessibility of guest molecules. Compared to the hosts G0 and G1, the association constant for the binding of L-5 in the dendritic host G3′ is a factor of 7 higher, which can be attributed to the number of H-bonds involved in the complexation. IR spectra of G3′ containing guest 5 showed N–H stretching vibrations at 3333 and 3306 cm−1, confirming that the binding is based on H-bonding interactions involving the amide bonds (a non-hydrogen bonded N–H stretching vibration belonging to free 5 was found at 3437 cm−1).
The two enantiomers of 5 are bound in the dendritic host G3′ with different association constants, demonstrating that the chiral core can discriminate between the two enantiomers through diastereomeric interactions. The calculated ΔG0 for the formation of the G3′·L-5 and G3′·D-5 host–guest complexes are −3.34 kcal mol−1− and −3.17 kcal mol−1, respectively, corresponding to ΔΔG (ΔGL − ΔGD) = −0.17 kcal mol−1. In a racemic mixture of 5, 57% of the bound guest is the L-5 enantiomer, indicating that the enantioselective recognition of the first guest molecule is small. Interestingly, the binding of the first guest molecule enhances the enantioselective binding of the second guest molecule, which is expressed by a larger ΔΔG of 1.4 kcal mol−1 calculated for the formation of the 1∶2 host–guest complexes (ΔGL = −2.1 kcal mol−1; ΔGD = −0.70 kcal mol−1). Consequently, in a racemic mixture of 5, 92% of the L-5 would be bound as the second guest molecule, which can be considered significant.40 This enhanced enantioselective recognition in the binding of the second guest molecule might be attributed to the presence of the first guest molecule preorganizing the binding site for the binding of the second guest molecule, leading to a higher energy barrier between the ‘match’ and ‘mismatch’ diastereomeric interactions.
Conclusions
Carbosilane dendritic wedges both with a bromine or amine focal point have been prepared and shown to be useful building blocks for the construction of core-functionalized carbosilane dendrimers that are otherwise difficult to obtain. Substitution of the bromide focal point or using standard peptide chemistry with the amine focal point can be used to synthesize carbosilane dendrimers in a convergent manner. We have synthesized two types of core-functionalized carbosilane dendrimers, which can be used for the (enantioselective) molecular recognition of guest molecules. The modular approach used enables the construction of carbosilane dendrimers containing a variety of binding sites, based on various amino acids, which might result in systems that can bind guest molecules with higher enantioselectivities and binding constants. These carbosilane dendrimers containing amino acids may be used as novel recyclable organocatalysts by binding and converting substrate molecules41 or as new materials for the resolution of amino acid derivatives.Experimental section
General remarks
All reactions were carried out in flame-dried glassware and under a dry nitrogen atmosphere using standard Schlenk techniques unless mentioned otherwise. Solvents were dried and distilled under nitrogen; Et2O from sodium–benzophenone and CH2Cl2 from CaH2. Bis(tetrabutylammonium)platinum hexachloride [(Bu4N)2PtCl6] was synthesized according to a literature procedure.24 Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and N-(benzyloxycarbonyl)-D-glutamic acid 5-tert-butyl ester were purchased from NovaBiochem. Other chemicals were purchased from Acros Organics or the Aldrich Chemical Company. Allyl bromide was distilled prior to use. Allylmagnesium bromide was prepared in Et2O according to modified literature procedures.42 The molarity of the Grignard was determined prior to use via a titration with a 0.50 M isopropanol solution in xylenes using 1,10-phenanthroline as an indicator. Silica 60 (SDS Chromagel, 70–200 µm) was used for column chromatography. Melting points were determined on a Gallenkamp apparatus. Optical rotations were measured with a Perkin Elmer 241 Polarimeter. [α]D values are given in 10−1deg cm2 g−1. IR spectra were measured on a BioRad FT-IR (FTS-7) spectrophotometer. 1H and 13C NMR spectra were collected on a Bruker AMX 300 spectrometer, a Varian Mercury 300 or a Varian Inova 500 spectrophotometer. The chemical shift values δ are given in ppm, the values of the coupling constants J in Hz. GC/MS spectra were recorded on a Hewlett Packard 5890/5971-GC/MS (split/splitless injector, ZB-5 15 m column, film thickness 0.25 µm, carrier gas: 0.2 bar He). FAB mass spectra were recorded on a JOEL JMS SX/SX102A four sector mass spectrometer coupled to a JOEL MS-MP7000 data system using 3-nitrobenzyl alcohol as a matrix. MALDI-TOF mass spectra were collected on a Voyager-DE Biospectrometry Workstation (PerSeptive Biosystems Inc., Framingham, MA, USA) mass spectrometer equipped with a nitrogen laser emitting at 337 nm (3 ns pulses). The samples were labeled using silver trifluoroacetate31 and analyzed using dithranol as the matrix. Gel permeation chromatography (GPC) was performed on a Shimadzu apparatus equipped with three Waters Styragel Columns (HR-1, HR-2 and HR-4) connected in series and with a RID-10A refractive index detector and a SPD-10A VP UV/Vis detector.CAUTION: Hydrosilylation reactions are highly exothermic and good care should be taken. It is our experience that the occurrence of a vigorous reaction seems to be completely unpredictable (if this happened, it was always between 10 and 90 minutes) and a dry ice–acetone bath should be kept at hand when adding the trichlorosilane as a precaution.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 4.88 (6H, m, SiCH2CHCH2), 3.37 (2H, t, J 6.9, BrCH2), 1.89 (2H, m, BrCH2CH2), 1.61 (6H, d, J 8.1, SiCH2CHCH2) and 0.71 (2H, m, BrCH2CH2CH2Si). δC (75 MHz, CDCl3) 135.3, 115.4, 38.2, 28.8, 20.9 and 12.2. m/z (GC/MS) 231 (M+
− allyl).
CH2), 4.91 (18H, m, SiCH2CHCH2), 3.38 (2H, t, J 6.9, BrCH2), 1.83 (2H, m, BrCH2CH2), 1.61 (18H, d, J 8.2, SiCH2CHCH2), 1.38 (6H, m, CH2Si(allyl)3), 0.69 (6H, SiCH2CH2CH2Si) and 0.61 (8H, m, CH2Si(CH2)3). δC (75 MHz, CDCl3) 134.5, 113.7, 37.2, 28.2, 19.9, 18.4, 17.5, 16.8 and 11.9. m/z (MALDI-TOF) 795 (M+
+ Ag − allyl).
CH2), 4.86 (54H, m, SiCH2CHCH2), 3.38 (2H, t, J 6.9, BrCH2), 1.82 (2H, m, BrCH2CH2), 1.59 (54H, d, J 8.1, SiCH2CHCH2), 1.35 (24H, m, (3-bromopropyl)Si(CH2)3 and CH2Si(allyl)3), 0.66 (24H, m, SiCH2CH2CH2Si) and 0.57 (26H, m, BrCH2CH2CH2 and {CH2Si(CH2)3}3). 13C NMR (75 MHz, CDCl3) δC 134.5, 113.7, 37.4, 28.3, 19.9, 18.8, 18.5, 18.0, 17.9, 17.7, 16.8 and 11.8. m/z (MALDI-TOF) 2164 (M+
+ Ag − allyl).
CH2), 4.78 (6H, m, SiCH2CHCH2), 2.59 (2H, t, J 7.0, NH2CH2), 2.57 (2H, broad s, NH2), 1.51 (6H, d, J 8.2, SiCH2CHCH2), 1.41 (2H, m, NH2CH2CH2) and 0.50 (2H, m, CH2Si). δC (75 MHz, CDCl3) 134.2, 113.8, 45.1, 27.0, 19.6 and 8.6. m/z (GC/MS) 168 (M+
− allyl).
CH2), 4.87 (18H, m, SiCH2CHCH2), 2.65 (2H, t, J 7.0, NH2CH2), 1.57 (18H, d, J 8.0, SiCH2CHCH2), 1.34 (10H, m, NH2CH2CH2, NH2 and SiCH2CH2CH2Si), 0.65 (8H, m, CH2Si(allyl)3), 0.56 (6H, m, Si(CH2)3) and 0.46 (2H, m, NH2CH2CH2CH2). δC (75 MHz, CDCl3) 134.5, 113.7, 45.9, 28.4, 19.8, 18.4, 17.6, 16.7 and 9.65. m/z (FAB) 666.472 (M+
+ H. Calc. for C39H72NSi4 666.474).
CH2), 4.89 (54H, m, SiCH2CHCH2), 2.66 (2H, t, J 7.0, NH2CH2), 1.59 (54H, d, J 8.2, SiCH2CHCH2), 1.35 (28H, m, NH2CH2CH2 and (3-aminopropyl)Si(CH2)3, NH2 and CH2Si(allyl)3), 0.66 (24H, m, SiCH2CH2CH2Si), 0.56 (24H, m, {CH2Si(CH2)3}3) and 0.50 (2H, NH2CH2CH2CH2). δC (75 MHz, CDCl3) 134.5, 113.7, 45.8, 28.5, 19.9, 18.8, 18.5, 18.1, 18.0, 17.7, 16.8 and 9.5.
CH2), 4.90 (18H, m, SiCH2CHCH2), 3.45 (6H, J 6.7, NHCH2), 1.63 (6H, m, NHCH2CH2), 1.62 (18H, d, J 8.0, SiCH2CHCH2) and 0.66 (6H, m, CH2Si). δC (75 MHz, CDCl3) 165.8, 135.4, 134.3, 128.3, 114.2, 43.7, 24.0, 19.7 and 9.0. m/z (MALDI-TOF) 890 (M+
+ Ag). m/z (FAB) 784.476 (M+
+ H. Calc. for C45H70N3O3Si3 784.472).
CH2), 4.88 (54H, m, SiCH2CHCH2), 3.44 (6H, q, J 6.7, NHCH2), 1.58 (6H, m, NHCH2CH2), 1.58 (54H, d, J 8.0, SiCH2CHCH2), 1.36 (18H, m, CH2Si(allyl)3), 0.67 (18H, m, SiCH2CH2CH2Si), 0.59 (18H, m, CH2Si(CH2)3) and 0.54 (6H, m, NHCH2CH2CH2). δC (75 MHz, CDCl3) 165.4, 135.3, 134.6, 128.0, 113.7, 43.9, 24.6, 19.9, 18.4, 17.5, 16.8 and 10.1. m/z (MALDI-TOF) 2261 (M+
+ Ag).
CH2), 4.89 (162H, m, SiCH2CHCH2), 3.42 (6H, q, J 5.8, NHCH2), 1.59 (168H, d, J 8.1, NHCH2CH2CH2 and SiCH2CHCH2), 1.36 (72H, m, NHCH2CH2CH2Si(CH2)3 and CH2Si(allyl)3), 0.66 (72H, m, SiCH2CH2CH2Si) and 0.61 ppm (78H, m, NHCH2CH2CH2 and {CH2Si(CH2)3}3). δC (75 MHz, CDCl3) 165.2, 135.2, 134.9, 134.6, 113.8, 44.2, 24.7, 19.6, 18.5, 18.3, 17.9, 17.7, 16.8 and 10.2. m/z (MALDI-TOF) 6375 (M+
+ Ag).
CH2), 4.86 (54H, m, SiCH2CHCH2), 3.62 (2H, t, J 7.5, NCH2), 1.57 (56H, d, J 8.1, SiCH2CHCH2 and NCH2CH2), 1.33 (24H, m, {3-(Pht)propyl}SiCH2 and CH2Si(allyl)3), 0.64 (24H, m, SiCH2CH2CH2Si) and 0.56 (26H, m, PhtCH2CH2CH2 and {CH2Si(CH2)3}3). δC (125.8 MHz, CDCl3) 168.4, 134.7, 134.0, 132.5, 123.3, 113.8, 41.4, 23.6, 19.9, 18.8, 18.5, 18.1, 17.9, 17.7, 16.9 and 9.9. m/z (MALDI-TOF) 2126.4 (M+
− C3H6).
CH2), 4.88 (6H, m, SiCH2CHCH2), 3.37 (2H, t, J 6.9, BrCH2), 1.89 (2H, m, BrCH2CH2), 1.61 (6H, d, J 8.1, SiCH2CHCH2) and 0.71 (2H, m, BrCH2CH2CH2Si). δC (75 MHz, CDCl3) 135.3, 115.4, 38.2, 28.8, 20.9 and 12.2. m/z (GC/MS) 231 (M+
− allyl).
CH2), 4.91 (18H, m, SiCH2CHCH2), 3.38 (2H, t, J 6.9, BrCH2), 1.83 (2H, m, BrCH2CH2), 1.61 (18H, d, J 8.2, SiCH2CHCH2), 1.38 (6H, m, CH2Si(allyl)3), 0.69 (6H, SiCH2CH2CH2Si) and 0.61 (8H, m, CH2Si(CH2)3). δC (75 MHz, CDCl3) 134.5, 113.7, 37.2, 28.2, 19.9, 18.4, 17.5, 16.8 and 11.9. m/z (MALDI-TOF) 795 (M+
+ Ag − allyl).
CH2), 4.86 (54H, m, SiCH2CHCH2), 3.38 (2H, t, J 6.9, BrCH2), 1.82 (2H, m, BrCH2CH2), 1.59 (54H, d, J 8.1, SiCH2CHCH2), 1.35 (24H, m, (3-bromopropyl)Si(CH2)3 and CH2Si(allyl)3), 0.66 (24H, m, SiCH2CH2CH2Si) and 0.57 (26H, m, BrCH2CH2CH2 and {CH2Si(CH2)3}3). 13C NMR (75 MHz, CDCl3) δC 134.5, 113.7, 37.4, 28.3, 19.9, 18.8, 18.5, 18.0, 17.9, 17.7, 16.8 and 11.8. m/z (MALDI-TOF) 2164 (M+
+ Ag − allyl).
CH2), 4.78 (6H, m, SiCH2CHCH2), 2.59 (2H, t, J 7.0, NH2CH2), 2.57 (2H, broad s, NH2), 1.51 (6H, d, J 8.2, SiCH2CHCH2), 1.41 (2H, m, NH2CH2CH2) and 0.50 (2H, m, CH2Si). δC (75 MHz, CDCl3) 134.2, 113.8, 45.1, 27.0, 19.6 and 8.6. m/z (GC/MS) 168 (M+
− allyl).
CH2), 4.87 (18H, m, SiCH2CHCH2), 2.65 (2H, t, J 7.0, NH2CH2), 1.57 (18H, d, J 8.0, SiCH2CHCH2), 1.34 (10H, m, NH2CH2CH2, NH2 and SiCH2CH2CH2Si), 0.65 (8H, m, CH2Si(allyl)3), 0.56 (6H, m, Si(CH2)3) and 0.46 (2H, m, NH2CH2CH2CH2). δC (75 MHz, CDCl3) 134.5, 113.7, 45.9, 28.4, 19.8, 18.4, 17.6, 16.7 and 9.65. m/z (FAB) 666.472 (M+
+ H. Calc. for C39H72NSi4 666.474).
CH2), 4.89 (54H, m, SiCH2CHCH2), 2.66 (2H, t, J 7.0, NH2CH2), 1.59 (54H, d, J 8.2, SiCH2CHCH2), 1.35 (28H, m, NH2CH2CH2 and (3-aminopropyl)Si(CH2)3, NH2 and CH2Si(allyl)3), 0.66 (24H, m, SiCH2CH2CH2Si), 0.56 (24H, m, {CH2Si(CH2)3}3) and 0.50 (2H, NH2CH2CH2CH2). δC (75 MHz, CDCl3) 134.5, 113.7, 45.8, 28.5, 19.9, 18.8, 18.5, 18.1, 18.0, 17.7, 16.8 and 9.5.
CH2), 4.90 (18H, m, SiCH2CHCH2), 3.45 (6H, J 6.7, NHCH2), 1.63 (6H, m, NHCH2CH2), 1.62 (18H, d, J 8.0, SiCH2CHCH2) and 0.66 (6H, m, CH2Si). δC (75 MHz, CDCl3) 165.8, 135.4, 134.3, 128.3, 114.2, 43.7, 24.0, 19.7 and 9.0. m/z (MALDI-TOF) 890 (M+
+ Ag). m/z (FAB) 784.476 (M+
+ H. Calc. for C45H70N3O3Si3 784.472).
CH2), 4.88 (54H, m, SiCH2CHCH2), 3.44 (6H, q, J 6.7, NHCH2), 1.58 (6H, m, NHCH2CH2), 1.58 (54H, d, J 8.0, SiCH2CHCH2), 1.36 (18H, m, CH2Si(allyl)3), 0.67 (18H, m, SiCH2CH2CH2Si), 0.59 (18H, m, CH2Si(CH2)3) and 0.54 (6H, m, NHCH2CH2CH2). δC (75 MHz, CDCl3) 165.4, 135.3, 134.6, 128.0, 113.7, 43.9, 24.6, 19.9, 18.4, 17.5, 16.8 and 10.1. m/z (MALDI-TOF) 2261 (M+
+ Ag).
CH2), 4.89 (162H, m, SiCH2CHCH2), 3.42 (6H, q, J 5.8, NHCH2), 1.59 (168H, d, J 8.1, NHCH2CH2CH2 and SiCH2CHCH2), 1.36 (72H, m, NHCH2CH2CH2Si(CH2)3 and CH2Si(allyl)3), 0.66 (72H, m, SiCH2CH2CH2Si) and 0.61 ppm (78H, m, NHCH2CH2CH2 and {CH2Si(CH2)3}3). δC (75 MHz, CDCl3) 165.2, 135.2, 134.9, 134.6, 113.8, 44.2, 24.7, 19.6, 18.5, 18.3, 17.9, 17.7, 16.8 and 10.2. m/z (MALDI-TOF) 6375 (M+
+ Ag).
CH2), 4.86 (54H, m, SiCH2CHCH2), 3.62 (2H, t, J 7.5, NCH2), 1.57 (56H, d, J 8.1, SiCH2CHCH2 and NCH2CH2), 1.33 (24H, m, {3-(Pht)propyl}SiCH2 and CH2Si(allyl)3), 0.64 (24H, m, SiCH2CH2CH2Si) and 0.56 (26H, m, PhtCH2CH2CH2 and {CH2Si(CH2)3}3). δC (125.8 MHz, CDCl3) 168.4, 134.7, 134.0, 132.5, 123.3, 113.8, 41.4, 23.6, 19.9, 18.8, 18.5, 18.1, 17.9, 17.7, 16.9 and 9.9. m/z (MALDI-TOF) 2126.4 (M+
− C3H6).
The binding constants K1 and K2 of host G3′ with both enantiomers of guest N-CBZ-glutamic acid 1-methyl ester (5) in CDCl3 (T = 24 °C) were determined by varying the guest concentration between 0.76–14 mM while keeping the host concentration constant at approximately 3.4 mM. The downfield shifts of the G3′ aromatic, Ala-CH and C(O)NHCH2 proton resonances were followed. The association constants were calculated from the data using a non-linear least-squares fitting program.45 The best fits were obtained assuming a 1∶2 host–guest binding stoichiometry. The titration experiments were performed in duplo.
Acknowledgements
The authors would like to thank Dr Roel H. Fokkens for measurement of the MALDI-TOF MS spectra. The Netherlands Organization for Scientific Research (NWO/STW) is acknowledged for financial support (RvH).References
- For some general reading on dendrimers see: Dendrimers and Other Dendritic Polymers, ed. J. M. J. Fréchet and D. A. Tomalia, John Wiley & Sons, Ltd., West Sussex, 2001 Search PubMed; G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendrimers and Dendrons, Wiley-VCH, Weinheim, 2001 Search PubMed; G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendritic Molecules, Verlag Chemie, Weinheim, 1996 Search PubMed; F. Vögtle, Top. Curr. Chem., 1998, 197, vii Search PubMed; J. P. Majoral and A.-M. Caminade, Chem. Rev., 1999, 99, 845 Search PubMed; A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665 Search PubMed.
- For reviews on gene therapy and/or drug delivery see: J. M. Loutsch, D. Ong and J. M. Hill, Drugs Pharm. Sci., 2003, 130, 467 Search PubMed; P. K. Tripathi, A. J. Khopade, T. Dutta, R. B. Umamaheshewari, S. Jain, D. K. Saraf and N. K. Jain, Indian J. Pharm. Sci., 2003, 65, 15 Search PubMed; K. Sadler and J. P. Tam, Rev. Mol. Biotechnol., 2002, 90, 195 Search PubMed; T. Sakthivel and A. T. Florence, Drug Delivery Technol., 2003, 3, 52 CrossRef CAS; K. Kono, Drug Delivery Syst., 2002, 17, 462 Search PubMed; M. J. Cloninger, Curr. Opin. Chem. Biol., 2002, 6, 742 Search PubMed; A. K. Patri, I. J. Majoros and J. R. Baker, Curr. Opin. Chem. Biol., 2002, 6, 466 CrossRef CAS; S.-E. Stiriba, H. Frey and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 1329 CrossRef CAS; A. T. Florence and N. Hussain, Adv. Drug Delivery Rev., 2001, 50, S69 CrossRef CAS; M. Liu and J. M. J. Fréchet, Pharm. Sci. Technol. Today, 1999, 2, 393 CrossRef CAS; L. G. Schultz and S. C. Zimmerman, Pharm. News, 1999, 6, 25 CrossRef CAS; R. Gandhi, A. J. Khopade and N. K. Jain, Indian Drugs, 1997, 34, 593 Search PubMed.
- For reviews on supramolecular chemistry and dendrimers see: P. J. Gittins and L. J. Twyman, Supramol. Chem., 2003, 15, 5 Search PubMed; M. W. P. L. Baars and E. W. Meijer, Top. Curr. Chem., 2000, 210, 131 CrossRef CAS; F. W. Zeng and S. C. Zimmerman, Chem. Rev., 1997, 97, 1681 CAS.
- V. J. Pugh, Q.-S. Hu, X. Zuo, F. D. Lewis and L. Pu, J. Org. Chem., 2001, 66, 6136 CrossRef CAS; L.-Z. Gong, Q.-S. Hu and L. Pu, J. Org. Chem., 2001, 66, 2358 CrossRef CAS.
- A. W. van der Made, P. W. N. M. van Leeuwen, J. C. de Wilde and R. A. C. Brandes, Adv. Mater., 1993, 5, 466 CrossRef CAS.
- C. Schlenk and H. Frey, Monatsh. Chem., 1999, 130, 3 CrossRef CAS.
- A. W. van der Made and P. W. N. M. van Leeuwen, J. Chem. Soc., Chem. Commun., 1992, 1400 RSC.
- G. Dantlgraber, U. Baumeister, S. Diele, H. Kresse, B. Luehmann, H. Lang and C. Tschierske, J. Am. Chem. Soc., 2002, 124, 14852 CrossRef CAS; A. Y. Bobrovsky, A. A. Pakhomov, X.-M. Zhu, N. I. Boiko, V. P. Shibaev and J. Stumpe, J. Phys. Chem. B, 2002, 106, 540 CrossRef CAS; S. A. Ponomarenko, E. V. Agina, N. I. Boiko, E. A. Rebrov, A. M. Muzafarov, R. M. Richardson and V. P. Shibaev, Mol. Cryst. Liq. Cryst., 2001, 364, 93 CrossRef CAS; X.-M. Zhu, N. I. Boiko, E. A. Rebrov, A. M. Muzafarov, M. V. Kozlovsky, R. M. Richardson and V. P. Shibaev, Liq. Cryst., 2001, 28, 1259 CrossRef CAS; N. Boiko, X. Zhu, A. Bobrovsky and V. Shibaev, Chem. Mater., 2001, 13, 1447 CrossRef CAS; S. Ponomarenko, N. Boiko, E. Rebrov, A. Muzafarov, I. Whitehouse, R. Richardson and V. Shibaev, Mol. Cryst. Liq. Cryst., 1999, 332, 2553 CAS; D. Terunuma, R. Nishio, Y. Aoki, H. Nohira, K. Matsuoka and H. Kuzuhara, Chem. Lett., 1999, 565 CrossRef CAS; R. M. Richardson, S. A. Ponomarenko, N. I. Boiko and V. P. Shibaev, Liq. Cryst., 1999, 26, 101 CrossRef CAS; E. I. Ryumtsev, N. P. Evlampieva, A. V. Lezov, S. A. Ponomarenko, N. I. Boiko and V. P. Shibaev, Liq. Cryst., 1998, 25, 475 CrossRef CAS; D. Terunuma, T. Kato, R. Nishio, K. Matsuoka, H. Kuzuhara, Y. Aoki and H. Nohira, Chem. Lett., 1998, 59 CrossRef CAS; S. A. Ponomarenko, E. A. Rebrov, A. Y. Bobrovsky, N. I. Boiko, A. M. Muzafarov and V. P. Shibaev, Liq. Cryst., 1996, 21, 1 CrossRef CAS; K. Lorenz, D. Hölter, B. Stühn, R. Mülhaupt and H. Frey, H., Adv. Mater., 1996, 8, 414 CAS; H. Frey, K. Lorenz, D. Hoelter and R. Mülhaupt, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1996, 37, 758 CAS.
- M. Q. Slagt, J. T. B. H. Jastrzebski, R. J. M. Klein Gebbink, H. J. van Ramesdonk, J. W. Verhoeven, D. D. Ellis, A. L. Spek and G. van Koten, Eur. J. Org. Chem., 2003, 1692 CrossRef CAS; R. Roesler, B. J. N. Har and W. E. Piers, Organometallics, 2002, 21, 4300 CrossRef CAS; L. Ropartz, R. E. Morris, D. F. Foster and D. J. Cole-Hamilton, J. Mol. Catal. A: Chem., 2002, 182–183, 99 CrossRef CAS; D. de Groot, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Eur. J. Org. Chem., 2002, 1085 CrossRef CAS; G. Rodriguez, M. Lutz, A. L. Spek and G. van Koten, Chem.–Eur. J., 2002, 8, 45 CrossRef CAS; A. W. Kleij, R. J. M. Klein Gebbink, M. Lutz, A. L. Spek and G. van Koten, J. Organomet. Chem., 2001, 621, 190 CrossRef CAS; M. Benito, O. Rossell, M. Seco and G. Segales, J. Organomet. Chem., 2001, 619, 245 CrossRef CAS; A. W. Kleij, R. J. M. Klein Gebbink, P. A. J. van den Nieuwenhuijzen, H. Kooijman, M. Lutz, A. L. Spek and G. van Koten, Organometallics, 2001, 20, 634 CrossRef CAS; D. de Groot, P. G. Emmerink, C. Coucke, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Inorg. Chem. Commun., 2000, 3, 711 CrossRef CAS; E. B. Eggeling, N. J. Hovestad, J. T. B. H. Jastrzebski, D. Vogt and G. van Koten, J. Org. Chem., 2000, 65, 8857 CrossRef CAS; H. Beerens, F. Verpoort and L. Verdonck, J. Mol. Catal. A: Chem., 2000, 159, 197 CrossRef CAS; H. Beerens, P. Wijkens, J. T. B. H. Jastrzebski, F. Verpoort, L. Verdonck and G. van Koten, J. Organomet. Chem., 2000, 603, 244 CrossRef CAS; H. Beerens, F. Verpoort and L. Verdonck, J. Mol. Catal. A: Chem., 2000, 151, 279 CrossRef CAS; N. J. Hovestad, J. T. B. H. Jastrzebski, G. van Koten, S. A. F. Bon, C. Waterson and D. M. Haddleton, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1999, 40, 393 CAS; D. de Groot, J. C. de Wilde, R. J. van Haaren, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, E. B. Eggeling, D. Vogt, H. Kooijman, A. L. Spek and A. W. van der Made, Chem. Commun., 1999, 1623 RSC; N. J. Hovestad, J. L. Hoare, J. T. B. H. Jastrzebski, A. J. Canty, W. J. J. Smeets, A. L. Spek and G. van Koten, Organometallics, 1999, 18, 2970 CrossRef CAS; J. W. J. Knapen, A. W. van der Made, J. C. de Wilde, P. W. N. M. van Leeuwen, P. Wijkens, D. M. Grove and G. van Koten, Nature, 1994, 372, 659 CrossRef CAS.
- M.-C. Daniel, J. Ruiz, S. Nlate, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 2003, 125, 2617 CrossRef CAS; M. Zhou and J. Roovers, Macromolecules, 2001, 34, 244 CrossRef CAS.
- B. V. Lebedev, T. G. Kulagina, M. V. Ryabkov, S. A. Ponomarenko, E. A. Makeev, N. I. Boikov, V. P. Shibaev, E. A. Rebrov and A. M. Muzafarov, J. Therm. Anal. Calorim., 2003, 71, 481 CrossRef CAS; S. W. Krska and D. Seyferth, D., J. Am. Chem. Soc., 1998, 120, 3604 CrossRef CAS; E. V. Getmanova, E. A. Rebrov, N. G. Vasilenko and A. M. Muzafarov, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1998, 39, 581 CAS.
- For a recent review on transition-metal containing carbosilane dendrimers see: O. Rossell, M. Seco and I. Angurell, C. R. Chim., 2003, 6, 803 Search PubMed.
- Z. J. Yu, Q. Z. Zhang and Q. F. Zhou, Chin. Chem. Lett., 2003, 14, 465 CAS; L. Ropartz, R. E. Morris, D. F. Foster and D. J. Cole-Hamilton, J. Mol. Catal. A: Chem., 2002, 182–183, 99 CrossRef CAS; X. Zhang, K. J. Haxton, L. Ropartz, D. J. Cole-Hamilton and R. E. Morris, J. Chem. Soc., Dalton Trans., 2001, 3261 RSC; C. Kim and S. Kang, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 724 CrossRef CAS; M. M. K. Boysen and T. K. Lindhorst, Org. Lett., 1999, 1, 1925 CrossRef CAS; C. Kim and K. An, J. Organomet. Chem., 1997, 547, 55 CrossRef CAS; C. Kim and K. An, Bull. Korean Chem. Soc., 1997, 18, 164 CAS.
- C. J. Hawker and J. M. J. Fréchet, J. Chem. Soc., Chem. Commun., 1990, 1010 RSC; C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- For reviews on core-functionalized dendrimers see: S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1047 Search PubMed; L. J. Twyman, A. S. H. King and I. K. Martin, Chem. Soc. Rev., 2002, 31, 69 CrossRef CAS; C. S. Cameron and C. B. Gorman, Adv. Funct. Mater., 2002, 12, 17 RSC; S. K. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819 CrossRef CAS; S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS; C. B. Gorman and J. C. Smith, Acc. Chem. Res., 2001, 34, 60 CrossRef CAS.
- C. Lach, R. Hanselmann, H. Frey and R. Mulhaupt, Macromol. Rapid Commun., 1998, 19, 461 CrossRef CAS.
- R. A. Gossage, E. Muñoz-Martínez, H. Frey, A. Burgath, M. Lutz, A. L. Spek and G. van Koten, Chem.–Eur. J., 1999, 5, 2191 CrossRef CAS; R. A. Gossage, E. Muñoz-Martínez and G. van Koten, Tetrahedron Lett., 1998, 39, 2397 CrossRef CAS.
- G. E. Oosterom, S. Steffens, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Top. Catal., 2002, 19, 61 CrossRef CAS; G. E. Oosterom, R. J. van Haaren, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. Commun., 1999, 1119 RSC.
- C. Müller, L. J. Ackerman, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2004, 126, 14960 CrossRef.
- R. van Heerbeek, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Tetrahedron Lett., 1999, 40, 7187.
- J. J. L. M. Cornelissen, R. van Heerbeek, P. C. J. Kamer, J. N. H. Reek, N. A. J. M. Sommerdijk and R. J. M. Nolte, Adv. Mater., 2002, 14, 489 CrossRef CAS; J. J. L. M. Cornelissen, M. Fischer, R. van Waes, R., R. van Heerbeek, P. C. J. Kamer, J. N. H. Reek, N. A. J. M. Sommerdijk and R. J. M. Nolte, Polymer, 2004, 45, 7417 CAS.
- B. C. de Pater, PhD thesis, University of Amsterdam, Amsterdam, 2003, ch. 4.
- P. N. M. Botman, A. Amore, R. van Heerbeek, J. W. Back, H. Hiemstra, J. N. H. Reek and J. H. van Maarseveen, Tetrahedron Lett., 2004, 45, 5999 CrossRef CAS.
- I. G. Iovel, Y. S. Goldberg, M. V. Shymanska and E. Lukevics, Organometallics, 1987, 6, 1410 CrossRef CAS.
- U. Deschler, P. Kleinschmit and P. Panster, Angew. Chem., Int. Ed. Engl., 1986, 25, 236 CrossRef; E. Lukevics, Z. V. Belyakova, M. G. Pomerantseva and M. G. Voronkov, J. Organomet. Chem. Libr., 1977, 5, 1 CAS.
- For an extensive study on the Pt(0)-catalyzed hydrosilylation including the “oxygen-effect” see: J. Stein, L. N. Lewis, Y. Gao and R. A. Scott, J. Am. Chem. Soc., 1999, 121, 3693 Search PubMed , and references cited therein.
- The reproducibility is generally better for dichloromethylsilane than trichlorosilane.
- D. L. Kleyer, B. T. Nguyen, D. E. Hauenstein, S. P. Davern and W. J. Schultz, U. S. Patent 5359111, 1994; D. L. Kleyer, B. T. Nguyen, D. E. Hauenstein, S. P. Davern and W. J. Schultz, Chem. Abs., 1994, 122, 133409 Search PubMed.
- This method has been developed by A. W. van der Made at Shell (Amsterdam).
- T. Shimada, K. Aoki, Y. Shinoda, T. Nakamura, N. Tokunaga, S. Inagaki and T. Hayashi, J. Am. Chem. Soc., 2003, 125, 4688 CrossRef CAS.
- P. Timmerman, K. A. Jolliffe, M. C. Calama, J.-L. Weidmann, L. J. Prins, F. Cardullo, B. H. M. Snellink-Ruël, R. H. Fokkens, N. M. M. Nibbering, S. Shinkai and D. N. Reinhoudt, Chem.–Eur. J., 2000, 6, 4104 CrossRef CAS; K. A. Jolliffe, M. C. Calama, R. H. Fokkens, N. M. M. Nibbering, P. Timmerman and D. N. Reinhoudt, Angew. Chem., Int. Ed., 1998, 37, 1247 CrossRef CAS.
- C. Kim, S. K. Choi and B. Kim, Polyhedron, 2000, 19, 1031 CrossRef CAS; C. Kim and S. Son, J. Organomet. Chem., 2000, 599, 123 CrossRef CAS; S. Arévalo, E. de Jeśus, F. J. de la Mata, J. C. Flores and R. Gómez, R., Organometallics, 2001, 20, 2583 CrossRef CAS.
- M. S. Gibson and R. W. Bradshaw, Angew. Chem., 1968, 80, 986 CrossRef; F. Effenberger and S. Heid, Synthesis, 1995, 1126 CrossRef CAS.
- A paper on the supramolecular properties of different generations of carbosilane dendrimers which contain the extended core is forthcoming.
- For some examples of C3 symmetric host molecules containing an aromatic core and amides see: H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 1998, 120, 10622 Search PubMed; S. S. Yoon and W. C. Still, Angew. Chem., Int. Ed. Engl., 1994, 33, 2458 CrossRef CAS.
- Similar observations have been made for N,N′,N″-1,3,5-tris((S)-3,7-dimethyloctyl)benzenetricarboxamide: L. Brunsveld, A. P. H. J. Schenning, M. A. C. Broeren, H. M. Jansen, J. A. J. M. Vekemans and E. W. Meijer, Chem. Lett., 2000, 292 Search PubMed . At concentrations higher than 15 mM self-association of G0, forming a columnar aggregate, in chloroform-d was observed. A self-association constant of 26 M−1 was calculated from an NMR dilution experiment using a non-linear curve-fitting procedure assuming the formation of indefinite stacks ([G0] = 17–43 mM, the shifts of the aromatic and amide proton resonances and methylene proton resonance adjacent to the amide were used as probes). For the fitting procedure see: M. Gardner, A. J. Guerin, C. A. Hunter, U. Michelsen and C. Rotger, New J. Chem., 1999, 23, 309 CrossRef CAS . Note: the factor 2 in equation 6 of this paper should be absent.
- In the absence of hosts it was not possible to prepare a 25 mM solution of 4 in chloroform-d due to insolubility.
- Y. Okada, Curr. Org. Chem., 2001, 5, 1 CrossRef CAS; N. L. Benoiton and F. M. P. Chen, Can. J. Chem., 1981, 59, 384 CAS . Racemization through tautomerization of an azlactone (5-(4H)-oxazolone) intermediate is less likely since the carbonyls in urethane protected amino acids are usually not nucleophilic enough to undergo the cyclization. If azlactone formation does occur, the 2-alkoxyoxazolones that form are stable towards racemization. See: J. H. Jones and M. J. Witty, J. Chem. Soc., Perkin Trans. 1, 1970, 3203 Search PubMed.
- K. A. Conners, Binding Constants, The Measurement of Molecular Complex Stability, Wiley-Interscience, New York, 1987 Search PubMed.
- B. Escuder, A. E. Rowan, M. C. Feiters and R. J. M. Nolte, Tetrahedron, 2004, 60, 291 CrossRef CAS.
- E. Delort, T. Darbre and J.-L. Reymond, J. Am. Chem. Soc., 2004, 126, 15642 CrossRef CAS; A. Clouet, T. Darbre and J.-L. Reymond, Adv. Synth. Catal., 2004, 346, 1195 CrossRef CAS; D. Lagnoux, E. Delort, C. Douat-Casassus, A. Esposito and J.-L. Reymond, Chem.–Eur. J., 2004, 10, 1215 CrossRef CAS.
- W. L. Gilliland and A. A. Blanchard, J. Am. Chem. Soc., 1926, 48, 410 CrossRef CAS; G. M. Whitesides and J. D. R. Nordlander, Discuss. Faraday Soc., 1962, 34, 185 RSC.
- Specific optical rotation measured for N-CBZ-L-5: [α]20D = + 7.9° (c 1.0 in CHCl3).
- GraFit 4.0, Erithacus Software.
- A. P. Bisson, C. A. Hunter, J. C. Morales and K. Young, Chem.–Eur. J., 1998, 4, 845 CrossRef CAS . Note: the equations 6 and 9 in this paper are wrong.
| This journal is © The Royal Society of Chemistry 2006 |