Dopamine selective molecularly imprinted polymers via post-imprinting modification
Toshifumi
Takeuchi
*ab,
Nobuo
Murase
a,
Hideshi
Maki
a,
Takashi
Mukawa
a and
Hideyuki
Shinmori
a
aGraduate School of Science and Technology, Kobe University, Kobe, 657-8501, Japan. E-mail: takeuchi@scitec.kobe-u.ac.jp; Fax: +81-788036158; Tel: +81-788036158
bPRESTO, Japan Science and Technology Agency (JST), Kawaguchi, 332-0012, Japan
First published on 5th January 2006
Abstract
A novel synthetic dopamine receptor bearing bidentate binding sites were prepared by covalent imprinting using a disulfide linkage which is cleaved and oxidized to a non-covalent sulfoxide recognition group. The used templates have dopamine-like structures connected to an allyl moiety via a disulfide and to a 4-vinylphenyl group via a cyclic boronic diester. After the polymerization, the ester bonds were hydrolyzed and the disulfide bond was reduced to remove the template moiety from the polymer matrix, followed by the oxidation to transform the thiol residues into sulfonic acid (post imprinted process). The imprinted polymer adsorbed dopamine selectively in aqueous solution with the two-point interaction, i.e. the formation of cyclic boronic diester and electrostatic interaction with the sulfonic acid residue.
Introduction
Molecular imprinting has been widely recognized as a technique for the construction of materials containing binding sites that can recognize a given target molecule.1 The most attractive aspect of this technique is the use of molecular templates to assemble functional monomers around them into complementary orientations, forming tailor-made binding sites for the target molecules within a synthetic polymer matrix. The resulting imprinted polymers show specific binding behaviors for the target molecules. Template analog–functional monomer complexes were fashioned using covalent and non-covalent interactions in which all or part of the template analog is removed after polymerization, yielding a three dimensional cavity. In this manner, not only molecular recognition but also catalytic and signalling functionality can be easily introduced into the synthetic polymers, using appropriately designed template molecules.2 Unlike low-molecular weight synthetic receptors, in which proper design of the binding site is the most crucial aspect, binding sites of imprinted polymers are automatically constructed according to the template molecules designed.Here, we report on a novel synthetic dopamine receptor bearing two-point binding sites prepared by a covalent imprinting system using disulfide templates followed by a post-imprinting treatment, in which the original functional groups are chemically transformed into other functional groups that can form strong non-covalent interactions.
The target molecule, dopamine, has 3,4-dihydroxyphenyl and amino groups in its structure that were utilized to form a cyclic diester with a boronic acid and non-covalent electrostatic interactions with acidic functional groups such as sulfonic acid. Therefore, we designed a molecularly imprinted polymer containing both a boronic acid and a sulfonic acid in the binding site by using covalent molecular imprinting. This polymer differs from previous reported catecholamine-imprinted polymers prepared by non-covalent imprinting using organic3 and inorganic materials.4 Because an excess of functional monomers, usually used in non-covalent imprinting, may generate nonspecific binding sites upon being randomly grafted into the polymer matrix, we employed a covalent molecular imprinting strategy for the introduction of minimum amounts of boronic acid and sulfonic acid into the polymer matrices (Fig. 1, step 1).
 | ||
| Fig. 1 Schematic diagram of dopamine imprinting with the post-imprinting oxidation. | ||
Results and discussion
We designed a template molecules 5-[2-(allyldithio)ethyl]-2-(4-vinylphenyl)benzo[1,3,2]dioxaborole, (Template 1) that has a dopamine-like core connected to an allyl moiety via a disulfide group and to a cyclic boronic diester via 4-vinyl phenylboronic acid. For the introduction of the boronic acid moiety, we employed a conventional imprinting method as previously reported by Wulff et al. for imprinting carbohydrates.5 Disulfides are easily cleaved from polymer matrices under reducing conditions (Fig. 1, step 2)6 and the cyclic boronic diesters can be hydrolyzed under acidic conditions (Fig. 1, step 3). The cleavage would create a cavity complementary to dopamine and the imprinting process is completed at this point.7The corresponding reduced polymer contains the thiol residues located in the proper position in the binding site that can interact with the amino group of dopamine, after being transformed into a sulfonic acid by oxidation with H2O2 (post-imprinting treatment, Fig. 1, step 4).6 Finally, after completing all of the treatments, phenylboronic acid and sulfonic acid residues are assembled suitably for the binding of dopamine, meaning that the dopamine binding sites can be generated, in which dopamine will be captured with two-point binding. A reference template, 5-[2-(allyldithio)ethyl]-2-(phenyl)benzo[1,3,2]dioxaborole (Template 2) was prepared, in order to verify the formation of binding sites capable of two-point binding in the imprinted polymers prepared using Template 1.
The two imprinted polymers IP(T1) and IP(T2) were prepared by co-polymerizing styrene, divinylbenzene and either Template 1 or Template 2. A blank polymer (BP) was also prepared without any template. After all the post-imprinting chemical modifications were completed, the binding sites in the polymer prepared using Template 1 (IP(T1–SO3H)) consist of both boronic acid and sulfonic acid residues, and the polymer prepared using Template 2 (IP(T2–SO3H) has binding sites with only sulfonic acid residues. Styrene/divinylbenzene based polymers were used as the matrix because it was resistant to the redox reaction conditions that were employed. The numbers of thiol groups in the polymers (IP(T1-SH) and IP(T2–SH)) after the cleavage by the NaBH4 treatment were estimated to be 175 µmol g−1 for the both polymers, which corresponds to about 70% of the templates used in the imprinting process. Following oxidation of the thiol groups into sulfonic acid residues6 the loading of sulfonic acid residues in the polymers were 120 µmol g−1 IP(T1–SO3H) and 110 µmol g−1 IP(T2–SO3H), respectively, meaning that about 70 to 80% of the thiol residues were converted to sulfonic acids and the overall yields of the binding sites were about 50%.
In order to examine effects of the post-imprinting treatment, the binding of dopamine was investigated by HPLC method using columns packed with IP(T2–SO3H) and IP(T2–SH), which have no boronic acid residues. The retention factors were 13.08 ± 0.24 for IP(T2–SO3H) and 0.03 ± 0.01 for IP(T2–SH) (n = 3) at pH 9.5. The binding of dopamine became dramatically strengthened after the post-imprinting treatment. This reveals that the sulfonic acid residues in the binding sites of IP(T2–SO3H) can form strong electrostatic interactions with the positively charged amino group of dopamine under the testing conditions. From these results, the effectiveness of the post-imprinting treatment was clearly demonstrated by the enhancement in affinity of the post-modified polymer.
With IP(T2–SO3H), dopamine and tyramine showed the strongest binding at pH 9.5. pKa values of both amino and phenolic hydroxyl groups are around 10,8 therefore, at pH 9.5, the compounds may exist as zwitter ions and the cooperation of ion-exchange and hydrophobic effects could contribute to the stronger binding. At pH 6.8 and 11, the binding may occur via a simple ion-exchange process (Fig. 2). Catechol has no functional group capable of forming an electrostatic interaction with the sulfonic acid residues, therefore, the binding was weak and the pH dependence was not clearly observed.
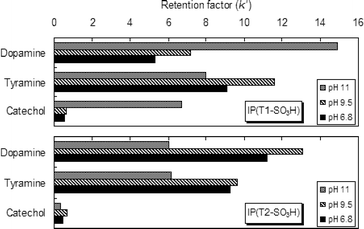 | ||
| Fig. 2 pH Dependence of the retention in IP(T1-SO3H) and IP(T2–SO3H). | ||
The introduction of boronic acid residues to the imprinted binding sites using Template 1 (IP(T1–SO3H)) provided an enhancement in binding affinity at higher pHs, because of the effective formation of cyclic boronic diester in alkaline solutions.9 Dopamine and catechol were strongly bound by IP(T1–SO3H) especially at higher pHs. In contrast, tyramine that has no dihydroxyl group showed a similar ion-exchange profile to that of IP(T2–SO3H). At pH 11.0, the binding of dopamine for IP(T1–SO3H) was the strongest among the tested compounds, and the k′ value of dopamine was approximately given by adding up those of tyramine (to a sulfonic acid binder) and catechol (to a boronic acid binder), suggesting that dopamine could be bound via two-point binding to the boronic acid and sulfonic acid residues. The blank polymer prepared without the templates showed almost no binding under all the conditions employed. Therefore, the imprinting effect clearly appears to be dominant, demonstrating that the template molecules worked well to generate binding sites selective for dopamine.
The relative selectivity of IP(T1–SO3H) and IP(T2–SO3H) at pH 11.0 is summarized in Table 1. In IP(T1–SO3H), dopamine showed the strongest binding because it has cis-diol and amino group. Tyramine has a primary amine but no cis-diol and catechol has no amine, and therefore, these two compounds had lower retention factors than dopamine. The compounds having carboxylic acid such as 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) showed lower affinity; of the two, DOPAC was better retained because it possesses a catechol structure. In IP(T2–SO3H), negatively charged compounds having no amino groups such as catechol, DOPAC and HVA showed almost no affinity due to the electrostatic repulsion. Finally, it was notable that IP(T1–SO3H) showed higher affinity for dopamine than IP(T2–SO3H).
|
|
||
|---|---|---|
| Sample | IP(T1–SO3H) | IP(T2–SO3H) |
| a The relative selectivity is expressed as ratios of retention factors of the tested samples to a retention factor of dopamine at pH 11.0. | ||
| Dopamine | 1.00 | 1.00 |
| Tyramine | 0.54 | 1.02 |
| Catechol | 0.45 | 0.05 |
| DOPAC | 0.36 | 0.02 |
| HVA | 0.08 | 0.00 |
| Epinephrine | 0.05 | 0.12 |
| Norepinephrine | 0.35 | 0.05 |
Although epinephrine and norepinephrine have also the both functional groups, the binding was weaker. Epinephrine showed especially low affinity to IP(T1–SO3H). The basicity of the secondary amine of epinephrine is higher than that of norepinephrine, thus epinephrine should be more strongly bound to IP(T1–SO3H) than norepinephrine, but the opposite trend was observed. In IP(T2–SO3H), epinephrine was more retained than norepinephrine, as expected. This trend can be explained by the binding sites in IP(T2–SO3H) being large enough to fit these compounds and can bind them by an ion-exchange mechanism, since it was generated by using Template 2, which generates a larger cavity than Template 1 due to the presence of unpolymerizable boronic acid moiety in Template 2. In contract, norepinephrine was retained longer than epinephrine by IP(T1–SO3H), suggesting that epinephrine does not fit as well into the imprinted cavity generated by Template 1 as norepinephrine has a less bulky primary amine. These results suggest that the selectivity is affected not only by the functional groups but also by the size of imprinted cavities.
Conclusions
The polymers with the post-imprinting treatment showed enhanced affinity without loss of the imprinting effect in aqueous solution. The present results prove that the proposed imprinting system involving the post imprinting oxidation can generate binding cavities as intended with two functional groups positioned in the binding site and work cooperatively. Tailoring the binding sites after constructing preferable molecularly imprinted binding sites by using organic chemistry shown here would open a new strategy to design more desirable molecular recognition and/or catalytic materials.Experimental
Preparation of Templates 1 and 2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.56 (2H, br s, 2 × OH), 5.83–5.94 (1H, m, CHCH2) and 6.63–6.83 (3H, m, Ar–H).
CH2), 5.35 (1H, d, CHCH2), 5.83–5.94 (2H, m, CHCH2 and CHCH2), 6.63–6.83 (4H, m, Ar–H and CHCH2), 7.51 (2H, d, Ar–H) and 8.15 (2H, d, Ar–H).
CH2), 5.78–5.94 (1H, m, CHCH2), 6.60–6.78 (3H, m, Ar–H) and 7.30–8.20 (5H, m, Ar–H).
CH2), 5.56 (2H, br s, 2 × OH), 5.83–5.94 (1H, m, CHCH2) and 6.63–6.83 (3H, m, Ar–H).
CH2), 5.35 (1H, d, CHCH2), 5.83–5.94 (2H, m, CHCH2 and CHCH2), 6.63–6.83 (4H, m, Ar–H and CHCH2), 7.51 (2H, d, Ar–H) and 8.15 (2H, d, Ar–H).
CH2), 5.78–5.94 (1H, m, CHCH2), 6.60–6.78 (3H, m, Ar–H) and 7.30–8.20 (5H, m, Ar–H).
Preparation of IP(T1) and IP(T2)
Either Template 1 or Template 2 (2 mmol) was dissolved in chloroform (5 mL) with divinylbenzene (50 mmol), styrene (10 mmol) and 2,2′-azobis(isobutyronitrile) (500 mg). The mixture was purged with nitrogen gas for 5 min. The glass tube was sealed and placed under UV light (XX-15L, UVP, Upland, CA) for 24 h at 5 °C, followed by heating for 3 h at 80 °C. A blank polymer (BP) was prepared without the template molecules.Removal of the template molecule to yield IP(T1–SH) and IP(T2–SH)
The obtained polymers were crushed roughly, then the particles were suspended in methanol (100 mL) with NaBH4 (20 mmol). The mixture was stirred for 12 h to cleave the disulfide bond of the template and the NaBH4 treatment was carried out three times. The particles were then treated with a diluted HCl solution containing 50% (v/v) methanol to hydrolyze the boronic acid ester of the template. The particles were washed twice with 50% (v/v) methanolic aqueous solution, then washed with methanol. Finally the particles obtained were dried in vacuo.To determine the amounts of thiol groups,10 polymer particles (200 mg) were suspended in 80% (v/v) methanolic aqueous solution containing 10 mM silver(I) nitrate (20 mL), and stirred for 3 h. After the filtration, the filtrate was adjusted to be 100 mL with water. An appropriate amount of iron (III) nitrate nonahydrate was added and a 10 mL aliquot was titrated with 2 mM potassium thiocyanate methanolic solution.
Post-imprinting treatment to yield IP(T1–SO3H) and IP(T2–SO3H)
Polymer particles (200 mg) were suspended in acetic acid containing ca. 15% hydrogen peroxide, and stirred for 12 h. After the filtration, the particles were washed twice with 50% (v/v) methanolic aqueous solution containing 50 mM sulfonic acid, then washed twice with methanol.The particles (200 mg) were treated with 1 M sodium chloride (100 mL) and pH in the supernatant was measured. This treatment was carried out three times and an amount of hydrogen released from sulfonic acid residues by ion exchange was calculated.
HPLC conditions
An HPLC used consisted of two pumps (Gilson model 305 and 306), an auto-injector (Gilson model 234) and a UV/VIS detector (Gilson model 119). The eluents used (1 mL min−1) were acetonitrile–10 mM phosphate buffer pH 11.0 (1 : 1 v/v), acetonitrile–10 mM glycine buffer pH 9.5 (1 : 1 v/v) or acetonitrile–10 mM phosphate buffer pH 6.8 (1 : 1 v/v). The detection was carried out at 254 nm. The sample size was 10 µL. Retention factors were calculated using the equation k′ = (tR − t0)/t0, where tR is the retention time of the solutes and t0 is the retention time of potassium nitrate used as a void marker. The measurement of each sample was performed in triplicate.Acknowledgements
The authors thank Prof. Ken Shimizu (University of South Calorina) for his helpful discussion.References
- (a) ACS Symp. Ser. 703: Molecular and Ionic Recognition with Imprinted Polymers, ed. R. A. Bartsch and M. Maeda, American Chemical Society, Washington, DC, 1997 Search PubMed; Molecularly Imprinted Polymers, ed. B. Sellergren, Elsevier, Amsterdam, 2001 Search PubMed; M. Komiyama, T. Takeuchi, T. Mukawa and H. Asanuma, Molecular Imprinting, WILEY-VCH, Weinheim, 2002 Search PubMed; G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812 Search PubMed; T. Takeuchi and J. Haginaka, J. Chromatogr., B: Biomed. Appl., 1999, 728, 1 Search PubMed; S. C. Zimmerman and N. G. Lemcoff, Chem. Commun., 2004, 5 Search PubMed.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495 CrossRef CAS; G. Wulff, Chem. Rev., 2002, 102, 1 CrossRef CAS; T. Takeuchi, T. Mukawa and H. Shinmori, Chem. Rec., 2005, 5, 263 CrossRef CAS.
- O. Ramström, C. Yu and K. Mosbach, J. Mol. Recognit., 1996, 9, 691 CrossRef CAS; R. Suedee, C. Songkram, A. Petmoreekul, S. Sangkunakup, S. Sankasa and N. Kongyarit, J. Pharm. Biomed. Anal., 1999, 19, 519 CrossRef CAS; A. B. Kharitonov, A. N. Shipway and I. Willner, Anal. Chem., 1999, 71, 5441 CrossRef CAS; S. A. Piletsky, E. V. Piletska, B. Chen, K. Karim, D. Weston, G. Barrett, P. Lowe and A. P. F. Turner, Anal. Chem., 2000, 72, 4381 CrossRef CAS; C. Liang, H. Peng, A. Zhou, L. Nie and S. Yao, Anal. Chim. Acta, 2000, 415, 135 CrossRef CAS; J. X. Du, L. H. Shen and J. R. Lu, Anal. Chim. Acta, 2003, 489, 183 CrossRef CAS; J. Matsui, K. Akamatsu, S. Nishiguchi, D. Miyoshi, H. Nawafune, K. Tamaki and N. Sugimoto, Anal. Chem., 2004, 76, 1310 CrossRef CAS.
- M. Morita, Y. Iwasaki, T. Horiuchi and O. Niwa, Denki Kagaku, 1996, 64, 1239 Search PubMed; R. Makote and M. M. Collinson, Chem. Commun., 1998, 425 RSC.
- G. Wulff and S. Schauhoff, J. Org. Chem., 1991, 56, 395 CrossRef CAS.
- T. Mukawa, T. Goto and T. Takeuchi, Analyst, 2002, 127, 1407 RSC ; R. D'Oleo et al. reported a similar approach for the preparation of ion-recognized polymer gels, however, the polymers were prepared with no template. Therefore, its concept was much different from our system in which the functional groups were introduced at appropriate positions inside the binding sites. See R. D'Oleo, C. Alvarez-Lorenzo and G. Sun, Macromolecules, 2001, 34, 4965 Search PubMed.
- Whitcombe et al. reported a covalent molecular imprinting system using 4-vinylphenyl carbonate ester to make non-covalent recognition sites for cholesterol. In this case, no post-imprinting treatment was performed. See M. J. Whitcombe, M. E. Rodriguez, P. Villar and E. N. Vulfson, J. Am. Chem. Soc., 1995, 117, 7105 Search PubMed.
- G. P. Lewis, Br. J. Pharmacol. Chemother., 1954, 9, 488 CAS . The pKa values were also calculated by pKalc 3.2 in Pallas for Windows (CompuDrug International, USA).
- P. J. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769 CrossRef CAS.
- C. R. Stahl and S. Siggia, Anal. Chem., 1957, 29, 154 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |