Mechanistic insights into triterpene synthesis from quantum mechanical calculations. Detection of systematic errors in B3LYP cyclization energies†
Seiichi P. T.
Matsuda
*ab,
William K.
Wilson
b and
Quanbo
Xiong
b
aDepartment of Chemistry, Rice University, 6100 Main Street, Houston, TX 77005, USA. E-mail: matsuda@rice.edu; Fax: 01 713 348 5154; Tel: 01 713 348 6158
bDepartment of Biochemistry and Cell Biology, Rice University, 6100 Main Street, Houston, TX 77005, USA
First published on 4th January 2006
Abstract
Most quantum mechanical studies of triterpene synthesis have been done on small models. We calculated mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G* energies for many C30H51O+ intermediates to establish the first comprehensive energy profiles for the cationic cyclization of oxidosqualene to lanosterol, lupeol, and hopen-3β-ol. Differences among these 3 profiles were attributed to ring strain, steric effects, and proton affinity. Modest activation energy barriers and the ample exothermicity of most annulations indicated that the cationic intermediates rarely need enzymatic stabilization. The course of reaction is guided by hyperconjugation of the carbocationic 2p orbital with parallel C–C and C–H bonds. Hyperconjugation for cations with a horizontal 2p orbital (in the plane of the ABCD ring system) leads to annulation and ring expansion. If the 2p orbital becomes vertical, hyperconjugation fosters 1,2-methyl and hydride shifts. Transition states leading to rings D and E were bridged cyclopropane/carbonium ions, which allow ring expansion/annulation to bypass formation of undesirable anti-Markovnikov cations. Similar bridged species are also involved in many cation rearrangements. Our calculations revealed systematic errors in DFT cyclization energies. A spectacular example was the B3LYP/6-311+G(2d,p)//B3LYP/6-31G* prediction of endothermicity for the strongly exothermic cyclization of squalene to hopene. DFT cyclization energies for the 6-311+G(2d,p) basis set ranged from reasonable accuracy (mPW1PW91, TPSSh with 25% HF exchange) to underestimation (B3LYP, HCTH, TPSS, O3LYP) or overestimation (MP2, MPW1K, PBE1PBE). Despite minor inaccuracies, B3LYP/6-31G* geometries usually gave credible mPW1PW91 single-point energies. Nevertheless, DFT energies should be used cautiously until broadly reliable methods are established.
Introduction
Triterpenes and their triterpenoid metabolites have important roles in many biological processes of plants, animals, and microorganisms.1 Triterpene synthases cyclize squalene (1) and oxidosqualene (7) to over 100 different carbon skeletons. Individual cyclases can selectively create 9 or more stereocenters and may produce a single triterpene with >99% accuracy. This combination of diversity, selectivity, and accuracy is achieved through numerous variations on the three fundamental pathways shown in Scheme 1.2,3 Vast effort has gone into studying these reactions, beginning with profound insights by Swiss4 and American5 workers 50 years ago. During the past decade, advances in molecular biology,6 molecular modelling,7 and protein crystallography8,9 have provided new tools for understanding this remarkable cyclization.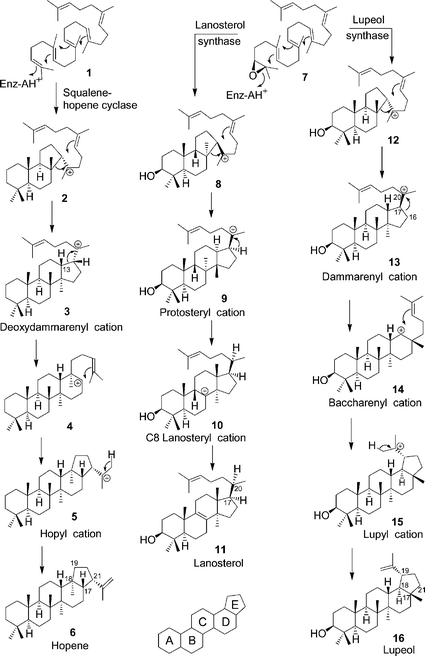 | ||
| Scheme 1 Three fundamental pathways of (oxido)squalene cyclization. | ||
Molecular modelling predicts that triterpene synthesis is a strongly exothermic process,2,7a,7b,7k and the high energy of cyclization correlates with properties of squalene-hopene cyclase (SHC).8,9c The low turnover rate of SHC allows for dissipation of energy, and the prominent QW motifs are thought to stabilize the enzyme against disintegration during the energetic reaction; the heat of reaction may melt the narrow exit channel to facilitate release of the bulky product from the active site cavity.8 Jenson and Jorgensen7a suggested that rings A and B form with an exothermicity of about 20 kcal mol−1 and that subsequent annulation should be exothermic by about 10 kcal mol−1.
Contrary to these authoritative estimates of strong exothermicity, some of our recent quantum mechanical calculations predicted endothermic annulations in triterpene synthesis.10 Notably, the B3LYP energies for the C25H43 models11 shown in Fig. 1 suggested that both D- and E-ring formation are endothermic for hopene and only slightly exergonic for lupeol. We have now calculated the overall energy of cyclization from squalene to hopene using the neutral C30H50 structures (see electronic supplementary information (ESI)†). Astonishingly, the ZPE-corrected B3LYP/6-311+G(2d,p)//B3LYP/6-31G* energy predicted this reaction to be endothermic.
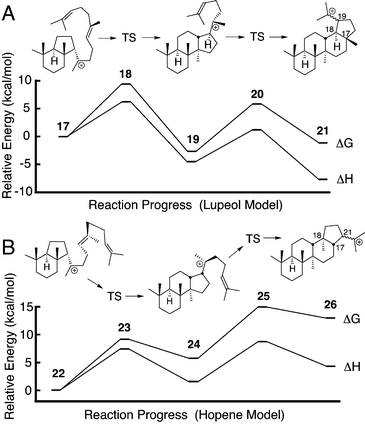 | ||
| Fig. 1 Energetics of D- and E-ring formation in lupeol (A) and hopene (B) synthesis predicted by B3LYP/6-311+G(2d,p)//B3LYP/6-31G(d) calculations for C25H43 models. Numerical data are given in Table S1. | ||
This result indicated either a massive error for the widely used B3LYP method or a major fallacy in the current understanding of triterpene synthesis. We thus sought a reliable estimate for the enthalpy of cyclization. Experimental heats of formation for triterpenes are unavailable, and we were deterred by the technical challenges of combustion calorimetry12 and by the insufficient accuracy of combustion energies for benchmark test samples sent to a major commercial analytical laboratory.13 Theoretical estimates of enthalpy differences using CBS–Q or G3 theory were far out of reach for C30H50 compounds with available computer hardware, as were CCSD(T) and other high-level ab initio methods.
We consequently resorted to empirical methods for estimating enthalpy differences. Heats of formation can be estimated from group additivity schemes,14 and accuracy is markedly improved by correcting for strain energies obtained from molecular mechanics15 or quantum mechanical calculations.16 For ordinary molecules, highly parameterized force fields like MM317 and MMX18 can usually predict heats of formation to within 1–2 kcal mol−1 of experiment.15,19 MM3-94 gave a heat of formation difference of −44 kcal mol−1 for cyclization of folded squalene to hopene, whereas B3LYP predicted endothermicity. These results validated current thinking about the exothermicity of squalene cyclization and pointed to a serious flaw in B3LYP energy predictions.
We show that the B3LYP errors are systematic and broad in scope. Unlike most DFT methods, mPW1PW91 gave reasonably accurate cyclization energies, and this method was used to generate the first reliable energy profiles for gas-phase (oxido)squalene cyclization. We also investigated whether the B3LYP errors affect geometry optimizations, activation energies, and transition state structures in triterpene synthesis. These studies provided the foundation for our primary objective, understanding the mechanism of triterpene synthesis. Described herein are detailed energetics for variations of (oxido)squalene cyclization, together with numerous mechanistic insights.
Results and discussion
Scope of errors in B3LYP cyclization energies. Comparison with other DFT methods
We searched for alternatives to B3LYP by comparing benchmark heats of formation with energies from various theoretical methods. Absolute DFT and ab initio energies are not readily converted to accurate heats of formation.14e–g However, energy differences among isomers benefit from a cancellation of errors and can be compared with differences in heats of formation from experiment or force-field calculations.We began by comparing energies among C10H18 isomers for which experimental heats of formation are known. Table 1 gives the energy differences between 5-decyne and various alicyclic hydrocarbons for many theoretical methods. (The kinetic improbability of these “cyclizations” does not undermine the validity of the thermodynamic energy differences.) Deviation of the predicted values from experiment is given by the RMSD for ΔH, and the MSE gives the direction of the error. The results indicate that B3LYP systematically underestimates the exothermicity of cyclization. Among DFT methods20 with the 6-311+G(2d,p) basis set, exothermicity of cyclization was low with B3LYP, O3LYP, B1LYP, HCTH, and TPSS and was overestimated with MPW1K, VSXC, and PBE1PBE. Good agreement21 was found for mPW1PW91, TPSSh (25% HF exchange), B3P86, and the MMX and MM3 force fields. The theoretical methods showed similar behavior with sets of C10H16 isomers and C10H18O isomers (Tables S3 and S4 in the ESI†),22 although the lack of accurate experimental combustion data limited the value of these comparisons.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Method | RMSD ΔEb | RMSD ΔHc | MSE ΔH | Energies relative to 5-decyne (34)/kcal mol−1 | ||||||
| 27 | 28 | 29 | 30 | 31 | 32 | 33 | ||||
| a Values for quantum mechanical methods are electron energies. Geometries and thermochemistry results are from B3LYP/6-31G* calculations. Energies are for the most stable conformer only. Additional data and references for the experimental heats of formation (Hf) are given in Table S2 in the ESI1. The RMSD (root mean square deviation) and MSE (mean signed error) calculations describe only cyclization enthalpies and thus exclude dihydromyrcene data. Deviation = theory − experiment. b RMSD of predicted electron energy differences relative to experimental heat of formation differences, without ZPE or thermal energy corrections. c RMSD of predicted enthalpy (or heat of formation) differences relative to experimental heat of formation differences. For quantum mechanical methods, enthalpies were obtained by adding the ΔH increment to the electron energy. Only ZPE and thermal vibrational contributions were scaled. d Estimated from Benson-type calculation (ref. 14d). | ||||||||||
| Experiment (Hf) | — | 0.0 | 0.0 | −11.4d | −48.0 | −44.9 | −39.1 | −30.9 | −35.8 | −35.6 |
| MM3-94 | — | 1.5 | 1.0 | −9.8 | −45.8 | −43.0 | −39.4 | −30.7 | −35.3 | −33.3 |
| MM3 (PCMODEL) | — | 1.5 | 0.9 | −10.2 | −45.8 | −43.0 | −39.4 | −31.0 | −35.3 | −33.3 |
| MMX (PCMODEL) | — | 1.2 | 0.6 | −10.4 | −46.4 | −43.6 | −39.4 | −31.9 | −35.1 | −33.7 |
| AM1 | 3.5 | 2.3 | −0.1 | −1.8 | −50.6 | −47.8 | −42.3 | −26.2 | −40.6 | −37.7 |
| HF/6–31G* | 3.6 | 4.8 | 4.4 | −3.8 | −45.2 | −41.8 | −36.4 | −25.9 | −32.6 | −31.4 |
| B3LYP/6–31G* | 2.9 | 4.3 | 3.9 | −7.0 | −45.2 | −41.9 | −36.8 | −28.5 | −32.5 | −32.4 |
| B3LYP/6-311+G(2d,p) | 8.2 | 9.2 | 8.4 | −7.3 | −39.1 | −35.7 | −31.2 | −25.2 | −27.5 | −26.8 |
| MPW1K/6-31G* | 11.7 | 9.1 | −8.3 | −6.6 | −60.9 | −57.7 | −52.6 | −36.4 | −47.5 | −47.3 |
| MPW1K/6-311+G(2d,p) | 7.5 | 5.3 | −4.8 | −7.4 | −56.2 | −52.9 | −48.1 | −34.3 | −43.4 | −42.9 |
| mPW1PW91/6-31G* | 7.4 | 5.2 | −4.7 | −6.9 | −56.1 | −52.9 | −48.0 | −34.2 | −43.2 | −42.9 |
| mPW1PW91/6-311+G(2d,p) | 3.1 | 1.3 | −1.1 | −7.5 | −51.2 | −47.9 | −43.4 | −31.8 | −39.2 | −38.5 |
| B3PW91/6-311+G(2d,p) | 0.7 | 2.0 | 1.7 | −7.1 | −47.5 | −44.2 | −39.8 | −29.6 | −36.0 | −35.1 |
| B1LYP/6-311+G(2d,p) | 8.6 | 9.6 | 8.8 | −7.1 | −38.8 | −35.3 | −30.7 | −24.9 | −27.1 | −26.4 |
| B3P86P/6-311+G(2d,p) | 2.2 | 0.6 | −0.4 | −8.0 | −50.3 | −47.0 | −42.4 | −31.6 | −38.0 | −37.6 |
| PBE1PBE/6-311+G(2d,p) | 5.1 | 3.1 | −2.8 | −7.9 | −53.4 | −50.1 | −45.6 | −33.2 | −41.1 | −40.6 |
| HCTH/6-311+G(2d,p) | 10.9 | 11.7 | 10.7 | −6.1 | −36.0 | −32.0 | −28.2 | −22.8 | −26.5 | −24.5 |
| VSXC/6-311+G(2d,p) | 8.6 | 6.7 | −5.2 | −19.2 | −53.8 | −53.9 | −52.4 | −42.0 | −38.8 | −40.0 |
| TPSS/6-311+G(2d,p) | 5.1 | 6.4 | 5.7 | −7.8 | −41.7 | −38.6 | −35.0 | −27.0 | −31.7 | −30.2 |
| TPSSh (25%)/6-311+G(2d,p) | 1.6 | 0.8 | 0.4 | −7.4 | −49.0 | −45.9 | −41.8 | −30.5 | −37.9 | −36.7 |
| O3LYP/6-311+G(2d,p) | 7.8 | 8.9 | 8.1 | −5.4 | −39.2 | −35.3 | −31.6 | −24.0 | −30.1 | −27.7 |
| MP2/6-31G* | 6.8 | 4.7 | −4.1 | −6.4 | −55.4 | −52.7 | −48.5 | −34.0 | −41.5 | −41.2 |
| CCSD(T)/6-31G* | 5.8 | 3.8 | −3.4 | −9.7 | −54.1 | −51.4 | −47.0 | −34.7 | −40.5 | −40.4 |
| ZPE increment | — | — | — | −0.7 | 4.6 | 4.8 | 4.3 | 1.4 | 3.5 | 4.4 |
| ΔH increment | — | — | — | −0.9 | 2.0 | 2.2 | 1.9 | 0.2 | 1.7 | 2.1 |
| ΔG increment | — | — | — | 0.2 | 10.0 | 10.3 | 9.2 | 4.8 | 5.7 | 9.1 |

The DFT results showed a marked dependence on basis set size and amount of exact HF exchange. Cyclization energies become more positive with larger basis sets (Table 1 and Tables S2b, and S7c in the ESI†) and smaller percentages of HF exchange (Table S7e in the ESI). Thus, small basis sets like 6-31G* reduce errors for DFT methods that underestimate the exothermicity of cyclization (e.g., B3LYP) and increase errors for methods that overestimate exothermicity (e.g., MPW1K). The best DFT method for cyclization energies depends on the preferred basis set. With faster computers, we would use a larger basis set like cc-pVTZ, and then mPW1PW91 and TPSSh cyclization energies would be somewhat underestimated. This problem could be solved expediently by increasing the amount of HF exchange (as in MPW1K) or by using a different functional, such as PBE1PBE.
Subsequent studies were focused on the B3LYP, mPW1PW91, and MPW1K hybrid functionals. The latter was chosen for its good performance in modelling transition states and activation energies.20c,d In the course of obtaining improved molecular mechanics parameters for epoxides, we determined DFT energies for isomers of ethylene oxide and its methyl, dimethyl, trimethyl, and tetramethyl derivatives (Table 2).23 For these 19 compounds,24 the G3B3 energies closely matched the available experimental heats of formation. The mPW1PW91/6-311+G(2d,p) energies approached the accuracy of CCSD(T)/6-31+G** and MP4/6-31+G** energies, and B3LYP/6-311+G(2d,p) performed better than in Table 1. B3LYP energies were somewhat too positive for the conversion of open-chain ketones to cyclic ethers or alcohols (Table S5 in the ESI†), and mPW1PW91 energies were slightly too negative, similar to trends observed in olefin cyclizations in Table 1.
| Method | ΔE RMSD | ΔH RMSD | ΔH MSE |
|---|---|---|---|
| a For quantum mechanical methods, geometries and frequencies were from B3LYP/6-31G* calculations. Energies of epoxide isomers (given in Table S5 in the ESI1) are relative to that of a keto or aldehyde isomer. RMSD and MSE are defined as in Table 1 except that electron energies (ΔE, italic values) or enthalpies (ΔH) are compared against G3B3 enthalpies instead of heat of formation (Hf). Deviation = theory − experiment. | |||
| G3B3 (taken as standard) | — | 0.0 | 0.0 |
| Experimental Hf | — | 1.3 | 0.6 |
| MM3 (PCMODEL) | — | 0.9 | −0.6 |
| MMX (PCMODEL) | — | 10.2 | 4.4 |
| HF/6-31G* | 4.1 | 4.5 | 4.1 |
| B3LYP/6-31G* | 2.9 | 3.4 | 2.2 |
| B3LYP/6-311+G(2d,p) | 1.8 | 2.4 | 2.2 |
| mPW1PW91/6-31G* | 2.4 | 2.0 | −0.9 |
| mPW1PW91/6-311+G(2d,p) | 2.3 | 1.6 | −1.2 |
| MP2/6-31+G** | 0.8 | 0.9 | 0.3 |
| MP4/6-31+G** | 1.0 | 1.2 | 0.9 |
| CCSD(T)/6-31+G** | 0.8 | 1.0 | 0.8 |
In addition to the transformations described above, we calculated energies for the (oxido)squalene cyclization models shown in Table 3. These reactions gave the same pattern of error, i.e., underestimation of exothermicity by B3LYP, overestimation by MPW1K and MP2, and good estimates by mPW1PW91/6-311+G(2d,p), as judged by comparisons with MM3 and G3MP2B3 enthalpies. Although the reaction of isobutylene with the t-butyl cation (R3) is not a cyclization, the DFT energies showed similar trends. As in Table 2, the B3LYP errors occur not only in olefin cyclizations but also in transformations of a double bond to two single bonds.
|
|
||||
|---|---|---|---|---|
| Reaction | ||||
| Method | R1 | R2 | R3 | R4 |
| a For quantum mechanical methods, geometries and frequency results are from B3LYP/6-31G* calculations. G3MP2B3 values are enthalpies. In reaction R1, the C4–C5 bond of the reactant (71) was frozen at 3.8 Å. Additional data are given in Table S6 in the ESI1. b Force-field energies were not calculated for cationic species. | ||||
| MM3-94 | —b | −27.5 | —b | −106.3 |
| MMX (PCMODEL) | —b | −30.3 | —b | −108.8 |
| AM1 | −11.3 | −17.6 | −16.8 | −109.2 |
| HF/6-31G* | −16.8 | −21.1 | −19.3 | −90.8 |
| B3LYP/6-311+G(2d,p) | −12.8 | −14.0 | −18.0 | −77.9 |
| MPW1K/6-311+G(2d,p) | −22.0 | −33.1 | −25.8 | −119.1 |
| mPW1PW91/6-311+G(2d,p) | −19.0 | −27.2 | −23.8 | −106.0 |
| MP2/6-31G* | −27.7 | −43.9 | −32.1 | −124.1 |
| MP4/6-31G* | −25.6 | — | −30.1 | — |
| G3MP2B3 | −21.6 | — | −26.3 | — |
| ZPE increment | 2.7 | 4.7 | 3.8 | 9.9 |
| ΔH increment | 2.1 | 2.2 | 2.5 | 5.5 |
| ΔG increment | 4.7 | 11.8 | 18.0 | 22.3 |
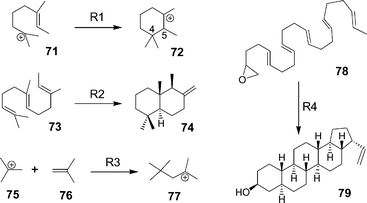
Energies for the cyclization of heptadesmethylsqualene 78 (Table 3, reaction R4) indicated that the B3LYP energy underestimations in triterpene synthesis are not attributable to the presence of angular methyl groups. This reaction produces roughly 20 kcal mol−1 for each of the five cation–olefin additions, similar to the energy predicted for reaction R3.7a In triterpene synthesis, effects of the angular methyl groups reduce this cyclization energy by about half. Simple Benson calculations14a–d largely neglect strain and thus overestimate triterpene heats of formation but match MM3 and mPW1PW91 predictions for reaction R4 rather closely, a sign of the lack of strain in the pentacyclic desmethyl product 79. Reaction R4 is kinetically improbable because the desired course of reaction would hard to control even if an enzyme could absorb the cyclization energy.
Despite the widespread application of the B3LYP hybrid functional to myriad problems in organic chemistry, the cited deficiencies in energy predictions have scarcely been recognized for several reasons. (1) These problems have been partially concealed by fortuitous cancellation of errors at lower levels of theory. Whereas ab initio predictions tend to improve with higher levels of theory, DFT results often do not. As seen in Table 1, when B3LYP is used with the 6-31G* basis set, the error is substantially smaller than with 6-311+G(2d,p). (2) The enthalpy underestimation by B3LYP is reduced when ZPE or thermal energy corrections are not performed. Notably, the uncorrected RMSD in Table 1 is much worse for mPW1PW91/6-31G* than B3LYP/6-31G* (7.4 vs. 2.9). (3) Cancellation of errors occurs for reactions with isodesmic character, which is decidedly lacking in the formation of two single bonds from a double bond in olefin cyclizations. (4) The error for B3LYP is modest for formation of a single ring, and small model compounds are still the norm for DFT studies. Only when several rings are formed from an acyclic precursor does the low B3LYP energy stand out.
The B3LYP underestimation of exothermicity in certain cyclization reactions has been noted by B. A. Hess25 and others.26,27a,b This trend is apparent in cycloaddition reactions between the hydroxyallyl cation and dienes,26 eneyne–allene cyclizations,27a cation–olefin cyclizations in triterpene synthesis,7n,10 and pericyclic reactions.27b–d The same B3LYP errors are manifest in energy comparisons among strained hydrocarbons28 and cyclic hydrocarbon conformers.29
The DFT problems with cyclization energies differ from known systematic errors in DFT. Most documented shortcomings of DFT14f,30 involve non-bonded interactions, charge transfer, bond dissociation, extended conjugation, excited states, free radicals, or transition states. Unlike most DFT problems, the errors for cyclization energies impact mainstream modelling of neutral organic molecules.
Some systematic DFT errors are superficially similar to the cyclization energy problem. For example, B3LYP predicts cumulenes to be more stable than polyynes.31 As with B3LYP cyclization energies, the errors accumulate with increasing carbon chain length and can be reduced by increasing the percentage of HF exchange.31b However, the cumulene–polyyne errors show little dependence on basis set size, and B3LYP and mPW1PW91 produce almost identical errors.31c The operational differences between the cumulene–polyene and cyclization energy problems suggest different underlying causes.
Systematic DFT errors are commonly mitigated by use of a specific method.30b For example, KMLYP gives reasonable cumulene–polyyne energies,31c BHLYP and KMLYP give correct minima for annulenes,30c MPW1K describes certain types of transition states well,20c,d and we use mPW1PW91 for cyclizations.
Much effort has been devoted to improving B3LYP, the prevalent hybrid method using the generalized gradient approximation (GGA).20b–n,32 Incremental improvements on classical hybrid methods include mPW1PW91,20b PBE1PBE,20j O3LYP,20e,20f KMLYP,32a X3LYP,32b and MPW1K.20c,20d The MPW1K method increases the amount of exact HF exchange in the mPW1PW91 method from 25% to 42.8% and gives improved transition-state geometries and activation energies.20c,20d In our results, MPW1K and B3LYP cyclization energies erred considerably in opposite directions, but both methods gave reasonable activation energies. Some other recent methods, e.g., VSXC20n and HCTH,20i avoid any mixing of exact HF exchange. The meta-GGA approach20k–n used with VSXC, TPSS, and PKZB32c includes the kinetic energy density along with the electron density and its gradient. Among many candidates, no clear successor to B3LYP has emerged.33 Our results suggest that more elaborate training sets will be needed for developing better DFT methods.
Energetics of triterpene synthesis
Overall energies for (oxido)squalene cyclization by three major pathways of triterpene synthesis are given in Table 4. Table 5 shows the incremental energy changes as each ring is formed. These energies have an uncertainty of several kcal mol−1, but greater accuracy can be expected for comparisons among different pathways or different stages of cyclization owing to cancellation of errors. As expected, the benchmark MM3 energies in Table 4 agreed much better with mPW1PW91 than B3LYP energies, which predicted endothermic C and D ring formation in lanosterol synthesis (Table 5).| Oxidosqualene | Squalene | Squalene-X | ||||
|---|---|---|---|---|---|---|
| Hopen-3β-ol | Lupeol | Lanosterol | Cycloartenol | Hopene | Hopene | |
a Values for quantum mechanical methods are electron energies; geometries and frequencies are from B3LYP/6-31G* calculations. Additional data are given in Table S7 in the ESI1. Energies are relative to the appropriately folded (oxido)squalene except that lanosterol and cycloartenol energies are compared to oxidosqualene folded for lupeol formation. Lanosterol and cycloartenol were modelled with the same non-extended side chain conformer (arbitrarily chosen before the lanosterol synthase crystal structure9a was reported). Energies are given for the C-ring boat conformer of cycloartenol. “Squalene-X” is a squalene conformer analogous to the 2-azasqualene conformer in an X-ray crystal structure of SHC;8 the proximal Me2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH-group was constructed manually, followed by B3LYP/6-31G* optimization in which carbon coordinates beyond C5 were frozen.
b MMX and MM3 are not well parameterized for substituted cyclopropanes.
c MM3 and MMX energies were not calculated for the Squalene-X conformer because fixing numerous coordinates in a force-field optimization is not practical or meaningful in this context. CH-group was constructed manually, followed by B3LYP/6-31G* optimization in which carbon coordinates beyond C5 were frozen.
b MMX and MM3 are not well parameterized for substituted cyclopropanes.
c MM3 and MMX energies were not calculated for the Squalene-X conformer because fixing numerous coordinates in a force-field optimization is not practical or meaningful in this context.
|
||||||
| MM3-94 | −55.6 | −66.8 | −61.3 | —b | −44.2 | —c |
| MM3 (PCMODEL) | −54.0 | −64.5 | −56.5 | —b | −42.7 | —c |
| MMX (PCMODEL) | −61.3 | −72.2 | −68.5 | —b | −54.7 | —c |
| AM1 | −50.5 | −59.7 | −58.5 | −46.0 | −24.9 | −24.9 |
| HF/6-31G* | −31.1 | −41.5 | −38.5 | −29.2 | −26.8 | −29.3 |
| B3LYP/6-311+G(2d,p) | −22.9 | −32.1 | −30.0 | −18.6 | −14.8 | −16.6 |
| MPW1K/6-311+G(2d,p) | −67.9 | −76.7 | −65.1 | −58.6 | −62.3 | −64.0 |
| mPW1PW91/6-311+G(2d,p) | −54.3 | −62.7 | −54.7 | −46.5 | −48.5 | −50.2 |
| ZPE increment | 10.4 | 10.8 | 7.6 | 8.5 | 11.1 | 11.4 |
| ΔH increment | 5.6 | 5.7 | 4.1 | 4.5 | 6.3 | 6.9 |
| ΔG increment | 26.8 | 26.4 | 19.9 | 21.4 | 26.6 | 26.5 |
| Number of rings | Hopen-3β-ol formation | Lupeol formation | Lanosterol formation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 3 | 4 | 5 | 2 | 3 | 4-C20 | 4-C8 | 4-C9 | |
| a Cyclization energies (kcal mol−1) or entropy (cal mol−1 K−1) relative to protonated oxidosqualene folded to form hopene or lupeol (also used for lanosterol energies). Geometries and frequencies are from B3LYP/6-31G* calculations. Energy differences of (protonated) oxidosqualene folded for lupeol and hopen-3β-ol synthesis were negligible. One bond was frozen at 3.8 Å for monocycles (C9–C10) and bicycles (C8–C14); other structures were energy minima. Lanosterol intermediates were modelled with the same non-extended side chain used for lanosterol calculations in Table 4. Additional data are given in Table S8 in the ESI1. For lanosterol formation, column headings 4-C8, 4-C9, and 4-C20 denote the lanosteryl C8, lanosteryl C9, and protosteryl cations, respectively. | |||||||||||||
| AM1 | −32.0 | −42.5 | −47.3 | −49.2 | −48.5 | −45.9 | −53.5 | −62.5 | −37.2 | −39.8 | −41.0 | −52.8 | −60.4 |
| HF/6-31G* | −19.7 | −32.1 | −35.7 | −35.2 | −36.2 | −36.1 | −42.7 | −49.1 | −23.9 | −25.2 | −25.9 | −40.0 | −45.5 |
| B3LYP/6-311+G(2d,p) | −16.9 | −27.6 | −30.0 | −29.1 | −27.1 | −30.0 | −32.5 | −36.6 | −19.8 | −18.0 | −16.4 | −28.4 | −32.1 |
| MPW1K/6-311+G(2d,p) | −23.8 | −43.8 | −54.2 | −63.2 | −70.4 | −54.4 | −67.3 | −81.1 | −35.9 | −43.8 | −51.4 | −64.2 | −68.1 |
| mPW1PW91/6-311+G(2d,p) | −21.5 | −38.9 | −47.2 | −53.7 | −57.7 | −47.3 | −57.2 | −67.9 | −30.7 | −37.6 | −43.6 | −53.2 | −56.7 |
| ZPE increment | 1.1 | 3.6 | 5.1 | 7.4 | 9.5 | 5.3 | 7.2 | 9.4 | 3.1 | 4.8 | 7.9 | 6.9 | 6.6 |
| ΔH increment | 0.3 | 1.7 | 2.5 | 3.7 | 4.9 | 3.1 | 4.1 | 5.2 | 1.8 | 2.8 | 4.5 | 3.9 | 3.5 |
| ΔG increment | 4.8 | 10.1 | 13.9 | 19.9 | 24.8 | 13.4 | 18.0 | 23.8 | 8.6 | 12.1 | 19.1 | 17.7 | 17.2 |
| Relative entropy | −15.4 | −28.0 | −38.5 | −54.2 | −66.7 | −34.5 | −46.3 | −62.3 | −22.8 | −31.0 | −48.9 | −46.3 | −45.9 |
The mPW1PW91 energies in Table 5 indicate that most of the reaction energy is released during AB ring formation, each annulation contributing close to the ca. 20 kcal mol−1 predicted in Fig. 2A and by the calculations of others.7a In protosteryl cation formation, the B-ring boat structure reduces the exothermicity of the second annulation to about 10 kcal mol−1. Formation of the 5-membered C ring produces only about 8 kcal mol−1 because the head-to-head construction of squalene from C15 precursors gives an arrangement of methyl groups that precludes a Markovnikov 6–6–6 product. At the tricycle stage, hopene and lupeol synthesis have each produced about 10 kcal mol−1 more energy than lanosterol synthesis. During D-ring formation, lupeol synthesis inches ahead in energy because hopene and lanosterol synthesis bear the cost of steric interactions between the side chain and C14 methyl. E-ring formation in hopenes suffers further from 1,3 axial interactions between the C14 and C17 angular methyls. The favorable series of 1,2-shifts in lanosterol synthesis propel the lanosteryl cation to the energy level of the hopyl cation. In lupeol synthesis, the last annulation is likewise favorable and makes the lupyl cation roughly 10 kcal mol−1 more stable than the other two. The large overall exothermicity of cyclization dwarfs the small energy differences among the substrate conformers.34
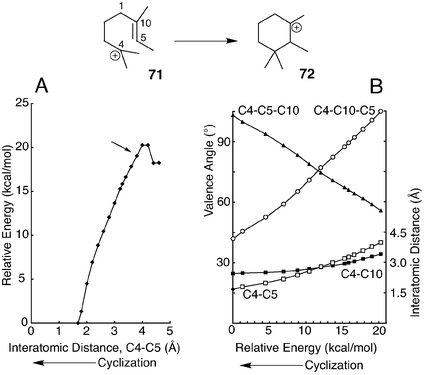 | ||
| Fig. 2 Relaxed PES scan (A) and geometry changes (B) during A-ring formation in hopene synthesis. The arrow in panel A indicates the point at which the C–C bond length was frozen for mono- and bicyclic intermediates of oxidosqualene cyclization. Data are from mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G* calculations. | ||
The ΔH and ΔG increments increase with each annulation and are similar among the three pathways with one exception. The absence of ring E in lanosterol makes its overall ΔH and ΔG increments lower than in the other pathways. Consequently, lanosterol synthesis is almost as exergonic as lupeol synthesis. Otherwise, the electron energy comparisons in Table 5 generally parallel the enthalpy and free energy differences.
Most cyclization energies for neutral species (Table 4) were somewhat lower than the corresponding energies of cation cyclization (Table 5). This is because the cost of initial oxidosqualene protonation is usually higher than the benefit of final deprotonation to the neutral cyclized triterpene.35 As judged by gas-phase proton affinities (Table S10 in the ESI†), the enthalpy deficit for hopene, lupeol, and cycloartenol synthesis is about 4, 5, and 11 kcal mol−1, respectively. Lanosterol showed an enthalpy surfeit of 1 kcal mol−1. The protonation/deprotonation enthalpy differences between lanosterol and cycloartenol (12 kcal mol−1) exceeded their neutral energy differences (8 kcal mol−1) because lanosterol is deprotonated from the C8 lanosteryl cation and cycloartenol from the more stable C9 cation.36,37Tables 4 and 5 also show that oxidosqualene cyclization is ∼6 kcal mol−1 more exothermic and exergonic than squalene cyclization.
The energies in Table 5 for formation of rings A, B, and C are somewhat arbitrary. Because we were unable to locate energy minima corresponding to formation of ring A or B,38 geometries were optimized by freezing the distance of the newly forming C–C bond in structures of the monocyclic and bicyclic cations. A value of 3.8 Å was chosen for this distance from the plot of energy vs. frozen bond distance for the simple model reaction R1 shown in Fig. 2A. The energy of 19 kcal mol−1 at this distance varied by only 1 kcal mol−1 over the range of 3.6–4.0 Å. Although some thermochemical increments in Table 5 are not fully valid because of the frozen bond distances, errors should be minor.
Results from Tables 4 and 5 were combined with activation energy data from Table S1 in the ESI† to give a summary of the overall gas-phase energetics of oxidosqualene pentacyclization (Fig. 3). Intermediates are protected only by low forward activation energies that are small relative to the overall energy of cyclization. About 70–80% of the cyclization energy is produced during the formation of rings A, B, and C. The remaining two annulations are much less energetic. In hopen-3β-ol formation (equivalent in this context to hopene formation), the free energy change from the tricyclic to pentacyclic cation is negligible. Enzymatic stabilization of the tricyclic or tetracyclic intermediate could further slow the reaction and invite byproduct formation. Along similar lines, Rajamani and Gao7b have proposed that the reported SHC byproducts39 stem from competition between kinetic and thermodynamic pathways in hopene synthesis. The results in Fig. 3 are compatible with calculations for related (oxido)squalene cyclizations by Jenson and Jorgensen7a and Hess7d–h and differ somewhat from energetics described by Rajamani and Gao.7b Protosteryl cation (9) formation follows the same general pattern, with some minor differences noted above.
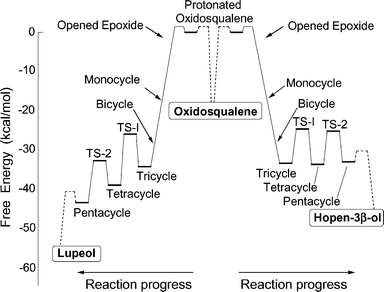 | ||
| Fig. 3 Predicted free energy profile for the cyclization of oxidosqualene to hopene or lupeol. These energies are based on gas-phase mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G* calculations. Energies involving the protonation of oxidosqualene and deprotonation of the pentacyclic cation to lupeol or hopene (shown by dashed lines) are indeterminate in this model, although the relative energies of protonation/deprotonation can be estimated (see text). | ||
The preceding discussion neglects the role of the enzyme in the initial protonation step, the stabilization of cationic intermediates, and the final deprotonation. Inclusion of these enzymatic effects drastically alters the energetics of A-ring formation but has little influence on conversion of the monocyclic cation to a tetra- or pentacyclic intermediate.40
Geometry optimization
The substantial underestimation of olefin cyclization energies by B3LYP raised concerns about the accuracy of the corresponding B3LYP geometries. We thus investigated geometries for several models of triterpene synthesis.Reactions R1 and R3 were studied using a variety of theoretical methods for geometry optimization (Table 6). As expected, β C–C bonds that aligned with the vacant cationic 2p orbital were elongated from hyperconjugation41 in all DFT and ab initio methods. For the hyperconjugated bond, B3LYP gave a loose geometry (long C–C bond). Geometries from the different methods showed the usual underestimation of cyclization energies by B3LYP and overestimation by MPW1K and MP2. However, single-point mPW1PW91 and B3LYP energies were essentially constant across all geometries except for the inexpensive AM1 and HF structures (Table 6).
| Optimization method | Reaction | ΔEa | ΔEb | ΔEb | C–Cc |
|---|---|---|---|---|---|
| Opt | mPW1PW91 | B3LYP | (Å) | ||
| a Electron energy differences (product − reactant(s)) from the method used for geometry optimization. For B3LYP/6-31G* thermochemical results, see Table 3; additional data are given in Table S12 in the ESI1. Gaussian 03 was used for reaction R3 and the HCTH data of reaction R1, in which the single-point energy was obtained from OmPW1PW91 (differing from the mPW1PW91 reaction energy by 0.1 kcal mol−1). b Electron energy differences from mPW1PW91/6-311+G(2d,p) or B3LYP/6-311+G(2d,p) single-point energy calculations. c Length of the C–C bond hyperconjugated to the product cation (C4–C5 for reaction R1). | |||||
| AM1 | R1 | −13.7 | −18.5 | −12.2 | 1.553 |
| HF/3-21G | R1 | −24.5 | −19.8 | −13.4 | 1.630 |
| B3LYP/6-31G* | R1 | −15.6 | −19.0 | −12.8 | 1.685 |
| B3LYP/6-31+G** | R1 | −14.1 | −19.0 | −12.8 | 1.683 |
| mPW1PW91/6-31G* | R1 | −21.8 | −19.2 | −12.6 | 1.658 |
| MPW1K/6-31G* | R1 | −25.1 | −19.1 | −12.4 | 1.633 |
| HCTH/6-31G* | R1 | −10.9 | −19.4 | −13.1 | 1.687 |
| MP2/6-31G* | R1 | −27.8 | −19.5 | −12.9 | 1.674 |
| AM1 | R3 | −20.5 | −22.4 | −15.3 | 1.557 |
| HF/3-21G | R3 | −25.4 | −24.2 | −18.1 | 1.627 |
| B3LYP/6-31G* | R3 | −22.7 | −23.8 | −18.0 | 1.668 |
| B3LYP/6-31+G** | R3 | −19.7 | −23.9 | −18.0 | 1.664 |
| mPW1PW91/6-31G* | R3 | −28.1 | −23.9 | −17.9 | 1.616 |
| MP2/6-31G* | R3 | −32.5 | −24.0 | −17.7 | 1.646 |
| MP2/6-31+G** | R3 | −31.5 | −24.0 | −17.7 | 1.645 |
| MP4(SDQ)/6-31G* | R3 | −28.4 | −24.0 | −17.8 | 1.624 |
| CCSD/6-31G* | R3 | −28.0 | −24.1 | −17.9 | 1.621 |
Constancy of the single-point energies is noteworthy because the hyperconjugated bonds were considerably longer in B3LYP optimizations than in MP4 and CCSD geometries. Thus, the underestimation of cyclization energies by B3LYP cannot be attributed to deficiencies of B3LYP/6-31G* geometries. In similar comparisons of geometries and energies among different theoretical methods, elongated B3LYP bonds were also observed for models of transition states (see below) and C-ring expansion/D-ring formation (Table S11 in the ESI†).
Lupeol geometries were compared using AM1, B3LYP, mPW1PW91, and MPW1K with the 6-31G* basis set. Relative to data from a crystal structure of lupeol acetate,42 average deviations for C–C bonds in rings A–D were 0.014 Å (B3LYP), 0.004 Å (mPW1PW91), −0.004 Å (MPW1K), and −0.009 Å (AM1). No corrections were made in comparing the quantum mechanical re values with the rα crystal values, which differ slightly due to room-temperature vibrations.43 Similarly, for oxidosqualene B3LYP C–C bond lengths were slightly longer than values for 2-azasqualene in an SHC crystal structure (Table S13 in the ESI†).8 Modelled oxidosqualene CC–C–C torsion angles differed somewhat from crystallographic values because of active-site constraints, but the folding for incipient rings B, C, and D was similar, as were the C–C distances corresponding to 5- and 6-membered ring formation (roughly 3.7 and 4.3 Å). The above results are consistent with the known ability of B3LYP and newer DFT methods to provide fairly accurate geometries for ordinary molecules.44
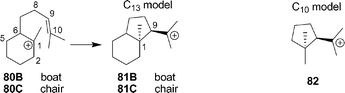
We studied a grossly distorted B3LYP geometry7e in order to understand how energy predictions are affected. Hess discovered an unusually long C–C bond (2.395 Å) in the B3LYP/6-31G* optimization of a small model (81B) for C-ring formation en route to the protosteryl cation.7e He found more reasonable bond lengths in MP2/6-31G* and HF/3-21G geometries (1.892 and 1.679 Å, respectively) and in the B3LYP/6-31G* optimization of the simpler analog 82.7e In an extension of his findings, we used a variety of methods to optimize the geometry of 80B, 81B, 81C, and 82. In these models, the 6-membered boat (80B, 81B) represents ring B in lanosterol synthesis. In dammarenyl cation formation, the corresponding ring is a chair (80C, 81C) and stereocenters in ring C are inverted. As noted by Hess,7e the folded conformer of 80B undergoes barrierless collapse to 81B, and we modelled 80B by freezing the C1–C10 distance at 3.8 Å.45
Geometries for 81B from several theoretical methods (Table 7) were fairly normal with two exceptions. B3LYP/6-31G* gave an energy minimum with a C1–C9 bond length of 2.40 Å, and HCTH/6-31G* showed a second minimum with a 2.81 Å bond length. Both methods gave low cyclization energies, suggesting a nearly flat potential energy surface (PES). Indeed, a B3LYP/6-31G* relaxed PES scan along the C1–C9 bond (Fig. 4) indicated a variation of only 0.5 kcal mol−1 in energy over the range 1.9–2.7 Å. Remarkably, relative mPW1PW91 single-point energies for corresponding points of the B3LYP and mPW1PW91 PES scans in Fig. 4 were almost identical (average deviation <0.04 kcal mol−1, Table S15 in the ESI†). Nevertheless, the results indicated that faulty B3LYP and HCTH energies can lead to highly inaccurate geometries, especially when conformational space is approximated as a series of discrete energy minima and saddle points, as we have done.
|
|
||||
|---|---|---|---|---|
| Method for geometry optimization | ΔEb (81B) | C1–C9 Bond distance | ||
| 81B | 81C | 82 | ||
| a Additional data are given in Table S14 in the ESI1. b Electron energies for cyclization of 80B to 81B. | ||||
| HF/6-31G* | −2.3 | 1.645 | 1.644 | 1.652 |
| MP2/6-31G* | −15.0 | 1.893 | 1.884 | 1.805 |
| B3LYP/6-31G* | −3.5 | 2.396 | 1.847 | 1.786 |
| MPW1K/6-31G* | −11.0 | 1.705 | 1.696 | 1.692 |
| mPW1PW91/6-31G* | −8.4 | 1.755 | 1.740 | 1.728 |
| HCTH/6-31G* | 0.8 | 1.850 | 1.815 | 1.747 |
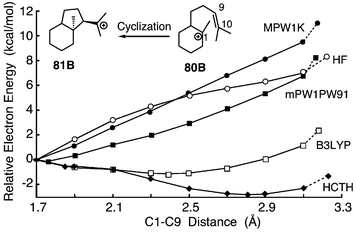 | ||
| Fig. 4 Relaxed PES scans for the cyclization of 80B to 81B from DFT methods with a 6-31G* basis (or HF/3-21G). Energies are from the geometry optimization method and are relative to the energy at 1.7 Å. Additional points included in the curves are energy minima and a geometry with C1–C10 frozen at 3.8 Å (point connected by a dashed line). Further details are given in Table S15 in the ESI†. | ||
The unusually wide range of bond lengths predicted for C1–C9 in 81C (1.64–1.88 Å) and 82 (1.65–1.81 Å) suggested that these structures are difficult to model accurately. Other examples are known in which B3LYP gives false minima29 or extremely long bond lengths.26 In a [3 + 2] cycloaddition of hydroxyallyl cation to cyclopentadiene, Cramer and Barrows26 attributed this problem to the excessive delocalization of charge46 by some DFT methods.
The occurrence of loose or grossly distorted B3LYP geometries in Table 7 and Fig. 4 raises doubts about the credibility of PES scans that rely on B3LYP optimizations. This concern is effectively addressed by calculating a PES scan of mPW1PW91 single-point energies for the B3LYP geometries. Data in Table S15 in the ESI† show that mPW1PW91 single-point energies correct the B3LYP distortions of the energy profile of Fig. 4, and Tables 6 and 8 indicate that relative mPW1PW91 single point energies are essentially constant for a variety of geometries. All PES scans herein were confirmed with mPW1PW91 single-point energies.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Method | ΔEb | ΔEc | Interatomic distance/Å | ||||
| Opt | mPW1PW91 | a | b | c | d | ||
| a Geometry optimizations were done by the method indicated with the 6/31G* basis set (or 6-31+G** for CCSD geometries). Electron energies are relative to 84 (for 83A, 83B), 86 (for 85B), or the preceding intermediate in Fig. 1 (for C25H43 models 18, 20, 23, and 25). Energies were referenced to 84 or 86, which, unlike the 1-propyl and 1-butyl cations, correspond to a unique energy minimum. Among the C25H43 models, only 20 contains an Hx atom (distance d). The longest cyclopropane bonds are the ones from which bond migration began. b Relative electron energies from the method used for geometry optimization. c Electron energies from mPW1PW91/6-311+G(2d,p) single-point energy calculations. d Hudson et al., Ref. 50b. e This bond was also long (2.363 Å) in the analogous B3LYP/6-31G* structure of the transition state in Fig. 2 of Ref. 10 but only 2.198 Å in the MPW1K/6-31G* geometry (Table S11 in the ESI1). | |||||||
| 83A | B3LYP | 10.4 | 8.6 | 1.798 | 1.790 | 1.388 | 2.085 |
| 83A | CCSD | 7.6 | 8.5 | 1.778 | 1.778 | 1.385 | 2.060 |
| 83B | B3LYP | 10.2 | 8.6 | 1.842 | 1.724 | 1.393 | 1.958 |
| 83B | HCTH | 10.2 | 8.5 | 1.775 | 1.659 | 1.402 | 1.860 |
| 83B | mPW1PW91 | 8.0 | 8.4 | 1.793 | 1.676 | 1.395 | 1.882 |
| 83B | MPW1K | 7.7 | 8.5 | 1.784 | 1.664 | 1.389 | 1.863 |
| 83B | MP2 | 4.1 | 8.4 | 1.795 | 1.708 | 1.395 | 1.912 |
| 83B | QCISDd | — | — | 1.823 | 1.709 | 1.393 | 1.933 |
| 83B | CCSD | 7.2 | 8.2 | 1.823 | 1.700 | 1.393 | 1.902 |
| 85B | B3LYP | 10.7 | 8.9 | 1.890 | 1.741 | 1.390 | 1.947 |
| 85B | mPW1PW91 | 9.0 | 8.9 | 1.822 | 1.656 | 1.400 | 1.806 |
| 85B | MPW1K | 8.5 | 8.7 | 1.806 | 1.635 | 1.396 | 1.768 |
| 85B | CCSD | 8.2 | 8.9 | 1.851 | 1.691 | 1.395 | 1.855 |
| 18 | B3LYP | 6.6 | 5.3 | 2.192 | 1.907 | 1.391 | — |
| 20 | B3LYP | 6.6 | 5.2 | 2.015 | 1.859 | 1.405 | 2.130 |
| 23 | B3LYP | 6.8 | 7.3 | 2.361e | 1.992 | 1.407 | — |
| 25 | B3LYP | 7.1 | 7.0 | 2.187 | 1.961 | 1.400 | — |
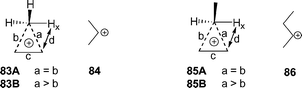
Our mechanistic conclusions about triterpene synthesis are based on energies and cation hyperconjugation rather than on precise bond lengths or angles. As judged by chemical intuition, pathological cases of B3LYP geometries were absent in our modelling of triterpene synthesis, and hyperconjugated C–C bond lengths were satisfactory (ca. 1.7 Å). Interestingly, the B3LYP/6-31G* geometry of tricycle 8, for which 81B is a model, had a reasonable C8–C13 bond length of 1.715 Å.
Transition states and activation energies
Deviant energy predictions by B3LYP were less pronounced in activation energies of (oxido)squalene cyclization. B3LYP, mPW1PW91, and MPW1K all gave activation energies of roughly 7 kcal mol−1 for the ring-expansion/annulation steps in hopene and lupeol formation (Table S1 in the ESI†). Both steps involve the formation of a cyclopropane/carbonium ion transition state from a tertiary carbenium ion bearing cyclopentyl, methyl, and alkyl groups (Scheme 2). The role of these non-classical ions in triterpene synthesis was recognized in 1955 by Swiss workers4 but, with a few notable exceptions,7b,d,e,n,47 has recently been overlooked.48 A bridged transition state in lanosterol synthesis was first described computationally by Hess as an alternative to an anti-Markovnikov intermediate.7d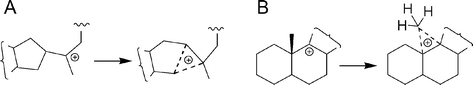 | ||
| Scheme 2 Cyclopropane/carbonium ions as transition states in ring enlargement (A) and 1,2-methyl shifts or cycloartenol formation (B). The long cyclopropane bonds to the pentavalent carbon are shown as dashed lines. The migrating carbon, which becomes pentavalent, can be CR3 (ring-C enlargement for lupeol and hopene), CHR2 (ring-D enlargement for hopene), CH2R (ring-D enlargement for lupeol), or CH3 (methyl migration). | ||
As shown in Scheme 2A, cyclopropane/carbonium ions can adopt various roles in triterpene synthesis. In addition to being transition states for D- and E-ring formation, protonated cyclopropane species are implicated in 1,2-methyl shifts leading to tetracyclic triterpenes like lanosterol and are presumably the immediate precursors of cycloartenol, phyllanthol, and some marine sterols with cyclopropane-containing side chains.49 Cyclopropane formation has also been observed in terpene-like biosynthesis with catalytic antibodies.47 However, aberrant enzymatic deprotonation of the pentavalent carbon to cyclopropanes has apparently47a not been detected for these short-lived species, even among mutants and substrate analogs.
We modelled some simple cyclopropane/carbonium ions in order to assess the accuracy of the B3LYP geometries and DFT energies calculated for D- and E-ring formation. Protonated cyclopropane50 and its methyl derivative50b,51,52 have been studied by ab initio and DFT methods. For both species, two nearly isoenergetic conformers are found, a symmetrical saddle point (83A, 85A) and an unsymmetrical energy minimum (83B, 85B).50,51 These corner-protonated cyclopropanes have energies 7–8 kcal mol−1 above the isomeric isopropyl and sec-butyl cations. Compared with QCISD/6-31G* and CCSD/6-31+G** structures of these simple carbonium ions, geometries were too loose with B3LYP/6-31G* and too tight with mPW1PW91 and MPW1K (Table 8). The geometrical differences among methods did not affect energies, the mPW1PW91 single point energies being almost constant, as in Table 7. B3LYP energies for cyclopropane/carbonium ion formation were too positive (except for 23 and 25), but this deviation was smaller than for net energies of annulation in triterpene synthesis.
We also studied protonated tetramethylcyclopropane 88 as a model of D-ring expansion in lupeol synthesis. We expected the carbonium ion to be a saddle point between cations 87 and 89 but could not find a stationary point corresponding to 88. A relaxed PES scan suggested a barrierless path linking 87 and 89 (Fig. 5).
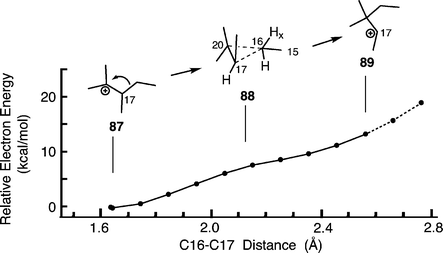 | ||
| Fig. 5 Relaxed PES scans (B3LYP/6-31G*//B3LYP/6-31G*) describing the conversion of tertiary to secondary carbenium ions via cyclopropane/carbonium ions (modelling ring-D enlargement in lupeol synthesis). The C16–C17 bond lengths above 2.6 Å represent unrealistic elongation of the C16–C20 bond (indicated by dashed lines). mPW1PW91 single-point energies give a similar curve (Table S16 in the ESI†). | ||
Vertical and horizontal cations in triterpene synthesis
As shown in Fig. 6, carbocation intermediates in triterpene synthesis can be oriented with the vacant 2p orbital aligned either in the plane of the ABCD ring system (horizontal cations) or perpendicular to the ABCD plane (vertical cations).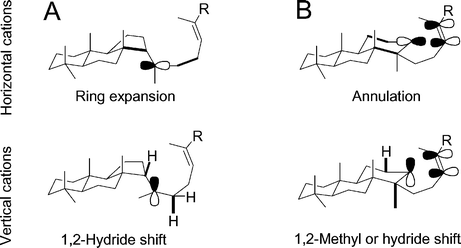 | ||
| Fig. 6 Illustration of vertical and horizontal cations for (A) 6–6–5 tricycle 2 and (B) its 6–6–6 counterpart. Thick bonds denote hyperconjugation with the cation. R = –CH2CH2CHCMe2. | ||
Horizontal and vertical cations have different reactivities, as noted by Nishizawa et al.7j,p,53 In vertical cations, orientation of the 2p orbital leads to hyperconjugation in axial substituents instead of ring bonds. The elongated substituent bonds readily undergo exothermic 1,2-shifts. Thus, the transition from horizontal to vertical cations transfers reactivity from bonds in the ABCD plane (promoting cyclization) to axial substituent bonds (promoting rearrangement). Horizontal cations prevail throughout the annulation portion of triterpene synthesis, and vertical cations dominate during the rearrangement phase. Vertical cations are typically only a few kcal mol−1 higher in energy than the corresponding horizontal cations.7j,p Enzymatic control and the dynamics of cyclization prevent a premature transition from horizontal to vertical cations.
Secondary cations in triterpene synthesis
Formation of rings C and D could proceed by anti-Markovnikov addition to generate secondary cations like 90 and 14. However, most evidence favors Markovnikov addition, which generates 5-membered rings such as 12 and 13.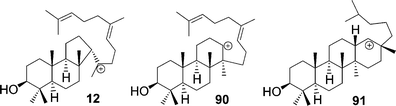
Evidence for the intermediacy of 6–6–5 tricycles (e.g.12) rather than 6–6–6 tricycles (e.g.90) is particularly strong. Many 6–6–5 tricycles occur in nature3 or arise from simple substrate analogs,2 whereas natural 6–6–6 triterpenes from (oxido)squalene are unknown.54,55 Molecular modelling shows a low-energy 6–6–5 pathway of C- and D-ring formation that circumvents the 6–6–6 tricycle.7d,10 A horizontal variant of 90 occurs transiently as a partially bridged structure56 in D-ring formation but is neither an intermediate nor transition state along the minimum-energy path (MEP).10 Our attempts at B3LYP/6-31G* geometry optimization of the vertical form of cation 90 led to D-ring annulation on the 13β face or to migration of the C14 methyl to C13. The absence of such aberrant products in enzymatic systems adds to the already strong evidence2 against 6–6–6 tricycles as cyclization intermediates.
Parallel reasoning would appear to exclude the intermediacy of the baccharenyl cation (14) in E-ring formation. Bridged transition states 20 and 25 avoid higher energy 6–6–6–6 paths, and many 6–6–6–5 tetracyclic triterpenes are known in nature. However, Hoshino et al.57 reported 6–6–6–6 triterpenes with hopene skeletons from an SHC mutant, and similar 6–6–6–6 natural products are known in the dammarane series.3 These structures are presumably derived from secondary cations 4 or 14. Cation 14 also appears to be a transition state in the biosynthesis of nepehinol and ursanes,3 and our preliminary calculations with C25H43 models (Fig. 1) indicate an activation energy of only about 8 kcal mol−1 from 19, i.e., only 3 kcal mol−1 higher than the bridged transition state 20.
Further evidence for 14 is the formation of small amounts of bacchar-12-en-3β-ol via the 6–6–6–6 cation 91 by incubation of 22,23-dihydro-2,3-oxidosqualene with β-amyrin synthase.11a The complex energy profile of this reaction was studied by PES scans58 starting with the C21H37 model 92 (Fig. 7). The reaction proceeds through a bridged transition state (92A), horizontal (93) and vertical (94) secondary cations, a 1,2-hydride shift to form 95, and elimination of H12 to terminate the cationic mechanism. Although D-ring expansion occurs without the benefit of cation stabilization by a terminal double bond, the activation energy is only modestly elevated, as in nepehinol synthesis. In contrast to the energy profile in Fig. 5, B3LYP/6-31G*//B3LYP/6-31G* calculations predicted shallow energy minima for the horizontal (93) and vertical (94) secondary cations, with transition states during ring expansion, horizontal–vertical cation conversion, and the 1,2-hydride shift. Only the first minimum survived in the mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G* energy profile. The energy profile for the 1,2-hydride shift suggests that a bridged C13–C17–H17 system derived from 9 may be a transition state (see Vrcek et al.7m) but not an intermediate, as was proposed for a similar cation rearrangement en route to cycloartenol.37
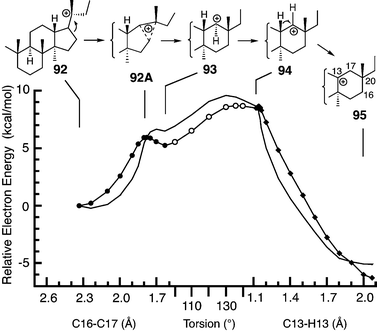 | ||
| Fig. 7 B3LYP/6-31G* PES scans modelling the conversion of the 17β-dammarenyl cation to the C13 cation en route to bacchar-12-en-3β-ol. Sequential PES scans were done for the C16–C20 bond (92 to 93, closed circles, the C14–C13–C17–H17 torsion angle (93 to 94, open circles), and the C13–H13 bond (94 to 95, diamonds). The line without points shows mPW1PW91/6-311+G(2d,p)//B3LYP/6-31G* energies. | ||
Another example of secondary cations in triterpene synthesis is concomitant E-ring expansion and methyl migration, as occurs in the synthesis of 6–6–6–6–6 pentacyclic triterpenes. Vrcek et al. modelled the conversion of the rearranged lupyl cation (model 96) to the taraxasteryl cation (model 98).7i Methyl migration and E-ring expansion occur sequentially, the transition state being a secondary carbenium ion (97) rather than a bridged carbonium ion.7i Energetically, each half of the reaction path resembles the interconversion between 87 and 89. Relaxed PES scans (Fig. 8) suggested that the cyclopropane/carbonium ion species 96A and 97A are neither transition states nor intermediates. The ring expansion mandated the unusual horizontal–vertical–horizontal–vertical series of interconversions.
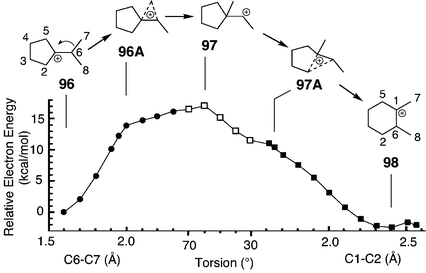 | ||
| Fig. 8 Relaxed PES scans (B3LYP/6-31G*//B3LYP/6-31G*) modelling the interconversion of tertiary and secondary carbenium ions via cyclopropane/carbonium ions. First, the C6–C7 bond was frozen at values from 1.6–2.3 Å (closed circles). A similar PES scan was obtained by freezing the C1–C2 bond length (closed squares). These two PES scans were linked by a third PES scan of the C7–C1–C6–C8 torsion angle in the vicinity of the transition state (open squares). mPW1PW91 single-point energies give a similar curve (Table S18 in the ESI†). | ||
Our modelling results indicate that secondary cations do have a modest role in triterpene synthesis. Dammarenyl cations probably have an extended side-chain conformation,8,40,59 and secondary cations may be significantly populated, especially when D-ring expansion is faster than side-chain folding. The 6–6–6–6 secondary cations typically react by undergoing deprotonation or E-ring annulation on the α or β face. The absence of minor 6–6–6 products can be rationalized but is still puzzling. As intimated by Jenson and Jorgensen,7a 6–6–6 tricycle derivatives may yet be detected as minor byproducts in cyclase mutants.
Conclusions
We established reliable procedures for calculating cyclization energies by DFT methods. These procedures were used to construct energy profiles for the cationic cyclization of oxidosqualene to lupeol, lanosterol, and hopen-3β-ol. The profiles indicated that rings A and B form more exergonically than rings C and D owing to a better arrangement of cation-stabilizing methyls in the early annulations. Other features of the energy profiles were interpreted similarly in terms of steric interactions, ring strain, and proton affinities.These gas-phase energy profiles for the bare substrate models provide a fundamental understanding of cyclization prior to adjustments for solvation or enzymatic effects. Enzymatic stabilization is important in (oxido)squalene protonation and A-ring formation,40 but most further annulations and rearrangements are amply exothermic and encounter only modest activation energy barriers. The common belief that the cationic intermediates require enzymatic stabilization is unfounded.
The cationic 2p orbital of triterpene intermediates can be horizontal (in the plane of the ABCD ring system) or vertical (perpendicular to this plane). Horizontal cations lead to annulation and ring expansion by fostering reactivity of bonds in the ABCD plane, whereas vertical cations promote 1,2-methyl and hydride shifts through hyperconjugative elongation of axial substituents. The transition from ring building to rearrangement can occur readily because vertical cations are almost as stable as their horizontal counterparts. In enzymatic reactions, substrate folding and the dynamics of annulation usually maintain the ring-building phase until completion of tetracyclization.
Mechanistically, the tri-, tetra-, and pentacyclic intermediates are connected via bridged cyclopropane/carbonium-ion transition states, which provide a low-energy pathway for ring-expansion/annulation. These bridged transition states maintain the horizontal cation orientation. The alternative anti-Markovnikov intermediates are somewhat higher in energy and readily become vertical cations, which engender rearrangements. Vertical 6–6–6–6 (but not 6–6–6) secondary cations occur in some biosynthetic pathways. Bridged cations are also involved in post-cyclization rearrangements leading to lanosterol, euphol, and β-amyrin; each 1,2-methyl or hydride shift appears to be a discrete step, followed by a pause for orbital realignment to facilitate hyperconjugation leading to the next 1,2-shift. The stepwise nature of the rearrangements undermines the presumption that the 1,2-shifts must proceed in an anti-periplanar manner and is consistent with the syn 1,2 hydride shift from C13 to C17 in lanosterol biosynthesis.40
Our molecular modelling results revealed systematic errors in energy calculations from B3LYP and other quantum mechanical methods. Substantial B3LYP errors for cation–olefin condensations were compounded in the five annulations of hopene synthesis and led to the absurd prediction that squalene cyclization to hopene is endothermic. This type of B3LYP error appears to affect many transformations involving the conversion of CC or CO bonds to two single bonds. This problem is manifest in the molecular modelling literature but has seldom been recognized. For medium-sized basis sets, the mPW1PW91 hybrid functional gives much improved cyclization energies.
The B3LYP functional is widely used for geometry optimizations, which are based on the same energy calculations that suffer from systematic errors. B3LYP geometries were slightly too loose as judged by comparisons with X-ray and CCSD structures, but this minor shortcoming was unrelated to the energy errors. Except in rare pathological cases, B3LYP geometries were acceptable for use with mPW1PW91 single-point energy calculations, which were remarkably insensitive to geometry differences among correlated methods.
The noted deficiencies in quantum mechanical methods would not have been detected if our calculations had been limited to the usual small models and modest basis sets. This work, which was done on inexpensive personal computers, illustrates that the full C30H50O triterpene structures can be modelled easily at a respectable level of theory.
Experimental
Molecular modeling
Quantum mechanical calculations were done mainly with Gaussian 98 (Linux version A.9 and Windows version A.11).60 Gaussian 03 (versions B.02, B.05 and C.02)61 generally gave geometries and energies identical to those from Gaussian 98 and was used for: G3 energies; HCTH (i.e., HCTH/407), PBE1PBE, O3LYP, BV5LYP, and TPSS (i.e., TPSSTPSS) calculations; methyl epoxides of Table 2; cyclopropane/carbonium ions; reaction R1 of Table 3; and PES scans of Figs. 5, 7, and 8. Comparison of MPW1K and mPW1PW91 energies between Gaussian 98 and 03 was avoided, and OmPW1PW91 was used when necessary, although deviations were usually <0.2 kcal mol−1.62 Geometry optimizations and energy calculations were done using ab initio, semi-empirical, and DFT,20,30b,32,63 methods. All models were considered as closed-shell systems (restricted calculations) with the frozen-core approximation. Zero-point energies and thermal energies at the B3LYP/6-31G* level were scaled by 0.96; no attempt was made to scale entropy data or to correct for the presence of low-frequency vibrations. Only the thermal energy contribution to enthalpies and free energies was scaled. All energy calculations were done with SCF = tight. The Gaussian implementation of B3LYP uses equation III of Vosko, Wilk, and Nusair64 (VWN) for local correlation, whereas VWN equation V or I is used in some other software packages. B3LYP cyclization energies differed little between equations III and V but were slightly more accurate with equation III (Tables S2a and S7a); geometries were nearly identical (Table S15 in the ESI†).Molecular mechanics calculations were done in PCMODEL version 8.5 or 9 (Serena Software, Inc., Bloomington, IN) using the MM3 and MMX force fields. In some cases, missing force-field parameters were taken from closely related structural moieties. Heats of formation were calculated from MM3 or MMX geometries. When parameterization permitted, MM3 heats of formation were also calculated with the MM3(94) program, which was purchased from the Quantum Chemical Program Exchange (Bloomington, IN). Computational strategies are described in the ESI†.
The molecular modelling techniques used herein have significant limitations despite the high level of theory relative to past calculations on triterpene synthesis. We have simplified the continuum of conformational space by studying only discrete energy minima and saddle points, whereas high-energy substrates are not confined to follow the MEP. Transition state structures were optimized as saddle points for electron energy, rather than for enthalpy or free energy. Our gas-phase energy calculations neglected enzyme–substrate interactions. Our models of open-chain and partially cyclized substrates are unlikely to correspond exactly to the conformations in the active site cavity, but energy differences among such conformers are generally minor. Our modelling relied heavily on DFT methods despite their serious shortcomings. However, except for the neglect of enzyme-substrate interactions40 and the lack of molecular dynamics studies, most of these deficiencies are either relatively minor or unavoidable.
Acknowledgements
We thank Paul S. Engel (Rice University) for advice on estimating and measuring heats of formation. Roger V. Moye (Rice University) configured the Linux computers for efficient use and set up a batch queue for running Gaussian jobs on multiple computers using Grid Computing. Dr Douglas J. Fox and Dr James Hess (Gaussian, Inc.) provided essential guidance on quantum mechanical calculations. Kevin E. Gilbert (Serena Software) gave many valuable suggestions on molecular modeling. We thank the National Science Foundation (MCB-0209769) and the Robert A. Welch Foundation (C-1323) for funding.References
- (a) W. R. Nes and M. L. McKean, Biochemistry of Steroids and other Isopentenoids, University Park Press, Baltimore, 1977 Search PubMed; (b) P. Harrewijn, A. M. van Oosten and P. G. M. Piron, Natural Terpenoids as Messengers: a Multidisciplinary Study of their Production, Biological Functions and Practical Applications, Kluwer Academic Publishers, Dordrecht, Netherlands, 2001 Search PubMed.
- K. U. Wendt, G. E. Schulz, E. J. Corey and D. R. Liu, Angew. Chem., Int. Ed., 2000, 39, 2812–2833 CrossRef CAS.
- R. Xu, G. C. Fazio and S. P. T. Matsuda, Phytochemistry, 2004, 65, 261–291 CrossRef CAS.
- A. Eschenmoser, L. Ruzicka, O. Jeger and D. Arigoni, Helv. Chim. Acta, 1955, 38, 1890–1904 CrossRef CAS.
- G. Stork and A. W. Burgstahler, J. Am. Chem. Soc., 1955, 77, 5068–5077 CrossRef CAS.
- For leading references, see: (a) M. J. R. Segura, B. E. Jackson and S. P. T. Matsuda, Nat. Prod. Rep., 2003, 20, 304–317 RSC; (b) T. Hoshino and T. Sato, Chem. Commun., 2002, 291–301 RSC; (c) I. Abe and G. D. Prestwich, in Isoprenoids Including Carotenoids and Steroids, ed. D. E. Cane, (vol. 2 in Comprehensive Natural Products Chemistry, ed. D. Barton, K. Nakanishi and O. Meth-Cohn), Pergamon/Elsevier, Amsterdam, 1999, pp. 267–298 Search PubMed.
- (a) C. Jenson and W. L. Jorgensen, J. Am. Chem. Soc., 1997, 119, 10846–10854 CrossRef CAS; (b) R. Rajamani and J. Gao, J. Am. Chem. Soc., 2003, 125, 12768–12781 CrossRef CAS; (c) D. Gao, Y.-K. Pan, K. Byun and J. Gao, J. Am. Chem. Soc., 1998, 120, 4045–4046 CrossRef CAS; (d) B. A. Hess, Jr., J. Am. Chem. Soc., 2002, 124, 10286–10287 CrossRef; (e) B. A. Hess, Jr., Org. Lett., 2003, 5, 165–167 CrossRef; (f) B. A. Hess, Jr., Collect. Czech. Chem. Commun., 2003, 68, 202–210 CrossRef; (g) B. A. Hess, Jr., Collect. Czech. Chem. Commun., 2004, 69, 261–266 CrossRef; (h) B. A. Hess, Jr. and L. Smentek, Org. Lett., 2004, 6, 1717–1720 CrossRef CAS; (i) V. Vrcek, M. Saunders and O. Kronja, J. Org. Chem., 2003, 68, 1859–1866 CrossRef CAS (Erratum 4126); (j) M. Nishizawa, A. Yadav, H. Imagawa and T. Sugihara, Tetrahedron Lett., 2003, 44, 3867–3870 CrossRef CAS; (k) T. Schulz-Gasch and M. Stahl, J. Comput. Chem., 2003, 24, 741–753 CrossRef CAS; (l) B. A. Hess, Jr., Eur. J. Org. Chem., 2004, 2239–2242 (Erratum 2747); (m) V. Vrcek, I. V. Vrcek and O. Kronja, Croat. Chem. Acta, 2001, 74, 801–813 CAS; (n) C. C. J. Fouillet and J. Mareda, J. Mol. Struct. (THEOCHEM), 2002, 589–590, 7–26 CrossRef CAS; (o) B. A. Hess, Jr. and L. Smentek, Mol. Phys., 2004, 102, 1201–1206 CrossRef; (p) M. Nishizawa, A. Yadav, Y. Iwamoto and H. Imagawa, Tetrahedron, 2004, 60, 9223–9234 CrossRef CAS.
- D. J. Reinert, G. Balliano and G. E. Schulz, Chem. Biol., 2004, 11, 121–126 CAS.
- (a) R. Thoma, T. Schulz-Gasch, B. D'Arcy, J. Benz, J. Aebi, H. Dehmlow, M. Hennig, M. Stihle and A. Ruf, Nature, 2004, 432, 118–122 CrossRef CAS; (b) K. U. Wendt, A. Lenhart and G. E. Schulz, J. Mol. Biol., 1999, 286, 175–187 CrossRef CAS; (c) K. U. Wendt, K. Poralla and G. E. Schulz, Science, 1997, 277, 1811–1815 CrossRef CAS; (d) A. Lenhart, W. A. Weihofen, A. E. W. Pleschke and G. E. Schulz, Chem. Biol., 2002, 9, 639–645 CrossRef CAS; (e) A. Ruf, F. Müller, B. D'Arcy, M. Stihle, E. Kusznir, C. Handschin, O. H. Morand and R. Thoma, Biochem. Biophys. Res. Commun., 2004, 315, 247–254 CrossRef CAS; (f) X. Zhu, A. Heine, F. Monnat, K. N. Houk, K. D. Janda and I. A. Wilson, J. Mol. Biol., 2003, 329, 69–83 CrossRef CAS; (g) K. U. Wendt, Angew. Chem., Int. Ed., 2005, 44, 3966–3971 CrossRef CAS.
- Q. Xiong, F. Rocco, W. K. Wilson, R. Xu, M. Ceruti and S. P. T. Matsuda, J. Org. Chem., 2005, 70, 5362–5375 CrossRef CAS.
- Previous calculations of D-ring formation were done with C21H37 models lacking the Δ24 double bond.10 Comparison of energies for C21H37 and folded C25H43 models suggests that the Δ24 double bond provides only 3–4 kcal mol−1 of cation stabilization (see Table S1 in the ESI†). This modest stabilization is lacking in tetracyclic cations with an extended side chain, e.g., the deoxydammarenyl cation (3) of the SHC active site8 and an intermediate in the formation of a 6–6–6–6 tetracycle from 22,23-dihydro-2,3-oxidosqualene: (a) I. Abe, Y. Sakano, H. Tanaka, W. Lou, H. Noguchi, M. Shibuya and Y. Ebizuka, J. Am. Chem. Soc., 2004, 126, 3426–3427 CrossRef CAS.
- (a) Experimental Thermochemistry; Measurement of Heats of Reaction, ed. F. D. Rossini, Interscience Publishers, New York, 1956, vol. 1 Search PubMed; (b) Combustion Calorimetry, ed. S. Sunner and M. Månsson, Pergamon Press, Oxford, 1979, vol. 1 Search PubMed.
- Details are given in the ESI†.
- (a) S. W. Benson, Thermochemical Kinetics: Methods for the Estimation of Thermochemical Data and Rate Parameters, Wiley, New York, 2nd edn., 1976 Search PubMed; (b) N. Cohen and S. W. Benson, Chem. Rev., 1993, 93, 2419–2438 CrossRef CAS; (c) J. Gasteiger, P. Jacob and U. Strauss, Tetrahedron, 1979, 35, 139–146 CrossRef CAS; (d) NIST Chemistry WebBook: http://webbook.nist.gov/; (e) L. A. Curtiss, P. C. Redfern and D. J. Frurip, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 2000, vol. 15, pp. 147–211 Search PubMed; (f) DFT-based methods: P. C. Redfern, P. Zapol, L. A. Curtiss and K. Raghavachari, J. Phys. Chem. A, 2000, 104, 5850–5854 Search PubMed; (g) P. Winget and T. Clark, J. Comput. Chem., 2004, 25, 725–733 CrossRef CAS.
- U. Burkert and N. L. Allinger, Molecular Mechanics, American Chemical Society, Washington, DC, 1982, pp. 169–194 Search PubMed.
- N. L. Allinger, K. Sakakibara and J. Labanowski, J. Phys. Chem., 1995, 99, 9603–9610 CrossRef CAS.
- (a) N. L. Allinger, H. Y. Young and J.-H. Lii, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS; (b) J.-H. Lii and N. L. Allinger, J. Am. Chem. Soc., 1989, 111, 8566–8575 CrossRef CAS; (c) J.-H. Lii and N. L. Allinger, J. Am. Chem. Soc., 1989, 111, 8576–8582 CrossRef CAS.
- J. J. Gajewski, K. E. Gilbert and J. McKelvey, Adv. Mol. Model., 1990, 2, 65–92 Search PubMed.
- Our studies of monocyclic, bicyclic, and acyclic C10 isomers indicated a similar accuracy range for differences in MM3 heats of formation. Errors for force-field cyclization energies, unlike those of B3LYP, appeared to be non-systematic and thus would not accumulate in comparing pentacyclic with acyclic species. However, other MM3 errors might accumulate with increasing molecular size, as is observed for B3LYP heats of formation.14f.
- B3LYP: (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; mPW1PW91: (b) C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS; MPW1K: (c) B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS; (d) B. J. Lynch and D. G. Truhlar, J. Phys. Chem. A, 2001, 105, 2936–2941 CrossRef CAS; O3LYP: (e) N. C. Handy and A. J. Cohen, Mol. Phys., 2001, 99, 403–412 CrossRef CAS; (f) V. A. Guner, K. S. Khuong, K. N. Houk, A. Chuma and P. Pulay, J. Phys. Chem. A, 2004, 108, 2959–2965 CrossRef CAS; B1LYP: (g) A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS; (h) C. Adamo and V. Barone, Chem. Phys. Lett., 1997, 274, 242–250 CrossRef CAS; HCTH: (i) F. A. Hamprecht, A. J. Cohen, D. J. Tozer and N. C. Handy, J. Chem. Phys., 1998, 109, 6264–6271 CrossRef CAS; PBE1PBE: (j) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS; (Erratum 78, 1396); TPSS: (k) V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS; (l) J. P. Perdew, J. Tao, V. N. Staroverov and G. E. Scuseria, J. Chem. Phys., 2004, 120, 6898–6910 CrossRef CAS; (m) J. P. Perdew, A. Ruzsinszky, J. Tao, V. N. Staroverov, G. E. Scuseria and G. I. Csonka, J. Chem. Phys., 2005, 123 Search PubMed; 062201/1-9; VSXC: (n) T. Van Voorhis and G. E. Scuseria, J. Chem. Phys., 1998, 109, 400–410 CrossRef CAS.
- Although TPSSh with 25% exact HF exchange gave the best agreement, this hybrid functional was optimized on a more comprehensive training set for 10% HF exchange.20k,l B3P86 also gave excellent agreement but performed poorly on G2 training sets: L. A. Curtiss, P. C. Redfern, K. Raghavachari and J. A. Pople, J. Chem. Phys., 1998, 109, 42–55 Search PubMed.
- The C10H16 isomers were adamantane (35), α-pinene (36), β-pinene (37), limonene (38), protoadamantane (39), 3-carene (40), camphene (41), ocimene (42), and myrcene (43). The C10H16O isomers were geraniol (44), cineole (45), isoborneol (46), borneol (47), myrtanol (48), nerol (49), 1-decalol (50), and isopinocampheol (51).
- The classical MM3-94 program and the MMX and MM3 force fields in version 8 of PCMODEL lacked certain bond increments and torsion parameters needed to estimate the contribution of the C–O epoxide bond in oxidosqualene to the heat of formation, and the built-in MM3-94 parameter estimator did not give useful values. Because of limited combustion data available for alkylated ethylene oxides and their isomers, we used G3B3 enthalpy differences as benchmark values for relative heats of formation. The resulting data provided a basis for revising the epoxide parameters of MM3 in PCMODEL.
- The 19 compounds comprised the following sets of isomers: C2H4O, acetaldehyde (52) and ethylene oxide (53); C3H6O, acetone (54), propylene oxide (55), propanal (56), and oxetane (57); C4H8O, 2-butanone (58), cyclobutanol (59), trans-2,3-dimethyloxirane (60), cis-2,3-dimethyloxirane (61), and tetrahydrofuran (62); C5H10O, methyl isopropyl ketone (63), trimethyloxirane (64), cyclopentanol (65), 2H-tetrahydropyran (66); C6H12O, methyl t-butyl ketone (67), tetramethyloxirane (68), cyclohexanol (69), and 1-methylcyclopentanol (70).
- In unpublished work, Hess calculated the overall energy changes for cyclization of oxidosqualene. He compared the B3LYP predictions with relevant experimental data and concluded that B3LYP markedly underestimates the energy of cyclization. B. A. Hess (Vanderbilt University), personal communication.
- C. J. Cramer and S. E. Barrows, J. Org. Chem., 1998, 63, 5523–5532 CrossRef CAS.
- (a) P. R. Schreiner and M. Prall, J. Am. Chem. Soc., 1999, 121, 8615–8627 CrossRef CAS; (b) G. O. Jones, V. A. Guner and K. N. Houk, J. Phys. Chem. A, 2005 DOI:10.1021/jp052055z; (c) V. Guner, K. S. Khuong, A. G. Leach, P. S. Lee, M. D. Bartberger and K. N. Houk, J. Phys. Chem. A, 2003, 107, 11445–11459 CrossRef CAS; (d) T. C. Dinadayalane, R. Vijaya, A. Smitha and G. N. Sastry, J. Phys. Chem. A, 2002, 106, 1627–1633 CrossRef CAS.
- (a) J. E. Walker, P. A. Adamson and S. R. Davis, J. Mol. Struct. (THEOCHEM), 1999, 487, 145–150 CrossRef CAS; (b) F. Jensen, Introduction to Computational Chemistry, Wiley, New York, 1999, pp. 289–295 Search PubMed.
- (a) K. B. Wiberg, J. Org. Chem., 2003, 68, 9322–9329 CrossRef CAS; (b) A. King, T. D. Crawford, J. F. Stanton and H. F. Schaefer, III, J. Am. Chem. Soc., 1999, 121, 10788–10793 CrossRef CAS.
- (a) J. R. Reimers, Z.-L. Cai, A. Bilic and N. S. Hush, Ann. N. Y. Acad. Sci., 2003, 1006, 235–251 CrossRef CAS; (b) W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, Germany, 2nd edn., 2000, pp. 117–259 Search PubMed; (c) C. S. Wannere, K. W. Sattelmeyer, H. F. Schaefer, III and P. von R. Schleyer, Angew. Chem., Int. Ed., 2004, 43, 4200–4206 CrossRef CAS.
- (a) D. A. Plattner and K. N. Houk, J. Am. Chem. Soc., 1995, 117, 4405–4406 CrossRef CAS; (b) H. L. Woodcock, H. F. Schaefer, III and P. R. Schreiner, J. Phys. Chem. A, 2002, 106, 11923–11931 CrossRef; (c) A. E. Pomerantz, J. H. Han and C. B. Musgrave, J. Phys. Chem. A, 2004, 108, 4030–4035 CrossRef CAS; (d) The popular belief that this problem stems from self-interaction errors deserves reconsideration: O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2004, 121, 8187–8193 Search PubMed.
- KMLYP: (a) J. K. Kang and C. B. Musgrave, J. Chem. Phys., 2005, 115, 11040–11051; X3LYP (b) X. Xu, Q. Zhang, R. P. Muller and W. A. Goddard, J. Chem. Phys., 2005, 122 Search PubMed . 014105/1-14; (c) PKZB: J. P. Perdew, S. Kurth, A. Zupan and P. Blaha, Phys. Rev. Lett., 1999, 82, 2544–2547 Search PubMed (Erratum 5179).
- Recent reviews of DFT strategies: (a) Ref. 20m; (b) A. Görling, J. Chem. Phys., 2005, 123, 062203 CrossRef; (c) R. J. Bartlett, V. F. Lotrich and I. V. Schweigert, J. Chem. Phys., 2005, 123, 062205 CrossRef. For a non-DFT approach, see: (d) P. M. W. Gill, D. L. Crittenden, D. P. O'Neill and N. A. Besley, Phys. Chem. Chem. Phys., 2005 10.1039/b511472a , published online.
- As detailed in the ESI† (Table S7b), energy differences among more stable substrate conformers are generally 1–3 kcal mol−1 and thus have little influence on the overall cyclization energy.
- For enzymatic reactions, we assume that no net chemical energy is transferred between substrate and the enzyme or cytosol by the overall protonation/deprotonation process. In hopene synthesis, where the sites of protonation and deprotonation are distant, the proton is ultimately returned to aspartate via cytosolic water.8 In lanosterol synthesis, the linkage may be through either cytosolic water or a series of hydrogen-bonded residues/waters.9a See: S. Lodeiro, M. J. R. Segura, M. Stahl, T. Schulz-Gasch and S. P. T. Matsuda, ChemBioChem, 2004, 5, 1581–1585 Search PubMed.
- Energies are based on the most stable forms of cycloartenol.37 Although the flat C9 lanosteryl cation is more stable than the arced C8 cation, deprotonation occurs from the C8 cation in lanosterol synthesis.9a Our analysis excludes the cyclopropane/carbonium ion en route to cycloartenol. Further discussion is given in comments to Table S8 in the ESI†.
- W. D. Nes, K. Koike, Z. Jia, Y. Sakamoto, T. Satou, T. Nikaido and J. F. Griffin, J. Am. Chem. Soc., 1998, 120, 5970–5980 CrossRef.
- Despite experimental2 and computational7a arguments favoring barrierless formation of rings A and B from protonated oxidosqualene, energy minima have been reported (see comments to Table S9 in the ESI†)7b,o.
- C. Pale-Grosdemange, C. Feil, M. Rohmer and K. Poralla, Angew. Chem., Int. Ed., 1998, 37, 2237–2240 CrossRef.
- S. P. T. Matsuda, W. K. Wilson and Q. Xiong, unpublished work on enzymatic stabilization in triterpene synthesis.
- (a) P. Buzek, P. von R. Schleyer and S. Sieber, Chemie Unserer Zeit, 1992, 26, 116–128 CrossRef; (b) T. Laube, Acc. Chem. Res., 1995, 28, 399–405 CrossRef CAS; (c) L. Radom, J. A. Pople and P. von R. Schleyer, J. Am. Chem. Soc., 1972, 94, 5935–5945 CrossRef CAS.
- A crystal structure of an apparent mixture of lupeol acetate and a related triterpene has been reported: G. Argay, A. Kalman, A. Kapor, B. Ribar, S. Petrovic and M. Gorunovic, J. Mol. Struct., 1997, 435, 169–179 Search PubMed.
- For a discussion of r values from different experimental and theoretical methods, see K. Kuchitsu in Accurate Molecular Structures. Their Determination and Importance, ed. A. Domenicano and I. Hargittai, Oxford University Press, Oxford, UK, 1992, pp. 14-46 Search PubMed.
- (a) C. J. Cramer, Essentials of Computational Chemistry, Wiley, New York, 2002, pp. 265–268 Search PubMed; (b) Ref. 28b, pp. 264–267; (c) Ref. 30b, pp. 119–126. Higher geometrical accuracy, which was not needed for our purposes, would require attention to effects of basis set size and the amount of exact HF exchange.
- A similar choice of 3.8 Å was made for reaction R1 based on data in Fig. 2A. Although reaction R1 involves 6-membered ring formation, Fig. 2B indicates that the initial valence angles and interatomic distances are poised for 5-membered ring formation and begin favoring 6-membered ring formation only about halfway into the reaction.
- (a) D. J. Lacks and R. G. Gordon, Phys. Rev. A, 1993, 47, 4681–4690 CrossRef CAS; (b) K. E. Laidig, Chem. Phys. Lett., 1994, 225, 285–292 CrossRef CAS; (c) Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Tanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS.
- (a) T. Li, K. D. Janda and R. A. Lerner, Nature, 1996, 379, 326–327 CrossRef CAS; (b) J. Hasserodt, K. M. Janda and R. A. Lerner, J. Am. Chem. Soc., 2000, 122, 40–45 CrossRef CAS; (c) X. Zhu, A. Heine, F. Monnat, K. N. Houk, K. D. Janda and I. A. Wilson, J. Mol. Biol., 2003, 329, 69–83 CrossRef CAS; (d) J. K. Lee and K. N. Houk, Angew. Chem., Int. Ed. Engl., 1997, 36, 1003–1005 CAS.
- Bridged structures are sometimes shown in mechanisms, e.g., (a) I. Abe and M. Rohmer, J. Chem. Soc., Perkin Trans. 1, 1994, 783–791 RSC; (b) D. Stach, Y. F. Zheng, A. L. Perez, A. C. Oehlschlager, I. Abe, G. D. Prestwich and P. G. Hartman, J. Med. Chem., 1997, 40, 201–209 CrossRef CAS.
- (a) J.-L. Giner, C. J. Silva and C. Djerassi, J. Am. Chem. Soc., 1990, 112, 9626–9627 CrossRef CAS; (b) J.-L. Giner, P. Buzek and P. von R. Schleyer, J. Am. Chem. Soc., 1995, 117, 12871–12872 CrossRef CAS; (c) L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625–1647 CrossRef CAS.
- (a) W. Koch, B. Liu and P. von R. Schleyer, J. Am. Chem. Soc., 1989, 111, 3479–3480 CrossRef CAS; (b) C. E. Hudson, S. Eapen and D. J. McAdoo, Int. J. Mass Spectrom. Ion Processes, 2003, 228, 955–967 Search PubMed.
- (a) S. Sieber, P. Buzek, P. von R. Schleyer, W. Koch and J. W. d. M. Carneiro, J. Am. Chem. Soc., 1993, 115, 259–270 CrossRef CAS; (b) P. Campomanes, D. Suarez and T. L. Sordo, J. Phys. Chem. A, 1999, 103, 5996–6002 CrossRef CAS.
- Carbonium ions 83B and 84B can be considered as complexes of methyl or ethyl cation with ethylene or as bridged forms of 1-propyl or 1-butyl cations51a.
- Nishizawa et al.7j,p described the concept of vertical and horizontal cations in different terms and without generalization.
- 6–6–6 Cyclization requires specially designed substrates employing either conformational constraint through a fused-ring substituent55a or a cation-stabilizing substituent, such as methyl,7j,p,55b–d fluorine,55e or tert-butyldimethylsilyloxy.55f Recent arguments for the intermediacy of 6–6–6 tricycles are discussed in the ESI†: (a) M. Seemann, G. Zhai, J.-W. de Kraker, C. M. Paschall, D. W. Christianson and D. E. Cane, J. Am. Chem. Soc., 2002, 124, 7681–7689 CrossRef CAS; (b) Ref. 7j; (c) Ref. 7p.
- (a) K. Berckmoes, P. J. De Clercq and D. Viterbo, J. Am. Chem. Soc., 1995, 117, 5857–5858 CrossRef CAS; (b) S. Nakano, S. Ohashi and T. Hoshino, Org. Biomol. Chem., 2004, 2, 2012–2022 RSC; (c) Q. Xiong, X. Zhu, W. K. Wilson, A. Ganesan and S. P. T. Matsuda, J. Am. Chem. Soc., 2003, 125, 9002–9003 CrossRef CAS; (d) T. Hoshino, Y. Kumai, I. Kudo, S.-I. Nakano and S. Ohashi, Org. Biomol. Chem., 2004, 2, 2650–2657 RSC; (e) W. S. Johnson, W. R. Bartlett, B. A. Czeskis, A. Gautier, C. H. Lee, R. Lemoine, E. J. Leopold, G. R. Luedtke and K. J. Bancroft, J. Org. Chem., 1999, 64, 9587–9595 CrossRef CAS; (f) A. X. Huang, Z. Xiong and E. J. Corey, J. Am. Chem. Soc., 1999, 121, 9999–10003 CrossRef CAS.
- Secondary cations have less effective hyperconjugation than tertiary cations and are commonly stabilized by bridging:41a,50,51 (a) V. Vrcek, M. Saunders and O. Kronja, J. Am. Chem. Soc., 2004, 126, 13703–13707 CrossRef CAS; (b) I. Alkorta, J. L. M. Abboud, E. Quintanilla and J. Z. Davalos, J. Phys. Org. Chem., 2003, 16, 546–554 CrossRef CAS; see also (c) D. Farcasiu and D. Hancu, J. Am. Chem. Soc., 1999, 121, 7173–7181 CrossRef CAS.
- T. Hoshino, T. Abe and M. Kouda, Chem. Commun., 2000, 441–442 RSC.
- The PES scans do not represent the MEP but did reveal several saddle points. Reaction paths were studied as PES scans since path calculations for our triterpene models usually failed to converge.
- H. Shan, M. J. R. Segura, W. K. Wilson, S. Lodeiro and S. P. T. Matsuda, J. Am. Chem. Soc., 2005, 127, 18008–18009 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98 (Revisions A.9 and A.11), Gaussian, Inc., Pittsburgh PA, 1998 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 (Revisions B.02, B.05, and C.02), Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- mPWPW91 of Gaussian 03 differs slightly from mPWPW91 of Gaussian 98 owing to an error in the original mPW exchange functional. Because the original functional was optimized with this error, the corrected functional is not fully optimized. Deviations from these differences are usually ≪1 kcal mol−1. See http://www.gaussian.com/g_tech/g03_rel.htm#updatedman or http://comp.chem.umn.edu/info/mpw1k.htm.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Clarendon Press, Oxford, UK, 1989 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: detailed results of molecular modelling, Tables S1-S18, and atomic coordinates. See DOI: 10.1039/b513599k |
| This journal is © The Royal Society of Chemistry 2006 |