Maleimide spacers as versatile linkers in the synthesis of bioconjugates of anthracyclines
Christine Le
Sann
*
School of Chemistry and Chemical Engineering, David Keir Building, Queen's University Belfast, Stranmillis Road, Belfast, BT9 5AG, Northern Ireland. E-mail: C.LeSann@qub.ac.uk; Fax: +44 (0)28 9097 4890; Tel: +44 (0)28 9097 4446
First published on 12th April 2006
Abstract
Covering: March 1990 to January 2006
This review covers the structural design and versatile use of maleimide spacers in the preparation of bioconjugates of the anthracyclines doxorubicin and daunorubicin. There are 66 references.
 Christine Le Sann | Christine Le Sann was born in 1974 in Brittany (France). She arrived at the University of Hertfordshire in the UK in 1994 and after three years study, which included a sandwich placement at SmithKline Beecham Pharmaceuticals (Harlow), she received her BSc Chemistry with Medicinal Chemistry. She then studied for a PhD at the University of Bristol under the supervision of Professor T. J. Simpson and in the field of polyketide intermediates synthesis. She subsequently took a postdoctoral fellowship position at the University of Canterbury (Christchurch, New Zealand) where she worked for two years with Professor Andrew Abell on the development of inhibitors of type II dehydroquinase. She is currently working with Professor John Mann on the synthesis of anticancer agents, at Queen's University Belfast and is an Associate Investigator in the new Centre for Cancer Research and Cell Biology (Belfast, Northern Ireland). |
1 Introduction
The anthracyclines, like doxorubicin (adriamycin) 1 and daunorubicin 2, constitute a very important class of antineoplastic agents used for many years in the treatment of leukaemia, breast carcinoma and other solid tumours.1 On the other hand, their clinical application has been limited by their toxic, dose-related side effects such as myelosuppression, gastrointestinal disorders, stomatitis, cumulative cardiotoxicity and extravasation.2,3 These side-effects may be linked to the pharmacokinetic properties of the anthracycline drugs. Indeed, the anthracycline drugs have a short half-life in the bloodstream and rapidly diffuse throughout the tissues, which results in an even distribution of the drug throughout the body, hence both the malignant and the normal tissues are affected.4
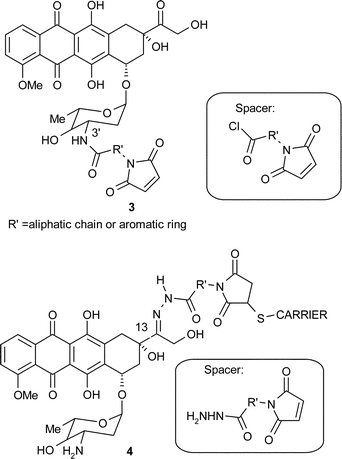
To overcome these limitations, drug delivery systems have been designed to improve the therapeutic index of the anthracycline drugs, by targeting the tumour with greater specificity. The delivery systems consist of the drug attached, via a particular linker or ‘spacer’, to a carrier that will bind to its specific receptor present on the cancer cell, and thereby direct the whole conjugate to the site of action of the drug. Interestingly, a number of bifunctional spacers include a maleimide functionality that allows linkage of the anthracycline drug to these carriers (e.g.3 and 4). Within each spacer a variety of aromatic rings and aliphatic chains have also been used in order to study the influence of the spacer length and polarity on the ability of the conjugate to deliver the drug to the cancer cell.5 In addition, spacers may be attached to the anthracycline molecule in different positions which also affects the potency of the drug.6
Initial bioconjugation studies included monoclonal antibodies7,8 as carriers, and it is also in this early work that most of the spacers were designed and assessed. Synthetic polymers were then used as a means to deliver the drug to malignant tissue. As the physiology of tumours was better understood and more overexpressed receptors were found on the cancerous cells, researchers went on to investigate various other carriers such as serum proteins and peptides for which specific receptors were found on the cancer cell, in order to further improve the specificity and efficacy of the anthracycline conjugates.
The object of this review is to present how the versatile maleimide spacers were designed and positioned on different anthracycline drugs and how they were then used in conjunction with different carriers to allow the drug to specifically destroy tumour cells. Although immunoliposomes9–11 were also successfully used as anthracycline carriers, they will not be covered in this review since, in these delivery systems, the maleimide moiety is used to modify the antibody rather than the anthracycline.
It is worth noting at this stage that most studies were carried out using doxorubicin 1 as the anthracycline of choice since it is found to have the widest spectrum of antitumour activity. However other anthracyclines such as daunorubicin 2 have also been investigated for bioconjugation. Furthermore, the anthracycline drugs are dark red in colour, which makes them ideal to visualise the trafficking of the prodrug and the free drug during the biological assays. This is of great help in developing a general methodology for bioconjugation of anthracyclines and has already led to conjugates of other anticancer drugs.
2 The maleimide spacer in anthracycline conjugation
Conjugates of anthracycline anticancer drugs have been synthesised and tested in order to overcome the side effects associated with the administration of the free drug. The conjugation method of the drug to a carrier should not affect the activity of the drug itself, but it should allow the facile and reproducible incorporation of the drug in the delivery system while avoiding aggregation of the conjugate and the formation of homopolymers of carrier or drug.8 Finally and in addition to biological and mechanistic considerations, the maleimide spacers are generally easy to prepare in a reproducible manner, and stable.12 From a synthetic point of view, they result from the initial condensation of the appropriate amine onto maleic anhydride followed by further functionalisation at the distal end of the amine chain for connection to the anthracycline drug at the required position. This drug derivative may then be attached to the desired carrier via the carbon–carbon double bond of the maleimide group.5 Additionally it is sometimes necessary to treat the carrier in order to generate thiol groups essential for linkage to the maleimide. Despite reports on the immunogenicity of the maleimide moiety, these spacers have been widely used in the synthesis of derivatives linked to a variety of new carriers.13,14Important aspects of the early studies on anthracycline conjugates have already been reviewed by Hermentin7 and Trail,15,16 including in particular the initial studies on the use of monoclonal antibodies (MAbs) as carriers, and the use of spacers within conjugates. The present review is more focused on the work that followed. However it is useful to recapitulate some of the results obtained with MAbs as carriers to understand the investigations on other types of vectors.
The 1980s saw the emergence of the first anthracycline–maleimido spacer–monoclonal antibody conjugates whereby monoclonal antibodies were selected as carriers for their ability to recognise antigens exposed on cancer cells, and were therefore used in an active targeting strategy to destroy the tumour. Hermentin7 observed that the direct binding of the drug to the monoclonal antibody should be avoided as it resulted in complete loss of activity of the drug, therefore the use of a spacer between the anthracycline and the carrier was very important. Furthermore, Fujiwara17 found that the maleimide functionality reacted readily with thiol groups in the carrier in order to create a covalent bond between the spacer and the carrier, and hence was one of the pioneers incorporating the maleimide moiety in such bifunctional spacers. This subsequently gave rise to a number of spacers bearing a maleimide functionality for specific conjugation to thiol-containing molecules, such as cysteine residues present in the carrier. Indeed, the maleimide group reacts only with cysteine thiol groups in preference to amines at neutral pH and below.18 Targeting thiol-containing residues as an anchoring point is an interesting strategy since these are usually only present in discrete numbers in the carriers. This contributes to the homogeneity of conjugates, for which the stoichiometry and site of attachment to the linkers can be easily predicted. Importantly, the activity and physical properties of the protein carrier are not affected by this mode of conjugation since their native pI remains unchanged.18
In addition, although most of the initial spacers were permanently attached to the drug via the amino sugar group, Hermentin observed that it was better for cytotoxic activity7 that the drug was liberated from the conjugate. Independently, Acton19 and Di Marco20 showed that the activity of the anthracycline drug was diminished by derivatisation of the amino sugar moiety. This led scientists at Bristol Myers Squibb, to investigate the rational design of bifunctional spacers that may be attached to doxorubicin in a labile manner via the C-13 keto position,21 and to monoclonal antibodies BR-64 or BR-96.22–27 They found that the most effective conjugates bore an acid-sensitive hydrazone bond between the anthracycline at C-13 and the spacer. This may be explained in terms of the mechanism by which the conjugate was taken up by the tumour cell. This phenomenon termed ‘receptor-mediated endocytosis’ gives rise to a change from pH 7.4 to pH 5.0 as the conjugate is taken up by the tumour cell, which causes the hydrazone bond to hydrolyse, and hence to liberate the drug where it is specifically required. This concept is illustrated further in Kratz's work,6 whereby a series of spacers were synthesised, and coupled to doxorubicin via a hydrazone or an amide bond. He tested the resulting conjugates for stability at pH 5.0 and 7.4. It was found that both the hydrazone bond and the amide bond were stable at pH 7.4, however only the hydrazone bond hydrolysed at pH 5.0. These results were then further related to conjugate activity tests with various carriers, where it was shown that the hydrazone derivatives were active whereas equivalent amide derivatives were not. Details are given in the next section of this review.
The hydrolysis rate of the spacer–drug bond may be further modified by using a variety of aromatic or aliphatic chains within the spacer. It is worth noticing in particular the maleimidocaproyl hydrazone spacer which was developed by Willner28 and was successfully used in a number of studies in the conjugates of monoclonal antibody BR-96 and doxorubicin to give 5 as well as two analogues 6 and 7.29

Alternative approaches for linking the drug to the spacer were also investigated. Indeed, although the hydrazone bond was found to be particularly effective to release the anthracycline drug from a number of conjugates, it is not directly applicable to other anticancer drugs. With the aim of extending their research to mitomycin C 8 and paclitaxel (Taxol®) 9, Dubowchik30 and co-workers set out to design a more complex spacer containing the caproyl maleimide linker for attachment to the monoclonal antibody as well as a peptide sequence sensitive to cathepsin B, a cysteine protease found in all mammalian lysosomes and also found to be overexpressed in tumours. Cathepsin B is also suspected to play an important role in cancer invasion. The peptide sequence in the spacer was quickly hydrolysed by cathepsin B but was not spontaneously hydrolysed in human plasma. Since doxorubicin was unlikely to fit in the active site of cathepsin B, a p-aminobenzyl carbonyl (PABC) linker was intercalated between the peptide sequence and the anthracycline, in order for this self-immolative unit to release the drug upon deacylation occurring in protic solvents.31,32 Two derivatives conjugated with BR-96 as the carrier to yield 10 and 11 were tested on the L2987 lung carcinoma cell line, and proved especially potent (IC50 0.025 µM MAb, 0.050 µM MAb respectively compared with 0.3 µM for the free doxorubicin.),33,34 hence demonstrating the potential of this complex spacer.


Furthermore, cell surface density of expressed antigen as well as the rate of endocytosis of a conjugate may be limiting factors in drug delivery strategies. To overcome this difficulty, the complexity of the spacer structure may be increased to improve the drug loading capacity of the conjugate. To this end, King and co-workers devised bifunctional spacers 12 and 13 that would increase the drug to carrier ratio, thereby improving the biological activity of the conjugate.18 Bivalent peptide linker 12 was also used in conjunction with a cathepsin B–cleavable linker and BR-96 monoclonal antibody as carrier and, after initial aggregation problems were solved by incorporating methoxytriethyleneglycol chains within the spacer, yielded the new conjugates 14 and 15.35,36 When tested on L2987 lung carcinoma cell line, 14 and 15 gave promising results and most importantly, were more potent than the single-branched conjugate 10. For example an IC50 value of 0.016 µM was obtained for 14 and 0.007 µM for 15, compared with a value of 0.025 µM for the single branched conjugate 10.37–39

Thus a number of maleimide spacers were developed in order to deliver the anthracycline drugs or other anticancer agents to the tumour cells. Promising biological results were obtained with different conjugates. There are, however, numerous disadvantages in choosing monoclonal antibodies as carriers in conjugates. For instance, the immunoconjugates may not readily undergo endocytosis in solid tumours or may be immunogenic.40 As was mentioned in the previous paragraph, the cell surface density of expressed antigens, and the antigens or the corresponding antibodies may lack specificity. For these reasons, other carriers such as polymers, serum proteins and peptides were investigated.
3 New carriers for anthracycline conjugates including a maleimide spacer
Although bioconjugation studies initially consisted in the development of chemoimmunoconjugates, a more recent approach has involved the synthesis of anthracycline prodrugs linked to carriers targeting specific receptors overexpressed on the cancer cells. In immunotargeting strategy, delivery of the drug is based on specific interactions between the MAb conjugate and their corresponding antigens, whereas passive targeting consists in the accumulation of prodrug in the tumour. The vasculature of the tumour is indeed often leaky hence allowing macromolecules to penetrate the malignant tissue. Furthermore, the macromolecular prodrugs are retained in the tumour cells since these do not generally have a lymphatic drainage system, and the combination of both these events results in the accumulation of the prodrug within the tumour cell. This phenomenon, termed ‘enhanced permeability and retention’ (EPR),41,42 may constitute an advantage in a conjugation strategy if vectors of suitable molecular weight are employed. In addition, these carriers should be taken up preferentially by the tumour tissue that expresses high amounts of specific receptors on their cell surface. Carriers should be non-immunogenic, biodegradable, and non-toxic, and readily available as a pure and uniform type.423.1 Polyethylene glycol
Accordingly, Kratz and co-workers investigated the use of polyethylene glycol (PEG) as a new way of delivering the anticancer drug to the tumour.43 PEGs are non-ionic, water-soluble synthetic polymers which show biocompatibility and have been found to increase the solubility and the half-life of other cytotoxic agents in the plasma. Consistent with these data and previous work on the design of maleimide spacers, doxorubicin was linked to PEGs of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 70000 Da via a hydrazone or an amide bond to a maleimido spacer (see for example structures 16–18). The effect of the different aromatic rings in each spacer involved was also investigated with respect to biological activity and cellular uptake of the conjugates. Only the conjugates bearing an acid-labile spacer possessed antitumour properties. The IC70 for the compounds tested on lung carcinoma cell line LXFL529 were found to be greater than that for free doxorubicin giving values of 0.04 µM, 0.09 µM and 0.02 µM respectively for compounds 16–18 compared with <0.001 µM for the free drug. However since the drug was distributed more specifically when conjugates were used, a much higher dose of these than the maximum tolerated dose (MTD) of free doxorubicin could be administered. Trafficking studies also showed that doxorubicin originating from the hydrazone-linked PEG was mostly found in the cytoplasm of tumour cell, by contrast to the free drug which was located in the cell nucleus.
000 and 70000 Da via a hydrazone or an amide bond to a maleimido spacer (see for example structures 16–18). The effect of the different aromatic rings in each spacer involved was also investigated with respect to biological activity and cellular uptake of the conjugates. Only the conjugates bearing an acid-labile spacer possessed antitumour properties. The IC70 for the compounds tested on lung carcinoma cell line LXFL529 were found to be greater than that for free doxorubicin giving values of 0.04 µM, 0.09 µM and 0.02 µM respectively for compounds 16–18 compared with <0.001 µM for the free drug. However since the drug was distributed more specifically when conjugates were used, a much higher dose of these than the maximum tolerated dose (MTD) of free doxorubicin could be administered. Trafficking studies also showed that doxorubicin originating from the hydrazone-linked PEG was mostly found in the cytoplasm of tumour cell, by contrast to the free drug which was located in the cell nucleus.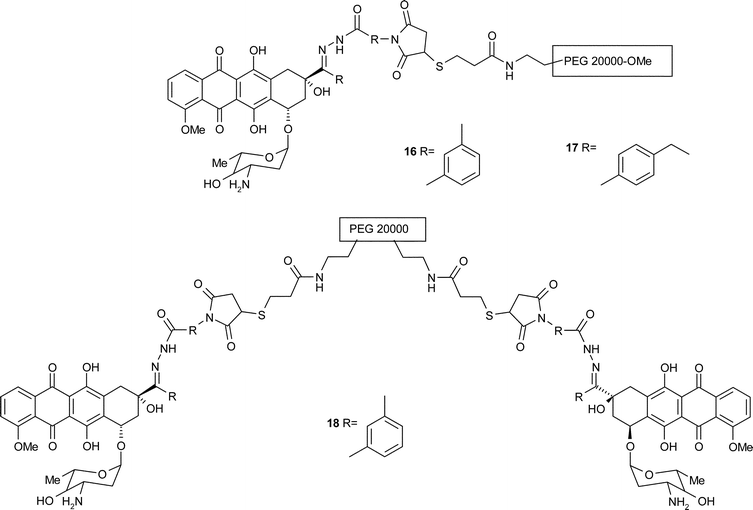
3.2 Transferrin
Investigations were also carried out on serum protein delivery systems which were compatible with the concept of passive targeting and EPR phenomenon owing to their high molecular weight. Transferrin is a β-glycoprotein (78000 Da) naturally used within the body for iron-transport, that fulfilled all of the required criteria for bioconjugates and therefore constituted an interesting lead for the development of a new type of anthracycline prodrugs.42 Initially, Kratz et al. prepared three transferrin conjugates of daunorubicin using derivatives 19–21.44 Each aromatic maleimide spacer was linked to daunorubicin to yield the hydrazone or the amide derivatives. As the drug was thought to be taken up by receptor-mediated endocytosis, it was important to test for the stability of the bond between the drug and the spacer, hence whether release of daunorubicin in the tumour cell was important for cytotoxic activity. The new conjugates were then tested for properties such as pH stability, and cytotoxic activity. All conjugates were stable at plasma pH (pH 7.4), however only the hydrazone derivatives hydrolysed in acidic conditions (pH 5). When tested on melanoma cell line MEXF 989, the amide conjugate 19 showed no cytotoxic activity. By contrast, the hydrazone conjugates 20 and 21 displayed IC70 values of 0.39 µM and 0.022 µM which, remarkably, was ten-fold lower than the IC70 for free daunorubicin, 0.26 µM. These encouraging results were confirmed by biological tests on lung cancer xenograft LXFL 529/11.
Kratz subsequently synthesised similar transferrin–doxorubicin conjugates,45 also analogous to immunoconjugates 5. When tested on the MDA-MB-468 breast cancer cell line, the amide derivatives 22 and 23 were not active, whereas the IC50 values for both hydrazone conjugates 24 and 25 were comparable to those obtained with the free drug (respectively 1.1 ± 0.04 µM, 2.0 ± 0.06 µM and 0.9 ± 0.03 µM), which again showed that there was a link between the stability of the chemical bond between the drug and the spacer and the biological activity of the conjugates. These results corroborated those obtained after tests on leukemia cell line U937. The trafficking of the conjugates was also compared to that of free doxorubicin. Interestingly, the drug originating from the conjugate 24 was situated in the cytoplasm of the tumour cell, by contrast to the free drug which is located in the nucleus of the cell. These results were similar to those previously obtained with PEG conjugates.

3.3 Albumin
Albumin is another key serum protein (66500 Da) that was extensively studied as a carrier in anthracycline conjugates.42 Albumin, like transferrin, is taken up significantly by malignant tissue, and possesses a molecular weight that is appropriate for the concept of EPR effect. Furthermore it is biodegradable, non-toxic, non-immunogenic and is available commercially in a pure form that can be thiolated in order to be bound to the maleimide spacer. Doxorubicin derivatives amides 22, 23 and hydrazones 24 and 25 that were previously tested with transferrin as vector were also assessed for biological properties with albumin as carrier. As was the case with transferrin, only the hydrazone derivatives had an antiproliferative activity on breast cancer MDA-MB-468 cell line (IC50 for 24: 1.9 ± 0.08 µM, 25: 2.1 ± 0.07 µM compared with free doxorubicin 0.9 ± 0.06 µM).4
Although exogenous albumin was successfully exploited in the initial production of prodrugs, Kratz discovered that the circulating albumin could also be used for the formation of conjugates ‘in situ’. Indeed approximately 70% of the circulating albumin is mercaptalbumin that bears a cysteine-34 residue.46 This amino acid potentially remained accessible to the anthracycline derivative since it is not hindered by endogenous thiol groups from cysteine or glutathione. In contrast, most of the other serum proteins do not exhibit free thiol groups. The doxorubicin hydrazone derivative 24 was hence employed in the investigations towards the formation of an in situ prodrug. It was found that the derivative bound rapidly and preferentially to endogenous albumin and showed superior cytotoxic activity to free doxorubicin on murine renal carcinoma (RENCA) cells, at equitoxic dose.46
In addition, the cellular distribution pattern, and the cytotoxicity of acid-sensitive albumin and transferrin conjugates 24 and 25, studied in the LXFL529 lung carcinoma cell line, were very similar whereby the predominant sites of accumulation of the conjugates were the Golgi apparatus and the mitochondria,47 and the corresponding IC50 values were, respectively, 0.3 µM, 0.4 µM, compared with 0.04 µM for free doxorubicin. The results on the trafficking studies were consistent with those obtained for the PEG carrier and transferrin carrier.
In an effort to further improve the efficacy of the albumin prodrug, Kratz48 set out to test analogous aliphatic maleimide spacers instead of the aromatic one so far employed. The binding properties and the in vitro and in vivo activities of the doxorubicin derivatives 26–30 were investigated. One of these, the maleimidocaproyl hydrazone albumin conjugate 29, when tested in three tumour cell lines provided IC50 values between 0.4 and 1 µM, that were approximately an order of magnitude higher than those of the free drug. In addition, the MTD for this conjugate was 3- to 4-times higher than that for doxorubicin, which meant that the therapeutic index of the conjugate was significantly enhanced. This conjugate therefore represented an interesting alternative to the equivalent monoclonal antibody conjugate 5 previously developed that showed limited toxicity towards breast cancer and unfortunately induced severe gastrointestinal side-effects.

A greater degree of specificity was achieved with the design of an albumin-binding doxorubicin prodrug that could be cleaved by matrix metalloproteinase MMP-2 and MMP-9, two metalloproteinases (gelatinases) that play an important role in the degradation and remodelling of basement membranes and the extracellular matrix.49,50 These proteases, in contrast to cathepsin B previously involved in the design of monoclonal antibody–doxorubicin conjugates, are found in the extracellular compartments where most of the body's albumin is also present. Two octapeptide-maleimide spacers that could be cleaved by MMP-2 and MMP-9 to liberate a doxorubicin tetrapeptide derivative were hence synthesised on solid phase. They were then bound to human serum albumin as the carrier, to yield conjugates 31 and 32 that were tested for cytotoxic activity against RENCA cells. Both conjugates were found to be active, in particular 32 with an IC50 of 0.2 µM compared with <0.01 µM for the free doxorubicin. Following these promising results, a modified version of the most cytotoxic conjugate 32 was designed. This new drug derivative 33 was water-soluble and could bind rapidly and selectively to cysteine-34 in circulating albumin to be then cleaved by MMP-2. Furthermore, it was hoped that in this case again the EPR phenomenon would be exploited advantageously for maximal drug supply within the tumour, since albumin was used as the macromolecular carrier. The prodrug was evaluated on a melanoma cell line (A375). Although the IC50 values for the prodrug was 10 ± 1.6 µM and 0.03 ± 0.01 µM for the free drug, the MTD was about four times higher than that of the free drug which meant that the conjugate was superior to the free drug at equitoxic dose.

Recently, emphasis has been placed on the increasingly specific treatment of tumours, therefore the next paragraphs are mostly concerned with on-going studies that show recent trends in this field.
As part of investigations for a cure for prostate cancer, studies were carried out towards the design of albumin conjugates of doxorubicin that would distinctively release the drug in the presence of prostate-specific antigen (PSA).51 Doxorubicin derivatives that incorporated a peptide sequence typically sensitive to PSA were linked to the aminosugar of doxorubicin. Each bifunctional maleimide spacer was designed to be water-soluble to further improve the therapeutic index of the conjugates. Hence a triethylene glycol chain was included in derivative 34, similarly to compound 33, and a caproyl peptidic chain incorporating a peptidic chain analogous to 34 with two additional arginine residues was used in the case of 35. Both anthracycline derivatives were coupled to human serum albumin (HSA) and tested for antitumour activity on PSA-positive and PSA-negative cell lines.52 Although the doxorubicin conjugates were less active than the free drug, they nevertheless presented good antitumour activity (e.g. IC50 values of 6.9 µM, 0.88 µM and 0.09 µM respectively on LNCAP, a PSA-positive line). Furthermore, the preliminary results for the specificity of the arginine derivative 35 towards PSA-positive cell lines were also promising.

The use of albumin conjugates was also investigated in the specific targeting of hepatocarcinoma.53 To increase the chemotherapeutic index of doxorubicin in the treatment of liver cancer, lactosaminated human albumin (L-HSA) was employed as the carrier in doxorubicin prodrug. Indeed it was found that this neoglycoprotein selectively delivers drugs to hepatocytes through the asiaglycoprotein receptor (ASGP-R), a lectin present only on hepatocytes and that mediates uptake and lysosomal digestion of galactosyl terminating macromolecules. ASGP-R is expressed on the large majority of the well-differentiated hepatocellular carcinomas, which incidentally may be easily identified by biopsies. Accordingly, the maleimidocaproyl hydrazone derivative of doxorubicin 36 was coupled to L-HSA, which was pre-treated in order to display free thiol groups for attachment to the maleimide double bond.54 This new prodrug was found to be considerably more specific than doxorubicin since the drug concentration in the liver was found to be about 10 times higher than that in extra-hepatic tissue. Furthermore, in contrast to the free drug, prodrug 36 did not damage the normal hepatocytes including the regenerating cells after surgical removal of the liver tumour. These results may be considered very promising in the treatment of liver cancer.

3.4 Peptides able to cross the blood brain barrier
Although specific treatment of tumours is most often problematic, treatment of a brain tumour presents yet another major obstacle in that the drug must be able to cross the blood brain barrier (BBB) to reach the tumour. This is usually difficult to achieve owing to the structure of the BBB that acts as a filter so that cancer drugs are unable to reach the brain, and indeed free doxorubicin was known not to penetrate the brain.To circumvent this problem, Chen et al.55,56 have synthesised doxorubicin conjugates of melanotransferrin 37 and 38 that were expected to get through the BBB. Melanotransferrin or p97 is a member of the transferrin family that has been found to localise in capillary endothelial cells of the human brain and to play an important role in the transport of iron across the BBB. Melanotransferrin as a carrier is therefore expected to allow doxorubicin to be carried through the BBB. No biological results have been published to this date, but it is hoped that the new conjugates will enable specific treatment of a brain tumour.
As an alternative approach to the melanotransferrin vector, it was found that a peptide derived from Antennapedia, a Drosophila transcription factor, may be employed as a carrier. Antennapedia includes a homeodomain, i.e. a specific sequence of 60 amino acids composed of three α-helices, that may bind to DNA and possesses translocation properties. Interestingly, it was shown that a specific peptide sequence of 16 residues mapped to the third helix of the homeodomain had translocation properties comparable to that of the entire homeodomain. A family of such ‘Trojan peptides’ named the penetratins and derived from this specific sequence, was then synthesised.57 One such peptide, D-penetratin, was hence selected as the carrier for a conjugate intended for brain tumour treatment.58 In this case, D-penetratin, was conjugated with doxorubicin via a short maleimide spacer connected to the aminosugar moiety of the drug to yield conjugate 39. It was also hoped that a prodrug incorporating this vector would evade the mechanism of multidrug resistance (MDR). Indeed, in early studies, the pharmacokinetic profile of the drug was determined, and it was found that the conjugate, as opposed to the free drug, could cross the BBB since it was not recognised by the efflux pump P-glycoprotein (P-gp) involved in MDR. The vector subsequently degraded, leading to drug release from the conjugate into the brain parenchyma. Interestingly, the distribution of the prodrug in the heart and lungs was less than that of the free doxorubicin. On the other hand, the brain seemed to accumulate the vectorised drug, which strongly contrasted with the fact that free doxorubicin was not able to cross the BBB. Additionally, it is worth pointing out that receptor-mediated endocytosis did not seem to be the mechanism of internalisation of such conjugate, although this was still under investigation. Additional studies showed that the cytotoxic effect of the doxorubicin conjugate 39 in the K562 cell line (human erytholeukemic) was less than that of the free drug, displaying an IC50 of 2.6 ± 0.94 µM compared with 0.42 ± 0.06 µM for doxorubicin. However when the conjugate was tested against K562/ADR resistant cells, it showed a potent and dose-dependent growth-inhibitory effect, displaying an IC50 of 3 ± 1.4 µM, corresponding to a 20-fold improvement compared to free doxorubicin.59 The initial results were encouraging and after optimisation of the spacer for a more efficient delivery of doxorubicin at the site of action, could lead to more conjugates of different anticancer drugs.

3.5 Miscellaneous peptidic chains
Peptidic chains may also serve as vectors in conjugates for other solid tumours. Recently, neuropeptide Y (NPY) was used by Kratz et al. as carrier in daunorubicin conjugation.60 NPY, a 36 amino acid peptide of the pancreatic polypeptide family was chosen as a model peptide because NPY receptors are overexpressed in a number of neuroblastoma tumours and the derived cells. The amino acids that were essential for retaining binding affinity to the receptor were determined, then, in order to enable attachment of the maleimide spacer to the peptide, glutamine at position 15 in the peptidic chain was replaced by a cysteine, to give the suitable carrier [C15]-NPY. The modified NPY showed little loss of binding affinity for the NPY-specific receptor Y1. As described for transferrin, albumin and PEG, doxorubicin and daunorubicin were covalently linked to [C15]-NPY via a hydrazone or a amide bond to give three different conjugates 40–42. Only the daunorubicin conjugate featuring a hydrazone bond 40 displayed cytotoxic activity on SK-N-MC neuroblastoma cells which was comparable to that of the free drug, whereby the cell growth was reduced by 66.9 ± 2.5% in comparison with 68.6 ± 0.4% for free daunorubicin. The specificity of the treatment was proven further by testing the same conjugate on NPY receptor-free glioblastoma cells. Lastly, conjugate 40 showed no cytotoxic activity on these cells, which only free daunorubicin could destroy, hence demonstrating that these conjugates can successfully provide treatment of the tumour without causing the toxic side-effects usually obtained with the free drug.
Similarly, a modified segment of human calcitonin (hCT) was also chosen as a carrier for daunorubicin conjugates.61 hCT is a peptide hormone consisting of 32 amino acids that is secreted by the C-cells of the thyroid gland and is involved in the regulation of the physiological calcium balance. It is already used in the treatment of osteoporosis and other bone-related diseases, and in the treatment of hypercalcemia. Interestingly, treatment by nasal application of hCT is effective, and therefore constitutes a more convenient, less traumatic alternative to intravenous administration. It is important to note that no allergic reactions were expected because of the physiological origin of the peptide carrier. In order to enable the coupling of the maleimide spacer to the selected fragment of hCT, a cysteine residue was introduced at the N-terminus of this peptide to yield [C8]-hCT(9–32). The translocation properties of this segment were not affected by the modification. The mechanism of cellular uptake of the calcitonin segment is not yet fully understood, therefore both hydrazone 43 and amide 44 were tested for biological activity. Only hydrazone 43 had a cytotoxic activity, which showed the drug was taken up via an endocytosis process that gave rise to a drop of pH. The daunorubicin hydrazone conjugate was found to have reduced the SK-N-MC cells viability by 57.2%, hence illustrating the potential of [C8]-hCT(9–32) as an effective carrier in anthracycline conjugation.

In a final approach, two potentially universal maleimide spacers were used that would allow attachment of anthracyclines to a variety of carriers, while directly enabling the topoisomerase II inhibition properties of the anthracycline drug derivative. Topoisomerases II are nuclear enzymes that regulate the topology of DNA in all living cells. Cellular topoisomerase II continuously creates transient protein-linked double strand breaks in DNA, and it is this conformation called the cleavable complex that is the target of many drugs such as the anthracyclines doxorubicin and daunorubicin. Consequently, Sun62 set out to design two universal doxorubicin–linker systems that retained the topoisomerase inhibition properties of the free drug. The mode of action of these new conjugates appears different to that illustrated in most of the previous paragraphs, and it is therefore interesting to examine how maleimide spacers may be advantageously employed in this innovative conjugation strategy. Indeed in previous conjugates bearing a spacer at C3′, it was essential to cleave the spacer and restore the basic nature of C3′-nitrogen to enable the cytotoxic activity of the drug. When these novel linkers were employed, the doxorubicin derivative did not require hydrolysis at the C3′-nitrogen, since this nitrogen retained its basic character in the drug derivative. The synthesis of the doxorubicin–maleimide derivatives was reported to be mild, facile, and to allow parallel attachment of a series of vectors. Each derivative was then linked to amino acid chains including a thiol group to yield conjugates 45–51. The amino acids in each carrier were selected for their common and representative peptide functionalities so as to investigate the Michael reaction of the maleimide and the thiol group in the presence of other common functional groups. Upon synthesis, these groups did not appear to interfere with the coupling reaction. Conjugates 45–51 were subsequently tested for enzyme inhibition and they were found to retain the free drug's activity towards topoisomerase II, with overall first effective concentrations for topoisomerase inhibition ranging from 1 to 10 µM. Conjugates 45–51 illustrate the first example of a new approach employing maleimide linkers in the tremendous challenge presented by the treatment of solid tumours.

Hence, since the initial monoclonal antibody carriers associated with simple aromatic or aliphatic maleimide spacers, a number of new carriers such as PEG, serum proteins or peptides have been used together with sometimes very elaborate and labile maleimide linkers to generate new, increasingly tumour-specific anthracycline conjugates.
4 Conclusions
Bioconjugation strategies were undertaken to circumvent the toxic dose-related side effects caused by anthracycline drugs in the treatment of tumours. Initial studies on the design of conjugates were focused on the development of monoclonal antibody-bound prodrugs for the delivery of the anthracycline drugs to the tumour itself, via a specific receptor. A suitable spacer bound to anthracycline molecule in a labile manner at one end and to the carrier at the other end was thus employed in order to liberate the drug at the receptor. Interestingly, the maleimide group was found to be a very useful component of such bifunctional spacer since it very cleanly bound to the free thiol groups present in the carrier. One very versatile spacer designed following these criteria was the maleimidocaproyl moiety that was linked via a hydrazone bond to the C13 of daunorubicin or doxorubicin. In order to broaden these conjugation methods to other antineoplastic agents, more complex spacers, including a protease-sensitive chain bound to the C3′ of the aminosugar of the anthracyclines, were also designed. Multifunctional spacers were also synthesised in order to maximise the amount of drug released at the receptor site. Different carriers such as PEG or serum proteins (albumin, transferrin, melanotransferrin), or peptide sequences (e.g.D-penetratin) were also employed to produce conjugates with improved pharmacokinetic properties and greater tumour specificity. Lastly, the properties of the maleimide moiety as a topoisomerase II inhibitor were also exploited in the design of doxorubicin conjugates following a different mode of action. Following the promising results obtained with the bioconjugates of doxorubicin and daunorubicin, other anticancer drugs such as mytomycin C30, camptothecin,63 tallysomycin S10b,64 carboplatin,65 or paclitaxel30,66 have been derivatised using a maleimidospacer and various carriers, and tested successfully for antiproliferative activity. The versatility of the maleimide spacers thus appears truly encouraging and hopefully will lead to some novel exciting advances in conjugation chemistry, towards a side-effect free treatment of tumours.5 Acknowledgements
CLS wishes to thank Professor John Mann for his helpful comments and for proof-reading this manuscript.6 References
- R. T. Dorr and D. D. von Hoff, Cancer Chemotherapy Handbook 2, ed. Appleton and Lange, Norwalt, 2nd edn, 1994 Search PubMed.
- S. K. Carter, J. Natl. Cancer Inst., 1975, 55, 1265 CAS.
- C. E. Myers and B. A. Chabner, Anthracyclins, in: Cancer Chemotherapy Principles and Practice, ed. B. A. Chabner and J. M. Collins, Lippincott, Philadelphia, 1990, p. 356 Search PubMed.
- F. Kratz, U. Beyer, P. Collery, F. Lechenault, A. Cazabat, P. Schumacher, U. Falken and C. Unger, Biol. Pharm. Bull., 1998, 21, 56 CAS.
- U. Beyer, P. Schumacher, C. Unger and F. Kratz, Monatsh. Chem., 1997, 128, 91 CrossRef CAS.
- M. Krüger, U. Beyer, P. Schumacher, C. Unger, H. Kahn and F. Kratz, Chem. Pharm. Bull., 1997, 45, 399.
- P. Hermentin and F. R. Seiler, Behing Inst. Mitt., 1988, 197 Search PubMed.
- P. Hermentin, R. Doenges, P. Gronski, K. Bosslet, H. P. Kraemer, D. Hoffmann, H. Zilg, A. Streinstraesser, A. Schwartz, L. Kuhlmann, G. Lüben and F. R. Seuler, Bioconjugate Chem., 1990, 1, 100 CrossRef CAS.
- T. M. Allen, E. Brandeis, C. B. Hansen, G. Y. Kao and S. Zalipsky, Biochim. Biophys. Acta, 1995, 1237, 99 CrossRef CAS.
- M. Mercadal, J. C. Domingo, J. Petriz, J. Garcia and M. A. de Madariaga, Biochim. Biophys. Acta, 1999, 1418, 232 CAS.
- T. M. Allen and P. R. Curtis, Science, 2004, 303, 1818 CrossRef CAS.
- A. Lau, G. Bérubé and C. H. J. Ford, Bioorg. Med. Chem., 1995, 3, 1299 CrossRef CAS; A. Lau, G. Bérubé, C. H. J. Ford and M. Gallant, Bioorg. Med. Chem., 1995, 3, 1305 CrossRef CAS.
- C. Boeckler, B. Frisch, S. Muller and F. Schuber, J. Immunol. Methods, 1996, 191, 1 CrossRef.
- J. M. Peeters, T. G. Hazendonk, E. C. Beuvery and G. I. Tesser, J. Immunol. Methods, 1989, 120, 133 CrossRef CAS.
- P. A. Trail, D. Willner and K. E. Hellström, Drug Dev. Res., 1995, 34, 196 CrossRef CAS.
- P. A. Trail, H. D. King and G. M. Dubowchic, Cancer Immunol. Immunother., 2003, 52, 328 Search PubMed.
- K. Fujiwara, M. Yashiro and T. Kitagawa, J. Immunol. Methods, 1981, 45, 195 CrossRef CAS.
- H. D. King, G. M. Dubowchik and M. A. Walker, Tetrahedron Lett., 2002, 43, 1987 CrossRef CAS.
- K. Yamamoto, E. M. Acton and P. W. Henry, J. Med. Chem., 1972, 15, 872 CrossRef CAS.
- A. Di Marco, Cancer Chemother. Rep., 1975, 6, 91–106 Search PubMed.
- R. S. Greenfield, T. Kaneko, A. Daues, M. A. Edson, K. A. Fitzgerald, L. J. Olech, J. A. Grattan, G. L. Spitalny and G. R. Braslawsky, Cancer Res., 50, 6600 Search PubMed.
- T. Kaneko, D. Willner, I. Monkovic, J. O. Knipe and G. R. Braslawsky, Bioconjugate Chem., 1991, 2, 133 CrossRef CAS.
- H. Zhao, D. Willner, J. S. Cleaveland, G. R. Braslawsky and J. P. Brown, Bioconjugate Chem., 1992, 3, 549 CrossRef CAS.
- G. R. Braslawsky, M. A. Edson, W. Pearce, T. Kaneko and R. S. Greenfield, Cancer Res., 50, 6608 Search PubMed.
- P. A. Trail, D. Willner, S. J. Lasch, A. J. Henderson, R. S. Greenfield, D. King, M. E. Zoeckler and G. R. Braslawsky, Cancer Res., 1992, 52, 5693 CAS; P. A. Trail, D. Willner, S. J. Lasch, A. J. Henderson, S. Hofstead, A. M. Casazza, R. A. Firestone, I. Hellström and K. E. Hellström, Science, 1993, 261, 212 CrossRef CAS.
- P. A. Trail, D. Willner, J. Knipe, A. J. Henderson, S. J. Lasch, M. E. Zoeckler, M. D. TrailSmith, T. W. Doyle, H. D. King, A. M. Casazza, G. R. Braslawsky, J. Brown, S. J. Hofsead, R. S. Greenfield, R. A. Firestone, K. Mosure, K. F. Kadow, M. B. Yang, K. E. Hellström and I. Hellström, Cancer Res., 1997, 57, 100 CAS.
- H. O. Sjögren, M. Isakson, D. Willner, I. Hellström, K. E. Hellström and P. A. Trail, Cancer Res., 1997, 57, 4530 CAS.
- D. Willner, P. A. Trail, S. J. Hofstead, H. D. King, S. J. Lasch, G. R. Braslawsky, R. S. Greenfield, T. Kaneko and R. A. Firestone, Bioconjugate Chem., 1993, 4, 521 CrossRef CAS.
- H. D. King, A. J. Staab, K. Pham-Kaplita, D. Yurgaitis, R. A. Firestone, S. J. Lasch and P. A. Trail, Bioorg. Med. Chem. Lett., 2003, 13, 2119 CrossRef CAS.
- G. M. Dubowchik, K. Mosure, J. O. Knipe and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 8, 3347 CrossRef CAS.
- G. M. Dubowchik and S. Radia, Tetrahedron Lett., 1997, 38, 5257 CrossRef CAS.
- G. M. Dubowchik, H. D. King and K. Pham-Kaplita, Tetrahedron Lett., 1997, 38, 5261 CrossRef CAS.
- G. M. Dubowchik and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 8, 3341 CrossRef CAS.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855 CrossRef CAS.
- H. D. King, D. Yurgaitis, D. Willner, R. A. Firestone, M. B. Yang, S. J. Lasch, K. E. Hellström and P. A. Trail, Bioconjugate Chem., 1999, 10, 279 CrossRef CAS.
- G. M. Dubowchik, S. Radia, H. Mastalerz, M. A. Walker, R. A. Firestone, H. D. King, S. J. Hofstead, D. Willner, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 12, 1529 CAS.
- H. D. King, G. M. Dubowchik, H. Mastalerz, D. Willner, S. Hofstead, R. A. Firestone, S. J. Lasch and P. A. Trail, J. Med. Chem., 2002, 45, 4336 CrossRef CAS.
- E. W. P. Damen, F. M. H. de Groot and H. W. Scheeren, Expert Opin. Ther. Pat., 2001, 11, 651 Search PubMed.
- F. M. H. de Groot, E. W. P. Damen and H. W. Scheeren, Curr. Med. Chem., 2001, 8, 1093 CAS.
- C. Monneret and J.-C. Florent, Bull. Cancer, 2000, 87, 829 Search PubMed.
- M. Fleiner, P. Benzinger, T. Fichert and U. Massing, Bioconjugate Chem., 2001, 12, 470 CrossRef CAS.
- F. Kratz, Expert. Opin. Ther. Pat., 2002, 12, 433 Search PubMed.
- P. C. A. Rodrigues, U. Beyer, P. Schumacher, T. Roth, H. H. Fiebig, C. Unger, L. Messori, P. L. Orioli, D. H. Paper, R. Mülhaupt and F. Kratz, Bioorg. Med. Chem., 1999, 7, 2517 CrossRef CAS; R. B. Greenwald, C. D. Conover and Y. H. Choe, Crit. Rev. Ther. Drug Carrier Syst., 2000, 17, 101 Search PubMed.
- F. Kratz, U. Beyer, P. Schumacher, M. Krüger, H. Zahn, T. Roth, H. H. Fiebig and C. Unger, Bioorg. Med. Chem. Lett., 1997, 7, 617 CrossRef CAS.
- F. Kratz, U. Beyer, T. Rith, N. Tarasova, P. Collery, F. Lechenault, A. Cazabat, P. Schumacher, C. Unger and U. Flaken, J. Pharm. Sci., 1998, 87, 338 CrossRef CAS.
- F. Kratz, R. Müller-Driver, I. Hofmann, J. Drevs and C. Unger, J. Med. Chem., 2000, 43, 1253 CrossRef CAS.
- U. Beyer, B. Rothen-Rutishauser, C. Unger, H. Wunderli-Allenspach and F. Kratz, Pharm. Res., 2001, 18, 29 CrossRef CAS.
- F. Kratz, A. Warnecke, K. Scheuermann, C. Stockmar, J. Schwab, P. Lazar, P. Drückes, N. Esser, J. Drevs, D. Rognan, C. Bissantz, C. Hinderling, G. Folkers, I. Fichtner and C. Unger, J. Med. Chem., 2002, 45, 5523 CrossRef CAS.
- F. Kratz, J. Drevs, G. Bing, C. Stockmar, K. Scheuermann, P. Lazar and C. Unger, Bioorg. Med. Chem. Lett., 2001, 11, 2001 CrossRef CAS.
- A. M. Mansour, J. Drevs, N. esser, F. M. Hamada, O. A. Badary, C. Unger, I. Fichtner and F. Kratz, Cancer Res., 2003, 63, 4062 CAS.
- F. Kratz, A. Mansour, J. Soltau, A. Warnecke, I. Fichtner, C. Unger and J. Drevs, Arch. Pharm. Chem. Life Sci., 2005, 338, 462 Search PubMed.
- G. Di Stefano, M. Lanza, F. Kratz, L. Merina and L. Fuime, Eur. J. Pharm. Sci., 2004, 23, 393 CrossRef CAS.
- G. Di Stefano, M. Derenzini, F. Kratz, M. Lanza and L. Fuime, Liver Int., 2004, 24, 246 Search PubMed.
- L. Fuime, L. Bolondi, C. Busi, P. Chieco, F. Kratz, M. Lanza, A. Mattioli and G. Di Stefano, J. Hepatol., 2005, 43, 645 CrossRef.
- Q. Chen, D. A. Sowa, J. Cai and R. Gabathuler, Synth. Commun., 2003, 33, 2377 CrossRef CAS.
- Q. Chen and R. Gabathuler, Synth. Commun., 2004, 34, 2407 CrossRef CAS.
- D. Derossi, G. Chassaing and A. Prochiantz, Trends Cell. Bol., 1998, 8, 84 Search PubMed.
- C. Rousselle, P. Clair, J.-M. Lefauconnier, M. Kaczorek, J.-M. Scherrmann and J. Temsamani, Mol. Pharmacol., 2000, 57, 679 CAS.
- M. Mazel, P. Clair, C. Rousselle, P. Vidal, J.-M. Scherrmann, D. Mathieu and J. Temsamani, Anti-Cancer Drugs, 2001, 12, 107 CrossRef CAS.
- U. Krauss, F. Kratz and A. G. Beck-Sickinger, J. Mol. Recognit., 2003, 16, 280 CrossRef CAS.
- M. Langer, F. Kratz, B. Rothen-Rutishauser, H. Wunderli-Allenspach and A. G. Beck-Sickinger, J. Med. Chem., 2001, 44, 1341 CrossRef CAS.
- C. Sun, S. E. Aspland, C. Ballatore, R. Castillo, A. B. Smith, III and A. J. Castellino, Bioorg. Med. Chem. Lett., 2006, 16, 104 CrossRef CAS.
- M. A. Walker, G. M. Dubowchik, S. J. Hofstead, P. A. Trail and R. A. Firestone, Bioorg. Med. Chem. Lett., 2002, 12, 217 CrossRef CAS.
- M. A. Walker, H. D. King, R. A. Dalterio, P. A. Trail, R. A. Firestone and G. M. Dubowchic, Bioorg. Med. Chem. Lett., 2004, 14, 4323 CrossRef CAS.
- A. Warnecke, I. Fichtner, D. Garmann, U. Jaehde and F. Kratz, Bioconjugate Chem., 2004, 15, 1349 CrossRef CAS.
- P. C. A. Rodrigues, K. Scheuermann, C. Stockmar, G. Maier, H. H. Fiebig, C. Unger, R. Mülhaupt and F. Kratz, Bioorg. Med. Chem. Lett., 2003, 13, 355 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |