Recent developments in enzymatic chlorination
Cormac D.
Murphy
*
UCD School of Biomolecular and Biomedical Sciences, Centre for Synthesis and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: cormac.d.murphy@ucd.ie
First published on 24th January 2006
Abstract
Covering: up to end of 2005
While the existence of chlorinated natural products has been known for over 100 years, our understanding of the enzymology of biological chlorination reactions has been limited to chloroperoxidases, which are now known not to play a significant role in chlorometabolite biosynthesis. The discoveries of new classes of halogenases, described in this Highlight, have shed new light on the mechanisms of enzymatic chlorination of aromatic and aliphatic compounds.
 Cormac D. Murphy | Dr Cormac Murphy was born in Newry, Northern Ireland and obtained his degree in Biochemistry from Queen's University Belfast (1994). He studied for his PhD under Professor David Harper, also at Queen's, investigating biological fluorination and defluorination. After a postdoctoral fellowship with Professor Robert White in Dalhousie University, Nova Scotia, he joined Professor David O'Hagan's group in Durham, and subsequently St. Andrews, to work on the biosynthesis of fluorometabolites in Streptomyces cattleya. In 2001 he moved back across the Irish Sea to University College Dublin where he is a College Lecturer in Microbiology. His current research interests include the biosynthesis and biodegradation of halogenated compounds in bacteria. |
1 Introduction
Until the mid-1990s it was largely accepted that most biological chlorination reactions were catalysed by chloroperoxidases. The ease by which this enzyme activity could be detected, using a convenient spectrophotometric assay developed by the researchers involved in the initial discovery of this class of enzyme,1 led to the discovery of many haloperoxidases from a variety of sources without the need to identify the natural substrates.2 However, the broad substrate specificity, apparent lack of regiospecificity and the difficulty in determining Michaelis constants for organic substrates were inconsistent with a role for these enzymes in the biosynthesis of chlorinated metabolites.3 During the 1990s there were several key findings that determined that chloroperoxidases did not play a prominent role in enzymatic chlorinations. Dairi et al.4 were the first to report the discovery of a gene that did not code for a chloroperoxidase, but was required for the halogenation step of chlortetracycline biosynthesis. Similarly the identification and functional analysis of the gene cluster for pyrrolnitrin biosynthesis in Pseudomonas fluorescens revealed two genes responsible for chlorination (prnA and prnC), neither of which coded for a chloroperoxidase.5,6 Furthermore, it was established that the halogenating reagent formed during the chloroperoxidase-catalysed reaction was hypochlorous acid,7,8 which upon release from the active site, reacts spontaneously with suitable organic compounds in the surrounding medium, thus regioselective chlorinations cannot be catalysed by this class of enzyme. This Highlight discusses the significant advances in our understanding of biological chlorination in the post-chloroperoxidase era.2 FADH2-dependent halogenases
2.1 Tryptophan halogenase
The pyrrolnitrin gene cluster in Pseudomonas fluorescens consists of four genes prnABCD that catalyse the conversion of tryptophan 1 to pyrrolnitrin 4 (Scheme 1).5,6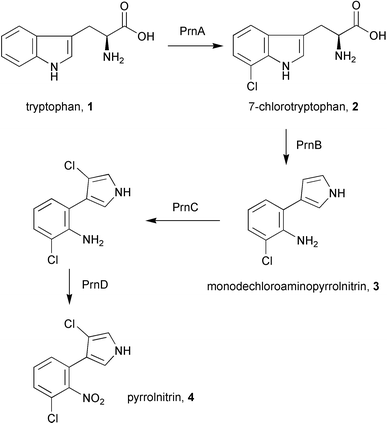 | ||
| Scheme 1 The biosynthesis of pyrrolnitrin in Pseudomonas fluorescens. | ||
Expression of plasmid-encoded prnA and prnC in a P. fluorescens mutant led to the first identification of in vitro chlorinating activity that was clearly distinct from chloroperoxidases, requiring NADH, O2 and chloride (but not H2O2) to regioselectively halogenate 1 and monodechloroaminopyrrolnitrin 3, respectively.9 When PrnA was subsequently purified it was found that in fact the enzyme required FADH2 for activity and that NADH was necessary for the reduction of the flavin by a non-specific reductase.10 The dependence of the enzyme for O2 indicated a monooxygenase-type reaction and initial speculation of the mechanism suggested that flavin hydroperoxide might activate the aromatic ring of 1 by forming an epoxide, which could be attacked by chloride ion and subsequent dehydration of the halohydrin could yield 7-chlorotryptophan 2. Christopher Walsh and co-workers, who purified a tryptophan 7-halogenase involved in rebeccamycin biosynthesis (RebH),11 proposed an alternative mechanism in which flavin hydroperoxide is attacked by chloride ion generating flavin hypochlorite as the halogenating reagent; this would subsequently react with the aromatic ring of 1, yielding the chlorinated product. Most recently the crystal structure of PrnA has been resolved by Jim Naismith's group in St Andrews, and has shed new light on the mechanism of tryptophan halogenation.12 The enzyme is a dimer and the subunits contain an FAD-binding module comprising of two large β-sheets, which is structurally similar to p-hydroxybenzoate hydroxylase, and a Cl− binding pocket nearby that contains threonine (T348) and glycine (G349) residues that hydrogen bond the anion. The Cl− is bound close to the isoalloxazine ring of FAD, but is more than 10 Å from the substrate binding module; this is too far for either a flavin hydroperoxide or flavin hypochlorite to react with 1. Paradoxically, a mechanism is proposed that involves the formation of hypochlorous acid by nucleophilic attack by Cl− on flavin hydroperoxide (Scheme 2a).
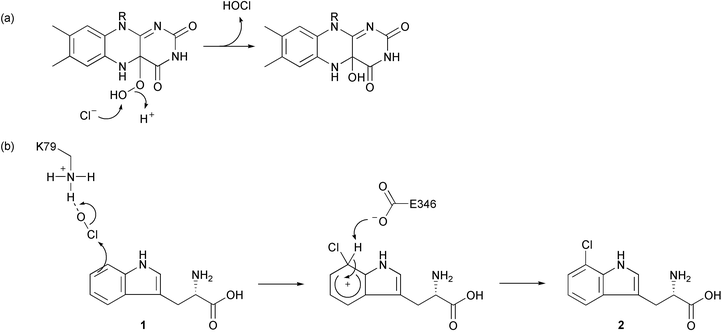 | ||
| Scheme 2 Mechanism of tryptophan chlorination catalysed by PrnA. Formation of HOCl by attack of Cl− on flavin peroxide (a) and electrophilic chlorination of tryptophan (b). | ||
The HOCl is not released into the solvent but rather travels along the 10 Å tunnel to the substrate. Since 1 does not react with HOCl in solution,11 it is likely that in PrnA the HOCl is made more electrophilic by hydrogen bonding to a lysine residue at the end of the tunnel (K79). Furthermore the Wheland intermediate formed during electrophilic addition of chlorine to 1 is stabilised by a glutamate residue in the substrate binding site (E346), which deprotonates the intermediate yielding 2 (Scheme 2b). Site directed mutagenesis experiments demonstrated the importance of these residues to the activity of PrnA: no activity was detected in a K79→A79 mutant, and the kcat was significantly diminished in an E346→Q346 mutant (Km remained the same).12
The genes of other tryptophan 7-halogenases have been identified,13 and sequence analysis has revealed two conserved motifs, one which is the FAD-binding site (GGGXXG) and another that contains two tryptophan residues (GWTWXIP).14 Similar motifs are also present in PrnC, and in all other FADH2-dependent halogenases identified thus far. A tryptophan halogenase with a different regioselectivity is involved in the biosynthesis of pyrroindomycin B in Streptomyces rugosporus LL-42D005.15 The gene coding for the enzyme (pyrH) was overexpressed in Ps. fluorescens and in vitro halogenating activity could be detected with the purified protein, yielding 5-chlorotryptophan. The enzyme has 56% similarity with PrnA and while no crystal structure is currently available, it could be reasoned that the orientation of the 1 relative to the hydrogen bonded HOCl determines the regioselectivity of the reaction.
2.2 Pyoluteorin biosynthesis
Pyoluteorin 5 is an antifungal compound produced by Pseudomonas fluroescens Pf-5, which also produces 4. It is composed of a resorcinol moiety, formed via polyketide biosynthetic reactions,16 linked to a bichlorinated pyrrole.
The biosynthetic gene cluster of 5 has been isolated and ten genes (plt) were identified in a 24 kb region of the Ps. fluorescens Pf5 genome. The deduced amino acid sequences of three of these genes (pltA, pltD and pltM) are similar to PrnC, but only PltA and PltM have the conserved FAD binding site motif, thus it is most likely that PltD does not have halogenating activity.17 Labelling studies demonstrated that proline is the precursor of the dichloropyrrole moiety of 5, and it was suggested that the gene product of pltE, which has sequence homology with acyl CoA dehydrogenases, was responsible for the formal oxidation of proline.17 It was subsequently shown that three proteins, PltF (L-prolyl-AMP ligase), PltL (peptidyl carrier protein) and PltE (dehydrogenase) are responsible for the conversion of L-proline to L-pyrrolyl-S-PltL (Scheme 3), and that a similar mechanism is responsible for the biosynthesis of the pyrrole moiety of undecylprodigiosin and chlorobiocin in S. coelicolor and S. roseochromogenes, respectively.18,19
 | ||
| Scheme 3 Biosynthesis of pyrrole moiety in pyoluteorin biosynthesis. | ||
This is a novel mechanism for the formation of pyrrole and possibly reflects a general strategy for channelling amino acids into secondary metabolism and ensuring that modification only occurs to the peptidyl carrier protein-bound amino acid and not those in the free cellular pool. Most recently, in vitro halogenating activity of PltA has been assayed with L-pyrrolyl-S-PltL as the substrate.20 Analysis of the chlorinated product revealed that PltA catalysed the incorporation of both chlorine atoms, which is the first example of enzyme-catalysed dichlorination; PltM, which has sequence similarity to other FADH2-dependent halogenases, is not required for the halogenating reaction and raises the question of the function of this protein. A mechanism was proposed for the dichlorination of the pyrrolyl-S-PltL involving electrophilic substitution of the pyrrole ring by flavin hypochlorite (Scheme 4). However, this might have to be revised to take into account the discovery of HOCl as the halogenating reagent in PrnA.
 | ||
| Scheme 4 Proposed mechanism of chlorination catalysed by PltA. | ||
2.3 Other FADH2-dependent halogenases
The discovery of FADH2-dependent halogenases has resulted in the identification of similar halogenase genes. Southern blot analysis of genomic DNA from microorganisms that produce halogenated antibiotics, using prnA and prnC as DNA probes, revealed that halogenase gene sequences are present in Pseudomonas pyrrocinia, Ps. aureofaciens ACN, Ps. aureofaciens Pa1, Ps. fluorescens CHA0, Actinoplanes sp., Kitasporia sp., Sacharothrix aerocolonigenes, Actinomadura melliaura and Streptomyces albrogriseolus.21 The genes responsible for the halogenating step of chloramphenicol 6, chlorobiocin and avilamycin A biosynthesis in Streptomyces venezuelae, S. roseochromogenes and S. viridochromogenes respectively, have been identified by mutational inactivation.22–24
However, it has proven more difficult to demonstrate activity of the halogenase enzymes in vitro; other than the enzymes detailed in 2.1 and 2.2, the activity of only one other FADH2-dependent halogenase has been detected, that of HalB from the pentachloropseudilin-producer Actinoplanes sp. ATCC 33002. When this gene was overexpressed in Pseudomonas aureofaciens ACN the crude cell extract catalysed the chlorination of the artificial substrate 2-(3,5-dichlorophenyl) pyrrole.25 The major obstacle in measuring in vitro halogenase activity is knowledge of the structure of the substrate. Thus, for a given halometabolite, an understanding of the likely biosynthetic pathway is required before cell-free investigations can be undertaken.
Through comparisons of the amino acid sequences of PrnA and PrnC with other putative halogenases it is clear that two classes of FADH2-dependent halogenases are emerging: one that halogenates tryptophan or indole (PrnA-type) and the other that halogenates phenyl or pyrrole (PrnC-type). However, it is worthwhile mentioning that the halogenase involved in the biosynthesis of 6 by S. venezuelae, CmlS, is approximately 30% similar to PrnC in Myxococcus fulvus, even though the structure of 6 does not suggest that a phenyl or pyrrole is the substrate for chlorination. Thus the categories as currently suggested may be too narrow for the diversity of halogenation reactions catalysed by FADH2-dependent halogenases.
The halide specificity of these halogenases has not been addressed in cell-free systems, although there are indications that in some cases chloride may be substituted with bromide, for example bromobalhimycin is produced by cultures of Amycolatopsis balhimycina when bromide salts are included in the fermentation medium.26 Interestingly, since brominated compounds are produced mainly by marine organisms, any halogenases involved in their biosynthesis must utilise bromide preferentially, since the ratio of chloride to bromide in sea-water is approximately 300 : 1.27
3 Non-heme FeII α-ketoglutarate- and O2-dependent halogenases
3.1 Syringomycin and barbamide
The mechanism of PrnA accounts for the regiospecific incorporation of chlorine into aromatic substrates, but not saturated aliphatic substrates. The mechanism of enzymatic chlorination of such aliphatic compounds has been examined in Pseudomonas syringae pv. syringae, which produces the phytotoxic lipodepsipeptide syringomycin E 7, and in Lyngbya majuscula, a cyanobacterium that produces the molluscicidal compound barbamide 8.28–32
Biosynthetic investigations using isotope labelled compounds indicated that the biosynthetic origin of the 4-chlorothreonine residue in 7 is threonine28 and the trichloromethyl group of 8 originates from the pro-S methyl group of leucine.29 Such precursors are not obvious substrates for FADH2-dependent halogenases. Feeding experiments with deuterated leucine demonstrated that C3 and C4 of leucine remain saturated during its incorporation into 8, thus the pro-S methyl group is not activated prior to chlorination, suggesting a halogenating mechanism involving radicals.29 Furthermore, no sequences having similarity with genes encoding FADH2-dependent halogenases were found in biosynthetic gene clusters of 7 and 8;30,31 however, similarities did emerge between the deduced amino acid sequences of BarB1/BarB2 in the barbamide cluster and SyrB2 in the syringomycin cluster. These proteins belong to a class of nonheme FeII, α-ketoglutarate-dependent enzymes and it was proposed that they were responsible for chlorination.30,31 Very recently Christopher Walsh and colleagues demonstrated the novel halogenating activity of SyrB2, which chlorinates L-Thr linked to the peptidyl carrier protein SyrB1.32 SyrB2 does not chlorinate free threonine, thus is a tailoring enzyme in the biosynthesis of 7. In FeII, α-ketoglutarate-dependent dioxygenases the FeII is coordinated with conserved His and Asp residues (also present in SyrB2) and α-ketoglutarate. Attack of FeII-bound oxygen on the ketone carbonyl of α-ketoglutarate results in decarboxylation and formation of an oxy-ferryl (FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) species, which generates a radical via homolysis of an unactivated C–H bond of the substrate. Hydroxylation of the substrate occurs via the resulting FeIII–OH acting as donor of an OH˙. It is proposed that in SyrB2 the succinate ligand of the FeIVO intermediate is replaced with chlorine, resulting in the transfer of Cl˙ to the substrate radical instead of OH˙ (Scheme 5). The formation of these reactive intermediates may also account for the observation that SyrB2 was inactivated after seven catalytic cycles. Presumably the BarB1 and BarB2 enzymes in L. majuscula catalyse the chlorination of leucine via a similar radical mechanism.
O) species, which generates a radical via homolysis of an unactivated C–H bond of the substrate. Hydroxylation of the substrate occurs via the resulting FeIII–OH acting as donor of an OH˙. It is proposed that in SyrB2 the succinate ligand of the FeIVO intermediate is replaced with chlorine, resulting in the transfer of Cl˙ to the substrate radical instead of OH˙ (Scheme 5). The formation of these reactive intermediates may also account for the observation that SyrB2 was inactivated after seven catalytic cycles. Presumably the BarB1 and BarB2 enzymes in L. majuscula catalyse the chlorination of leucine via a similar radical mechanism.
 | ||
| Scheme 5 Proposed mechanism of L-threonyl-PCP chlorination catalysed by SyrB2. | ||
3.2 Coronatine
A second non-heme iron halogenase was discovered by the Walsh group, and is involved in the biosynthesis of the non-halogenated phytotoxin coronatine 9 in Ps. syringae pv. tomato DC3000.33 The coronamic acid (CMA) residue of 9 derives from L-allo-isoleucine through the action of five proteins (Scheme 6) and describes a novel biosynthesis of cyclopropyl rings.
 | ||
| Scheme 6 Biosynthetic pathway yielding CMA in Ps. syringae. | ||
The amino acid is initially activated as the AMP ester by CmaA and installed on the thiolation (T) domain of this protein. An acyl transferase (CmaE) catalyses the aminoacylation of CmaD, a stand-alone T-domain, by CmaA. The tethered L-allo-Ile is chlorinated in the γ-position via CmaB, which is an enzyme similar to SyrB and BarB1/B2. This cryptic chlorination activates the Cγ for intramolecular attack to form the cyclopropyl ring, catalysed by CmaC, which has similarity to methylmalonyl-CoA epimerases and contains a mixture of divalent cations. This enzyme probably chelates the oxygen of the thioester, promoting base-catalysed abstraction of a proton from Cα, yielding a carbanion which attacks the chloromethyl group, displacing chlorine.
4 Nucleophilic chlorination
It has been known for several years that chloromethane production in plants, fungi and algae is catalysed by S-adenosyl methionine (SAM):Cl− methyltransferase.34 This was the only example of enzymatic nucleophilic chlorination that had been characterised. However, very recently it has been demonstrated that the fluorinase in Streptomyces cattleya, which catalyses the synthesis of 5′-fluoro-5′-deoxyadeonsine from SAM and fluoride via an SN2 reaction mechanism, can also utilise chloride ion as a substrate generating 5′ chloro-5′ deoxyadeonsine (Scheme 7).35 The reactions with both fluoride and chloride are reversible, but the equilibrium of the chlorination reaction lies substantially in favour of the substrates, thus it was only possible to detect the chlorinated product when the reaction was conducted in the presence of an amino acid oxidase to remove the methionine produced. | ||
| Scheme 7 Nucleophilic chlorination and fluorination of SAM catalysed by the fluorinase from S. cattleya. | ||
5 Biotechnological issues
The incorporation of chlorine in biological molecules can enhance their antibiotic properties,32 but since the chemical preparation of such compounds would be prohibitively expensive, manipulation of biosynthetic gene clusters of antibiotic-producing microorganisms to include halogenase genes offers potential for generating new antibiotic compounds. Sanchez et al.36 generated novel halogenated indolocarbazole compounds by coexpressing genes from the rebeccamycin (reb) and staurosporine (sta) biosynthetic clusters together with the halogenase genes involved in pyrrindomycin biosynthesis (tryptophan 5-halogenase) and thiendolin biosynthesis (tryptophan 6-halogenase) in Streptomyces albus protoplasts (Scheme 8). This work has demonstrated that such an approach to generating halogenated analogues of biologically active compounds is feasible, but it is currently limited to compounds that are biosynthesised from tryptophan. However, as more halogenases are discovered, the range of applications will increase in number, for example it might now be possible to generate novel halogenated derivatives of undecylprodigiosin by incorporating pltA from Ps. fluorescens into the undecylprodigiosin gene cluster from Streptomyces coelicolor. | ||
| Scheme 8 Production of bisindole compounds with novel halogenations via combinatorial biosynthesis. | ||
6 Acknowledgements
The author wishes to thank Professor David O'Hagan for helpful comments.7 References
- L. P. Hager, D. R. Morris, F. S. Brown and H. Eberwein, J. Biol. Chem., 1966, 241, 1769 CAS.
- S. L. Neidleman and J. Geigert, Biohalogenation: Principles, Basic Roles and Applications, Ellis Horwood Ltd., Chichester, 2006 Search PubMed.
- K. H. van Pée, Annu. Rev. Microbiol., 1996, 50, 375 CrossRef CAS.
- T. Dairi, T. Nakano, K. Aisaka, R. Katsumata and M. Hasegawa, Biosci., Biotechnol., Biochem., 1995, 59, 1099 CrossRef CAS.
- P. E. Hammer, D. S. Hill, S. T. Lam, K. H. van Pée and J. M. Lignon, Appl. Environ. Microbiol., 1997, 63, 2147 CAS.
- S. Kirner, P. E. Hammer, D. S. Hill, A. Altmann, I. Fischer, L. J. Weislo, M. Lanahan, K. H. van Pée and J. M. Ligon, J. Bacteriol., 1998, 180, 1939 CAS.
- H. A. Wagenknecht and W. D. Woggon, Chem. Biol., 1997, 4, 367 CAS.
- M. Sundaramoorthy, J. Terner and T. L. Poulos, Chem. Biol., 1998, 5, 461 CrossRef CAS.
- K. Hohaus, A. Altmann, W. Burd, I. Fischer, P. E. Hammer and K. H. van Pee, Angew. Chem., Int. Ed. Engl., 1997, 36, 2012 CrossRef CAS.
- S. Keller, T. Wage, K. Hohaus, M. Hölzer, E. Eichhorn and K. H. van Pée, Angew. Chem., Int. Ed., 2000, 39, 2300 CrossRef CAS.
- E. Yeh, S. Garneau and C. T. Walsh, Proc. Natl. Acad. Sci. USA, 2005, 102, 3960 CrossRef CAS.
- C. Dong, S. Flecks, S. Unversucht, C. Haupt, K. H. van Pee and J. H. Naismith, Nature, 2005, 309, 2216 CAS.
- P. E. Hammer, W. Burd, D. S. Hill, J. M. Lignon and K. H. van Pée, FEMS Microbiol. Lett., 1999, 180, 39 CrossRef CAS.
- K. H. van Pée and S. Zehner, in Handbook of Environmental Chemistry, ed. G. W. Gribble, Springer Verlag, Berlin, 2003, vol. 3, part P, p. 171 Search PubMed.
- S. Zehner, A. Kotzsch, B. Bister, R. D. Sussmuth, C. Mendez, J. A. Salas and K. H. van Pee, Chem. Biol., 2005, 12, 445 CrossRef CAS.
- D. A. Cupples, C. R. Howell, R. D. Stipanovic, A. Stossel and J. B. Stothers, Z. Naturforsch., C: Biosci., 1986, 41, 532.
- B. Nowak-Thompson, N. Chaney, J. S. Wing, S. J. Gould and J. E. Loper, J. Bacteriol., 1999, 181, 2166 CAS.
- M. G. Thomas, M. D. Burkart and C. T. Walsh, Chem. Biol., 2002, 9, 171 CrossRef CAS.
- S. Garneau, P. C. Dorrestein, N. L. Kelleher and C. T. Walsh, Biochemistry, 2005, 44, 2270.
- P. C. Dorrestein, E. Yeh, S. Garneau-Tsodikova, N. L. Kelleher and C. T. Walsh, Proc. Natl. Acad. Sci. USA, 2005, 102, 13843 CrossRef CAS.
- V. N. Burd and K. H. van Pée, Biochemistry (Moscow), 2003, 68, 1132 CrossRef CAS.
- M. Piraee, R. L. White and L. C. Vining, Microbiology, 2004, 150, 85 CrossRef CAS.
- A. S. Eustaquio, B. Gust, T. Luft, S. M. Li, K. F. Chater and L. Heide, Chem. Biol., 2003, 10, 279 CrossRef CAS.
- G. Weitnauer, A. Muhlenweg, A. Trefzer, D. Hoffmeister, R. D. Sussmuth, G. Jung, K. Welzel, A. Vente, U. Girreser and A. Bechthold, Chem. Biol., 2001, 8, 569 CrossRef CAS.
- I. Wyands and K. H. van Pée, FEMS Microbiol. Lett., 2004, 237, 363.
- B. Bister, D. Bischoff, G. J. Nicholson, S. Stockert, J. Wink, C. Brutani, S. Dunadio, S. Pelzer, W. Wohlleben and R. D. Süssmuth, ChemBioChem, 2003, 4, 649 CrossRef CAS.
- G. W. Gribble, Chem. Soc. Rev., 1999, 28, 335 RSC.
- I. Grgurina and F. Mariotti, FEBS Lett., 1999, 462, 151 CrossRef CAS.
- N. Sitachitta, J. Rossi, M. A. Roberts, W. H. Gerwick, M. D. Fletcher and C. L. Willis, J. Am. Chem. Soc., 1998, 120, 7131 CrossRef CAS.
- Z. Chang, P. Flatt, W. H. Gerwick, V.-A. Nguyen, C. L. Willis and D. H. Sherman, Gene, 2002, 296, 235 CrossRef CAS.
- E. Guenzi, G. Galli, I. Grgurina, D. C. Gross and G. Grandi, J. Biol. Chem., 1998, 273, 32857 CrossRef CAS.
- F. H. Vaillancourt, J. Yin and C. T. Walsh, Proc. Natl. Acad. Sci. USA, 2005, 102, 10111 CrossRef CAS.
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. E. O'Connor and C. T. Walsh, Nature, 2005, 436, 1191 CrossRef CAS.
- D. B. Harper, Nat. Prod. Rep., 2000, 17, 337 RSC.
- H. Deng, S. L. Cobb, A. McEwan, R. P. McGlinchey, J. H. Naismith, D. O'Hagan, D. A. Robinson and J. B. Spencer, Angew. Chem., Int. Ed., 2005, 45, 759.
- C. Sanchez, L. Zhu, A. F. Brana, A. P. Salas, J. Rohr, C. Mendez and J. A. Salas, Proc. Natl. Acad. Sci. USA, 2005, 102, 461 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |