DOI:
10.1039/B513734A
(Paper)
New J. Chem., 2006,
30, 197-207
Experimental and theoretical studies of a naturally occurring non-oligomeric steroidal supramolecular zipper†
Received
(in Durham, UK)
27th September 2005
, Accepted 2nd December 2005
First published on 10th January 2006
Abstract
Methanol extraction of the bulbs of Ornithogalum tenuifolium afforded a novel crystalline steroidal sapogenin (25R)-5β-spirostane-1β,3α-diol (compound 1). The structure and stereochemistry, but not the enantiomer, of this compound was unambiguously assigned using X-ray diffraction and multidimensional 1H and 13C NMR data. The crystal structure of 1 (monoclinic space group P21) is both novel and remarkable in that the extended lattice or “supramolecular” structure comprises an unusual one-dimensional hydrogen-bonded polymer with 21 symmetry (the polymer axis is co-linear with the b-axis of the unit cell). Each ladder-like hydrogen-bonded polymer stacks in an interlocking fashion with a neighbouring polymer along the c-axis of the unit cell. The self-recognition by hydrogen-bonding that leads to the formation of the polymer is characterized by discrete 10-membered supramolecular rings. Moreover, the steroid units of the one-dimensional polymer interlock perfectly to form a supramolecular zipper. The newly-discovered supramolecular synthon of the zipper has been elaborated on due to its possible use in crystal engineering applications. The supramolecular zipper system has also been simulated in vacuo using semi-empirical (AM1) quantum mechanics techniques, which correctly reproduce not only the experimental conformation but also the hydrogen-bonded extended structure of the compound. The simulations showed that formation of the supramolecular zipper structure of 1 most likely reflects charge-complementarity of its hydrogen-bonded hydroxy groups and that the electrostatically driven process is characterized by a favourable and cooperative in vacuo enthalpy of association. A further hallmark of the self-assembly of 1 was the lowering of the mean vibrational frequency of the hydrogen-bonded hydroxy groups upon formation of the zipper.
Introduction
Ornithogalum tenuifolium Delaroche (Hyacinthaceae) of the subfamily Ornithogaloideae (Fig. 1) is a widespread bulbous geophyte of bushveld and grassland in southern Africa, extending also to tropical Africa.1 Eight distinct chromosome races are known, including six from southern Africa.2 Several species of Ornithogalum including O. saundersiae Bak. and O. thyrsoides Jacq. have long been implicated in stock poisoning (severe purgation and blindness in cattle).3 This has been attributed to the cytotoxicity of cholestane glycosides isolated from these species.4,5 Perhaps more interesting from a medical standpoint, cholestane glycosides isolated from the bulbs of O. saundersiae (Fig. 1) exhibit a high cytostatic activity against human leukemia HL-60 cells.6,7 Although extra-African Ornithogalum species like O. nutans L. (syn. O. boucheanum Aschers)8,9 produce cardiac glycosides with digitalis-like activity, no such compounds have yet been found in the southern African Ornithogaloideae. However, some saponins have been reported to have stimulating effects on the uterus, and others to have an abortifacient effect when given to goats, rats and cows.10
 |
| | Fig. 1 Photographs of the flowering heads of (a) O. tenuifolium (by Dr N. R. Crouch) and (b) O. saundersiae (reproduced with the permission of the Trustees of the Royal Botanic Gardens, Kew, UK). | |
Herein we describe the isolation, purification, and characterization of the novel steroid (25R)-5β-spirostane-1β,3α-diol, 1, from O. tenuifolium (Fig. 2). The relative configuration of 1 has not been reported before and differs significantly from that of the methanol solvate of the related diastereomer (25R)-5α-spirostane-1β,3β-diol (compound 2) that was isolated from a methanol extract of an Ecuadorian plant Chamaedorea linearis Mart. (commonly known as the whale tail palm) by Patil and co-workers in 1993.11 (Patil et al. found that compound 2 was powerfully cytotoxic and, although beyond the scope of the present article, it will be of considerable future interest to establish the biological activity of 1 for comparison.) The molecular structure of 1 has been determined from X-ray diffraction data at ambient temperature and further analyzed using semi-empirical (AM1) quantum mechanics techniques, principally to gain an understanding of the relative stabilities of diastereomers 1 and 2 as well as the propensity of 1 for self-recognition by hydrogen-bonding in the solid-state. Importantly, inversion of the stereochemistry at C-5 (ring A) is shown to profoundly affect the conformation, shape, and supramolecular chemistry of 1, which is best described as a naturally occurring supramolecular zipper. Compound 2, in contrast, cannot form a supramolecular zipper because it does not have the correct stereochemistry.
 |
| | Fig. 2 Structural diagrams with numbering schemes and ring labels of the spirostane steroids relevant to this work. (It is important to note that the absolute structure of 1 could not be experimentally established from the X-ray data. The specific enantiomer of 1 for X-ray structure refinement was arbitrarily assigned with a (25R) stereogenic centre to allow facile comparison of 1 with 2. Compound 1 could be equally correctly inverted and named (25S)-5α-spirostane-1α,3β-diol.) | |
The formation of ladder-like supramolecular zippers12 (Fig. 3) from synthetic or natural oligomeric synthons (e.g., short single strands of DNA or RNA,13,14 nucleic acid derivatives,15 polypeptides/amides,16,17 coordination polymers,18,19 and metalloporphyrin dimers20) presents considerable design and synthetic challenges that define one of the cutting-edges of modern supramolecular chemistry. Such zippers are nevertheless architecturally conventional. More unconventional are the elegant non-oligomeric supramolecular zipper systems that have recently been reported that rely on the self-assembly of small organic and metal–organic building blocks.19,21,22 Exciting developments within the field also include the use of molecular simulations to determine the types of supramolecular synthon that might best yield self-assembling zipper target structures.23 It may even be argued that only oligomers like single-stranded DNA that undergo self-recognition processes generate true supramolecular zippers since the DNA bases (i.e., the recognition motifs or teeth of the zipper) are attached to a polynucleotide backbone in the same way that the two halves of a man-made zipper are attached to a fabric backbone. The self-assembly of non-oligomeric building blocks into a double-stranded zipper-like structure is thus more akin to self-assembly or closure of the interlocking teeth of a man-made zipper (clearly quite a formidable feat). Such supramolecular zippers will undoubtedly be harder to design than oligomeric ones unless, as we show in this paper, Nature provides examples of how best to do the job. Interestingly, none of the zipper systems reported to date involve steroids or their derivatives, despite the fact that steroids in general are growing in importance as relatively rigid scaffold components24,25 of supramolecular building blocks and architectures.26 Bile salts and related steroid derivatives, in particular, are known to form manifold useful supramolecular associations.27,28
 |
| | Fig. 3 Schematic illustration of synthon (building block) assembly into supramolecular zipper complexes for (a) single-stranded oligomeric synthons and (b) non-oligomeric synthons. | |
Experimental
Isolation
Fresh bulbs of flowering material of Ornithogalum tenuifolium Delaroche were collected in Ashburton, KwaZulu-Natal, and a voucher retained for verification purposes (Crouch 845, NH). The bulbs (567.3 g) were cut into pieces and extracted at room temperature with methanol for approximately 48 hours. The crude methanol extract (6.7 g) was subjected to column chromatography using hexane–ethyl acetate (8∶2) as the initial mobile phase. Increasing ratios of ethyl acetate were then introduced until a pure ethyl acetate mobile phase was reached. This was followed by the use of increasing ratios of methanol until a pure methanol mobile phase was reached. Five fractions were collected. Fraction 5 collected in ethyl acetate–hexane (9∶1) was further purified by silica gel column chromatography with ethyl acetate–hexane (8∶2) and compound 1 (12 mg), a steroidal sapogenin, was isolated.
General analysis
NMR spectra for 1 (Table 1) were recorded on a Varian Unity 400 MHz NMR spectrometer using standard 1D and 2D pulse sequences. IR spectra were recorded on a Shimadzu FTIR-4300 spectrophotometer and the mass spectra on a MAT CH7A mass spectrometer (Finnigan MAT, Bremen) coupled with a Varian type 1700 gas chromatograph. (25R)-5β-Spirostane-1β,3α-diol (1, 12 mg) formed colourless crystals with a mp of 195 °C. EIMS m/z 432 (M+, 4%), 139 (100), 289 (10). IR data (KBr pellet, νmax/cm−1) 3428 (O–H stretch); 2928, 2868 (C–H stretch); 1240 (CH2 and CH3 bend); 1061 (C–O stretch).
| Signal |
1H (ppm)a |
13C (ppm)b |
HMQCc |
NOESYc |
COSYc |
|
1H data at 400 MHz in CDCl3–CD3OD. Multiplicities are given in italics (e.g., d = doublet), br = broad, ax = axial, eq = equatorial, and numbers in parentheses are coupling constants in Hz.
13C data at 100 MHz in CDCl3–CD3OD.
Peak correlations are listed.
Signals overlap.
|
| 1 |
3.94 t-like (2.8) |
73.9 (CH) |
19 |
11, 19 |
2α, 2β |
| 2 |
1.59 α; 1.95 βd |
36.9 (CH2) |
— |
—; 1α |
1, 3; 1, 3 |
| 3 |
4.12 m |
66.5 (CH) |
4 |
2β, 5 |
2α, 2β, 4α, 4β |
| 4 |
1.55 β; 1.80 αd |
35.8 (CH2) |
— |
3β, 5; — |
3, 5; 3, 5 |
| 5 |
1.64d |
35.5 (CH) |
19 |
6β, 3 |
6α |
| 6 |
1.19 α; 1.47 βd |
26.1 (CH2) |
— |
—; 5 |
5; — |
| 7 |
1.63 α; 1.71 βd |
31.3 (CH2) |
— |
—; 6β |
8; — |
| 8 |
1.89d |
35.4 (CH) |
14 |
6β |
9, 14 |
| 9 |
1.42d |
41.8 (CH) |
19 |
12α |
8 |
| 10 |
— |
38.9 (C) |
19 |
— |
— |
| 11 |
1.33d |
20.6 (CH2) |
— |
1α |
12α, 12β |
| 12 |
1.12 α; 1.70 βd |
40.1 (CH2) |
18 |
17; — |
11; 11 |
| 13 |
— |
40.3 (C) |
14, 18 |
— |
— |
| 14 |
1.15d |
56.1 (CH) |
15β, 18 |
9 |
8, 15α, 15β |
| 15 |
1.29 β; 2.01 αd |
31.7 (CH2) |
14 |
18β; 16 |
14, 16; 14, 16 |
| 16 |
4.40 q-like (7.5,7.7) |
80.7 (CH) |
15α, 15β |
15α, 17 |
15α, 15β, 17 |
| 17 |
1.74d |
62.2 (CH) |
15α, 18, 21 |
16 |
16, 20 |
| 18 |
0.69 s |
16.4 (CH3) |
12, 14 |
8β |
— |
| 19 |
0.99 s |
18.3 (CH3) |
9 |
1α, 8, 5 |
— |
| 20 |
1.84d |
41.6 (CH) |
21 |
— |
21, 17 |
| 21 |
0.89 d (6.9) |
14.4 (CH3) |
20 |
20, 17 |
20 |
| 22 |
— |
109.2 (C) |
20, 21, 23 ax, 25, 26 ax, 26 eq |
— |
— |
| 23 |
1.46 eq; 1.66 axd |
28.7 (CH2) |
25, 27 |
24 eq; — |
—; — |
| 24 |
1.32 ax; 1.76 eqd |
26.5 (CH2) |
— |
—; 23 eq |
25; 25 |
| 25 |
1.62d |
30.2 (CH) |
27 |
26 eq, 21α |
24 (ax, eq), 26 (ax, eq) |
| 26 |
3.47 eq t-like (10.9); 3.39 ax br d (10.7) |
66.8 (CH2) |
27 |
27 eq, 25 ax; 27 eq |
25; 25 |
| 27 |
0.81 d (6.2) |
17.1 (CH3) |
24 |
25 ax |
25 |
X-Ray crystallography
Crystals of 1 were obtained from CD3OD–CDCl3 by slow evaporation in the NMR tube. X-Ray diffraction data for a faintly yellow (practically colourless) crystal of 1 (0.35 × 0.35 × 0.65 mm3) were collected at room temperature with an Enraf-Nonius CAD4 X-ray diffractometer (employing the ω
− 2θ scan technique) operating at 2.8 kW (graphite-monochromated Mo Kα radiation, λ = 0.71073 Å, long fine-focus sealed tube). Three reflections were monitored at 7200 s intervals to monitor decay of the crystal specimen (1%). The data were reduced using Lorentz and polarization correction factors; an absorption correction was not applied to the data (no heavy elements present). A total of 5466 reflections were collected with 2310 being unique and 2104 having I > 2σ(I); the internal R-factor for the data set was good at 2.04%. The structure was solved by direct methods in the monoclinic space group P21 using SHELXS-9729 running in WinGX V1.70.01.30 All non-H atoms were cleanly located in the E map, assigned, and refined anisotropically against F2 with SHELXL-97.29 All H atoms were located in a final difference Fourier map; however, only those of the hydroxy groups were refined independently as isotropic atoms. All other H atoms were included as standard idealized contributors with the normal SHELXL-97 idealization parameters in the least-squares refinement of the structure model. The refinement converged to the discrepancy indices given below. The low maximum and minimum electron densities of the final difference Fourier map indicated complete assignment of the electron density. Not unexpectedly for a light atom structure, the absolute structure parameter was unreliable and the correct enantiomer of 1 could not be determined from the experimental data. CCDC reference number 283813. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513734a
Crystal data
C27H44O4, M = 432.62 g mol−1, T = 293(2) K, λ = 0.71073 Å, monoclinic, P21, a = 11.4271(10) Å, b = 6.9958(19) Å, c = 15.189(3) Å, β = 98.054(11)°, V = 1202.3(4) Å3, Z = 2, ρcalc = 1.195 g cm−3, reflections collected = 5466, unique reflections = 2310 (Rint = 0.0204), goodness-of-fit on F2 = 1.080, R1 = 0.0380 [I > 2σ(I)], wR2 = 0.1006 [I > 2σ(I)], R1 = 0.0414 (all data), wR2 = 0.1038 (all data),31 absolute structure parameter = 10(10), largest difference peak and hole = 0.266 and −0.183 e Å−3.
Molecular simulations
Semi-empirical quantum mechanics calculations (AM1 method, RHF wave function, singlet ground state)32 were performed with Spartan ‘0433 using the X-ray coordinates of the monomeric structure of 1 or its hydrogen-bonded extended structures comprising up to six molecular units as input. Full geometry optimizations (SCF energy convergence = 1.0 × 10−6 au, maximum gradient tolerance = 4.5 × 10−4 au Bohr−1, distance tolerance = 1.8 × 10−3
Å) of up to six hydrogen-bonded asymmetric units were used to investigate the hydrogen-bonding between molecules of 1 in the crystal lattice. Frequency calculations (298.15 K) were performed on each optimized structure; no imaginary vibrational modes were found, suggesting that each structure was a true minimum on the potential energy surface for the system. Calculations on compound 2 were effected similarly.
We also carried out a high-level electronic structure theory calculation on 1 that employed a hybrid (ONIOM34) DFT/AM1 simulation of the trimeric supramolecular structure with Gaussian 03W.35 All atoms belonging to steroid ring A (as well as the substituent atoms) for the trimer (starting from the X-ray coordinates) were treated at the B3LYP/6-31+G** level of theory. All remaining atoms of the extended structure were treated at the AM1 level of theory. Whereas we could carry out a full geometry optimization of the trimer structure of 1 with AM1 (504 basis functions, 528 valence electrons) in around 60 h on an Intel Pentium IV system operating at 3.4 GHz, the analogous ONIOM calculation only converged after 13 days and offered no statistically significant improvement in the geometric accuracy of the simulated structure (supporting information†) to warrant continued use of this computationally more expensive method and basis set.
Results and discussion
Spectroscopic studies
We had difficulties growing X-ray quality crystals of 1 due to the lack of material and for quite some time had to rely on spectroscopic data for structure elucidation. As will become evident from the following section, the spectroscopic data are partly ambiguous and full structure elucidation became possible only once we had solved the X-ray structure of the compound. Given the nontrivial nature of some of the NMR data for 1, spectroscopic analysis of the system is described in some detail below.
The mass spectrum of 1 exhibited a parent ion peak at m/z 432 which was consistent with the proposed molecular formula, C27H44O4. The double bond equivalence calculated indicated the presence of six aliphatic rings as no double bond resonances were present in the 1H NMR spectra of the compound (Table 1). Thus, apart from the normal four rings associated with a triterpenoid skeleton, two extra rings were clearly evident. The resonances of two tertiary methyl groups were observed as singlets at δ = 0.69 ppm and δ = 0.99 ppm in the 1H-NMR spectrum and were assigned to 3H-18 and 3H-19, respectively. The HMQC spectrum showed correlations between the carbon resonance at δ = 73.9 ppm (CH) assigned to C-1 and the 3H-19 proton resonance. Correlations between the corresponding H-1 methine proton resonance at δ = 3.94 ppm and the 2H-2 proton resonances which, in turn, correlated to another methine proton resonance at δ = 4.12 ppm, assigned to H-3, were observed in the COSY spectrum. Hydroxy groups were thus placed at C-1 and C-3. The NOESY spectrum showed correlations between the 3H-19 resonance and the H-1, H-5β and H-8β resonances. The H-5β resonance in turn correlated to the H-3β resonance and revealed that the hydroxy group at C-3 must be α-oriented. A model showed that a correlation between the 3H-19 and H-1 resonances was possible in the NOESY spectrum, whether H-1 was α- or β-oriented. However, X-ray analysis (vide infra) confirmed that the hydroxy group at C-1 had a β configuration.
The methine carbon resonance at δ = 62.2 ppm was assigned to C-17 and showed correlations with the 3H-18 resonance and the 3H-21 doublet at δ = 0.89 ppm in the HMQC spectrum. An acetal carbon resonance at δ = 109.2 ppm also showed a correlation with the 3H-21 resonance in the HMQC spectrum and was assigned to C-22. The COSY spectrum showed correlations between the 3H-21 resonance and H-20 (δ = 1.84 ppm), between H-20 and H-17 (δ = 1.74 ppm), and between H-17 and H-16 (δ = 4.40 ppm). The chemical shift of the H-16 resonance indicated that an oxygen group was attached to C-16. The two extra rings required by the molecular formula could be explained by a spirostane skeleton with C-16, C-22 and C-22, C-26 ether linkages. The C-22 resonance showed correlations with the methylene proton resonances at δ = 3.39 ppm and δ = 3.47 ppm assigned to 2H-26 in the HMQC spectrum. The chirality at C-22 is fixed during biosynthesis by the stereospecificity in the formation of the ketal.36
Correlations between the 2H-26 resonances (δ = 3.47 and 3.39 ppm) and the H-25 resonance at δ = 1.62 ppm were observed in the COSY spectrum. The doublet integrating to three protons at δ = 0.81 ppm (J = 6.2 Hz) was assigned to 3H-27 and also showed correlations to the H-25 resonance in the COSY spectrum. The NOESY spectrum showed correlations between the H-26 resonance at δ = 3.47 ppm and the 3H-27 and H-25 resonances. The H-26 resonance at δ = 3.39 ppm correlated only with 3H-27. This indicated that H-26 (δ = 3.47 ppm) and 3H-27 are possibly equatorial protons. If 3H-27 is equatorial, then H-25 and H-26 (δ = 3.39 ppm) must be trans-diaxial. Importantly, the X-ray data for 1 confirmed these stereochemical deductions made from the NOESY spectrum of the compound.
X-Ray crystallography
Molecular and crystal structure of 1.
The ambient temperature X-ray crystal structure of 1 is shown in Fig. 4; the bond lengths and bond angles of the compound were normal and are available from the electronic supporting information (CIF file) for the structure. Although the X-ray data were not suitable for assigning the absolute configuration of 1 (no heavy atoms coupled with low-intensity Mo Kα radiation leading to a meaningless Flack parameter37), the molecular structure of 1 clearly shows that the stereochemistry of the axial substituents appended to C-5 (hydrogen atom 5) and C-10 (carbon atom 19) is cis-diaxial. This is the single most important stereochemical feature of the molecule as it ensures that ring A of the steroid skeleton tips down relative to the mean plane of ring B (and thus the majority of the steroid’s fused ring system, see Fig. 5). The consequences of this conformational architecture will be discussed further below as it engenders a unique extended or “supramolecular” structure for 1. Inspection of the 21 spirostane steroids that have been crystallographically characterized and included in the Cambridge Structure Database (CSD, August 2005 release), reveals that the 5β/10β configuration of 1 is known for only one other crystallographically characterized spirostane natural product, namely 1β,2β,3β,4β,5β,7α-hexahydroxy-spirost-25(27)-en-6-one (CSD code HXSPEO) isolated from the rhizomes of Campylandra aurantiaca Bak. and Rohdea japonica Roth. (Convallariaceae).38
 |
| | Fig. 4 Thermal ellipsoid plot (40% electron density ellipsoids) of the molecular structure of 1 with the crystallographic atom labelling scheme. H atoms are shown as spheres of arbitrary diameter. | |
 |
| | Fig. 5 Diagram depicting the relative orientations of the mean planes through rings A and B of the steroid skeleton of 1. The angle between the planes measures 75.1(1)°. | |
As might be anticipated, all of the six-membered rings of the steroid skeleton of 1 adopt a chair conformation (i.e., the energetically preferred ring conformation). In the case of ring A, the hydroxy groups appended to C-1 and C-3 are axial and equatorial, respectively. As noted above, we had no problem discerning from the NMR data that the hydroxy group attached to C-3 was equatorial (α configuration). However, the NMR data were unable to delineate the stereochemistry of the C-1 hydroxy group due to the ambiguity of the cross-peaks in the NOESY spectrum of 1. The X-ray data thus provide the crucial conformational information required for complete elucidation of the molecular configuration of 1.
The unique conformation of 1, coupled with the specific disposition of hydrogen bond donor and acceptor groups, results in the formation of an interesting 10-membered macroring structure at the supramolecular level (Fig. 6). Each hydrogen-bonded macroring involves three neighbouring molecules in the crystal lattice and two unique hydrogen bonds of 2.00(3) Å (O2⋯H300a; symmetry code a: −x, y
+ 0.5, −z) and 2.22(6) Å (O3⋯H200b; symmetry code b: x, y
− 1, z). As shown in Fig. 7(a), the one-dimensional hydrogen-bonded polymer which results from these hydrogen-bonding interactions runs co-linear with the b-axis of the unit cell. Each steroid molecule thus forms a “rung” of the ladder-like structure. Since the crystallographic b-axis defines the 2-fold screw axis of the unit cell, the one-dimensional hydrogen-bonded polymer has 21 symmetry (as illustrated in part (b) of Fig. 7). Each hydrogen-bonded polymer chain thus has a nominal width of 30.9 Å if we measure from an H atom appended to C-27 of one molecule to its symmetry-related counterpart in a neighbouring molecule (H-27A⋯H-27A′, where the primed atom belongs to the nearest hydrogen-bonded neighbour) and is thus about three times the diameter of a 10-nm wide single-walled carbon nanotube. Interestingly, there are relatively few short H⋯H contacts between the one-dimensional hydrogen-bonded polymer chains such that the step-like polymers apparently interlock in an efficient manner to give discrete sheets of steroid molecules lying in planes parallel to the ac plane of the crystal lattice.
 |
| | Fig. 6 Close-up view of the hydrogen-bonding arrangement in the crystal lattice of 1 that leads to the formation of a 1D polymer. | |
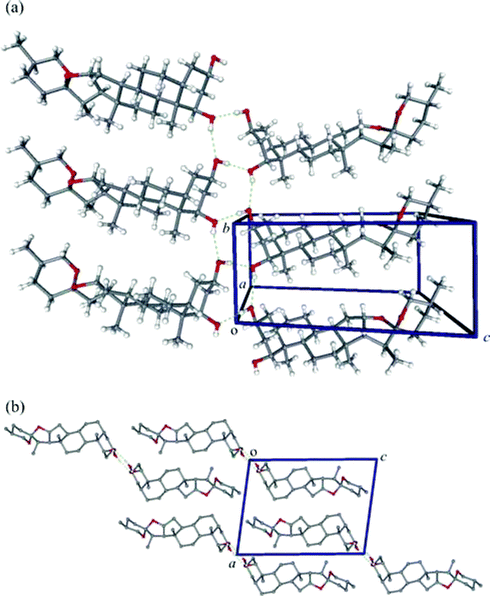 |
| | Fig. 7 (a) View approximately down the a-axis of the unit cell of 1 illustrating the hydrogen-bonded extended or “supramolecular” structure. The axis of the 1D hydrogen-bonded polymer is co-linear with the b-axis of the unit cell. (b) View down the b-axis of the unit cell illustrating the interlocking of the 1D polymer chains (non-polar H atoms have been removed for clarity). | |
Importantly, a similar type of hydrogen-bonding complementarity to that observed in the X-ray crystal structure of 1 is found in the crystal structure of 1β,2β,3β,4β,5β,7α-hexahydroxy-spirost-25(27)-en-6-one,38 which also crystallizes in the monoclinic space group P21 (Fig. S1, supporting information†). Although based on only two experimental observations at present, it appears that the 5β/10β configuration for ring A of the steroid skeleton, coupled with the presence of two or more hydroxy groups on the ring, may well constitute a type of naturally occurring molecular recognition motif or “supramolecular synthon” that favours self-recognition and the concomitant formation of effectively one-dimensional hydrogen-bonded polymer chains.
Supramolecular zipper architecture.
One of the most important observations from the crystal structure of 1 is that the steroid molecules within the system exhibit self-recognition by hydrogen bonding and thus have the intrinsic ability to form a self-assembled extended structure. Compound 1 is therefore an unusual example of a non-oligomeric natural product that forms a supramolecular zipper in the solid-state. As illustrated in Fig. 8, there are two key architectural features of the molecular structure of 1 that enable it to form a supramolecular zipper. These are (1) a rigid body with a relatively elongated form that encompasses a natural turn towards one end of the structure (i.e., ring A of the steroid skeleton) and (2) two hydroxy groups appended to roughly the top and bottom regions of the turned-over end of the molecule. Most importantly, the two hydroxy groups function both as H-bond donors and acceptors. This is crucial to the design of the zipper as hydrogen bonds link not only the adjacent molecules in the two halves of the structure related by the two-fold screw axis (21 symmetry element), but also those involving direct translation along the polymer axis (i.e., steroids related by two consecutive 21 symmetry operations). Thus, consistent with the design of a man-made zipper, each tooth on the left side makes contact with two teeth on the right side of the structure. Moreover, analysis of the intermolecular contacts and van der Waals interactions between the steroid molecules on either side of the zipper indicates that considerable space exists between neighbouring molecules in a stack (akin to the spacing between the teeth of a man-made zipper). A crucial question, of course, is whether one can prove that self-assembly of the steroid building blocks truly generates an energetically favoured supramolecular zipper. This is one question that we believe we have answered, at least in part, using in vacuo semi-empirical electronic structure theory calculations (vide infra).
 |
| | Fig. 8 (a) Illustration of the molecular recognition motif (supramolecular synthon) that is naturally built into the molecular structure of 1. (b) Illustration of the basic two-fold screw (21) symmetry of a man-made zipper and the 21 symmetry of the zipper assembled from the supramolecular synthon 1. (c) Space-fill diagram (CPK model) of an octamer portion of the supramolecular structure of 1 illustrating the loose fit of the zipper teeth in the non-interlocking region of the structure. | |
Comparison of the X-ray structures of 1 and 2.
To fully illustrate the significant conformational differences between the X-ray structures of 1 and 2, we have used least-squares regression methods to fit the conformations in two ways (Fig. 9). The fit that most readily displays the conformational variations of the two structures is that produced by comparing the geometries of rings B, C, and D of the steroid skeleton, i.e., Fig. 9(b). Clearly the most rigid part of the steroid comprises these three rings, while considerable conformational variance is evident for the spiroketal ring system (rings E and F). More importantly, the structural consequences of the 5β/10β configuration for ring A of 1 are immediately apparent and clearly result in the downward projection of ring A relative to the mean plane of ring B. This, in turn, facilitates the hydrogen-bonded supramolecular structure of 1 by allowing the optimum interaction of hydroxy groups on rings A of neighbouring molecules. Compound 2, in contrast, does not display self-recognition to form a one-dimensional hydrogen-bonded polymer even though it carries hydroxy groups at the same positions on ring A as 1. The X-ray crystal structure of 2,11 in fact, is a methanol solvate with the solvent molecule hydrogen-bonded to the hydroxy group appended to C-3 of the steroid skeleton. While this does lead to a hydrogen-bonded chain of alternating steroid and solvent molecules in the crystal lattice, this cannot be regarded as true molecular recognition between steroid molecules as it includes a non-steroidal component.
 |
| | Fig. 9 Least-squares fits comparing the conformations of the X-ray structures of 1 and 2. In (a) steroid rings B through F were used in the fit, while in (b) steroid rings B, C, and D were used for fitting. (H atoms omitted for clarity.) | |
Molecular simulations
Two significant structural phenomena worthy of further investigation emerge from an analysis of the X-ray structures of 1 and 2. First, the 5β/10β configuration for ring A of 1 leads to a somewhat folded conformation for this stereoisomer relative to the shape of stereoisomer 2. Second, only compound 1 undergoes self-recognition in the solid state to form a one-dimensional hydrogen-bonded polymer. Our objectives were to use computational methods to delineate (1) the energetics of stereoisomerism in this system, (2) the underlying reasons for the formation of the elegant hydrogen-bonded supramolecular structure of 1, and (3) the gas phase energetics of the self-assembly process. Since semi-empirical quantum mechanics methods like AM132 are known to correctly model hydrogen bonding39,40 in diverse organic compounds (including dimeric species41) and can be reliably used on larger systems up to the size of small proteins,42 we have simulated the in vacuo supramolecular structure of 1 using AM1 to investigate self-organization in this system. We have also analyzed the relative energies of the in vacuo structures of stereoisomers 1 and 2 with the same method.
Supramolecular structure of 1.
There are various supramolecular structures of 1 that can be considered depending on the number of hydrogen-bonded units that one wishes to include in the simulation. We chose to investigate structures with up to six steroid molecules (monomer through hexamer) in the hydrogen-bonded polymer chain for computational efficiency. In order to illustrate some of the key outcomes of these simulations concisely, data obtained for the trimer are discussed in more detail below since it is the smallest unit of the supramolecular structure of 1 that contains a fully representative set of hydrogen bonds between the left and right sides of the zipper-like structure. As shown in Fig. 10, the overall agreement between the calculated and observed trimer structures is acceptable (root mean square difference <1.2 Å), particularly for the important region as far as molecular recognition is concerned, namely ring A of the steroid. (Similar agreement was observed for all fragments of the supramolecular structure of 1 up to the hexamer.) It is also evident from the simulations that the spiroketal ring system of the X-ray structure (rings E and F) is conformationally the most variable region of the molecule [Fig. 10(b)], since it differs appreciably in orientation from that of the AM1-simulated structure. Visual inspection of the geometries of the 5-membered ring E of the calculated and crystallographic structures suggests that this deviation mostly relates to the fact that the experimental structure exhibits a twisted half chair ring conformation as opposed to a more nearly flat chair-like conformation in the calculated structure.
 |
| | Fig. 10 Least-squares fit comparing three hydrogen-bonded molecules of the X-ray structure of 1 with the AM1-simulated “supramolecular” structure of the steroid. In (a) all non-H atoms of the three steroid skeletons were used in the fit. In (b) steroid rings A-D of one selected (top right) molecule of the trimer were used for fitting. (H atoms omitted for clarity.) | |
The calculated geometry of the hydrogen-bonded 10-membered macroring has been more closely compared in Fig. 11, which also shows the partial charge distributions and calculated hydrogen bond distances for key functional groups involved in the intermolecular interactions. Relative to the gas-phase monomeric structure, formation of the hydrogen-bonding network favoured an increase in charge polarization for the hydroxy groups appended to ring A of 1. Specifically, the oxygen atom natural43 charges become more negative in magnitude by some 0.02 e units (−0.331(1) e in the monomer vs. −0.352(4) e in the trimer). This reflects a counterbalance to the loss of electron density for the hydroxy hydrogen atoms upon hydrogen bond formation (+0.200(1) e in the monomer vs. +0.228(3) e in the trimer). The charge complementarity between the oxygen atoms (partially negative) and the hydrogen atoms (partially positive) involved in the hydrogen bond network is also immediately apparent and strongly suggests that the molecular recognition or self-assembly process for 1 is probably mostly electrostatically driven, at least in the gas and solid states.
 |
| | Fig. 11 Natural partial charge43 distributions for the hydroxy groups of the AM1-calculated hydrogen-bonded trimer unit of the extended or “supramolecular” structure of 1. | |
There is always the question of how well a semi-empirical SCF MO method actually does at calculating key bond distances and angles in the structure of interest. Indeed, the literature reports both pros39,40 and cons44,45 to the use of AM1 for simulating hydrogen-bonded systems. However, given the sheer size of the system presently under consideration, AM1 is clearly the only viable quantum chemical approach to use for systems as large as hexamers of 1. Moreover, we found that higher-level DFT methods (an ONIOM34 calculation using two layers, B3LYP/6-31+G**//AM1) were not only frightfully slow, but afforded no major improvement in geometric accuracy over a simple AM1 simulation (see supporting information for additional information†). Notwithstanding the limitations of a MO model which treats only the valence electrons of the system then, the calculated OH⋯O distances average 2.14(2) Å for the fragment shown in Fig. 11 and compare favourably with the mean value of 2.11(11) Å for the X-ray structure of 1. However, the experimental hydrogen bond distances clearly come in two sets and this distinction is not evident in the calculated hydrogen bond distances. Specifically, the hydrogen bonds between the left and right halves of the experimental zipper structure are short (2.00 Å), while those between steroid molecules within either the left or the right hand stacks of the zipper structure are long (2.22 Å). Interestingly, this limitation was also observed at the B3LYP/6-31+G** level of theory (supporting information†), where all hydrogen bonds were calculated to be 1.90 Å in length. Given the satisfactory fits between the calculated and observed structures of 1 (monomer through hexamer), including the intermolecular hydrogen bonds, use of the AM1 method is not only computationally efficient, but entirely valid in this system.
Self-association energetics.
An inviting question was whether or not AM1 could be used to further delineate self-assembly in this system. We elected to calculate the in vacuo association energies for the dimeric through hexameric forms of 1 in an effort to answer this question. We first calculated the structure of the (steroid)n unit, where n = 2–6, using the X-ray coordinates as input. We then moved each of the n molecules apart to distances >10 Å and recalculated the geometries of the non-interacting molecules. The enthalpy of association, ΔHassoc, was then calculated from eqn (1), where ΔHf (n-mer) is the heat of formation at 298 K of the n-mer (e.g., hexamer with n = 6) and ΔHf# is the heat of formation of the non-interacting system (e.g., hexamer with six non-interacting steroid units).| | | ΔHassoc = ΔHf (n-mer) −
ΔHf# | (1) |
In effect, this is equivalent to a counterpoise correction46 for the basis set superposition error that typically affects the calculated binding energies of dimers and n-mers in all-electron basis set simulation methods. This exercise proved worthwhile and yielded some interesting data. In Fig. 12 we have plotted the enthalpy of association per steroid unit, i.e., ΔHassoc/n as a function of n. The data were then fit to a simple exponential decay function with ordinate and abscissa offsets, eqn (2), where k is the “rate” constant, A is the total span of ΔHassoc/n, and B is the value of ΔHassoc/n when n is infinitely large.| | | ΔHassoc/n = A e(−k(n−1))
+
B | (2) |
The “rate” constant in this case is best thought of as the parameter gauging the rate of change of ΔHassoc/n with n. The fit of eqn (2) to the data gave A = 5.78(3) kcal mol−1 steroid−1, B = −5.77(1) kcal mol−1 steroid−1, and k = 0.416(3) kcal mol−1 steroid−2. The graph may interpreted as reflecting a consistently favourable enthalpy of association for the self-assembly of 1 into a high-order one-dimensional system in the gas phase. Thus, the enthalpy of association is zero for a monomer. For the formation of the dimer, ΔHassoc/n = −1.904 kcal mol−1 steroid−1 and this value increases in magnitude at a decreasing rate as n
→
∞. The limiting value of the enthalpy of association per steroid unit is the value of B in eqn (2). It is important to note that the enthalpy values are all negative and thus the process is calculated to be exothermic in the gas phase. The value of ΔS for each step in the self-assembly of the one-dimensional hydrogen-bonded polymer cannot, unfortunately, be gleaned from the present simulation data. However, the values are likely to be negative for an associative process. In solution, the opposite scenario may well prevail for ΔS as self-association is likely to shed several hydrogen-bonded solvent molecules from each pair of interacting steroids. Finally, although it is clear that cooperativity in the ΔHassoc/n values exists for this system, and ΔH cooperativity is generally recognized as being pivotal for the formation of supramolecular assemblies47 including those reliant on hydrogen-bonding interconnects,48 there is a limiting value of ΔHassoc/n. Whether this is mirrored by the ΔG values for each associative step in this system remains an open and interesting question, especially since solvation should play a significant role in the self-assembly process in solution. However, unlike synthetic oligomeric zippers which can bind in increasingly complex ways as the molecular weight of the system gets larger,17 compound 1, being non-oligomeric, most likely self-assembles more simply such that a plot of ΔGassoc/n vs. n for the system should exhibit a similar shape to Fig. 12. For the system at hand then, the situation is akin to a cooperative ligand binding process for which K1 < K2 < K3⋯ < Kn. The only distinction here is that we have calculated gas-phase ΔH values (rather than association constants) such that ΔHassoc/n < ΔHassoc/(n
+ 1) < ΔHassoc/(n
+ 2) … < ΔHassoc/(n
+
m), where n
≥ 2, for compound 1.
 |
| | Fig. 12 Plot of the AM1-calculated in vacuo enthalpy of association as a function of the number of steroid units making up the one-dimensional hydrogen-bonded polymer for 1. | |
Relative energies of 1 and 2.
It is remarkable that the conformational architecture of steroid 1 turns out to be just right for self-assembly and the formation of an elegant supramolecular structure. Stereoisomer 2, on the other hand, is flatter and does not have the correct molecular recognition motif or synthon for self-association. Importantly, it is not so much the number of hydroxy groups on ring A of the steroid that matters most, but the overall conformation of the molecule in the region of ring A of the steroid skeleton. This is evidenced by the fact that 1β,2β,3β,4β,5β,7α-hexahydroxy-spirost-25(27)-en-6-one38 has the required conformation of ring A as well as the hydrogen bond donor and acceptor sites (hydroxy groups) to self-assemble into a one-dimensional supramolecular structure. Given the profound consequences that the stereochemistry of the compound has on its ability to self-assemble into a supramolecular structure, it is of some interest to determine which stereoisomer (1 or 2) is lower in energy. The AM1-calculated heat of formation for the monomer of 1 was −256.03 kcal mol−1; that for 2 was lower by 2.20 kcal mol−1 (−258.23 kcal mol−1). The origin of the lower energy for 2 may be traced to its flatter structure (Fig. 13). More specifically, the out-of-plane or folded projection of ring A of the steroid structure in 1 leads to several short intramolecular contacts and thus an increase in steric strain. The two key strain-increasing interactions H-4⋯H-9 and H-2B⋯H-9 measure 2.13 and 2.12 Å in the AM1-calculated structure of 1 (2.24 and 2.19 Å, respectively, in the X-ray structure). The flatter structure for 2 also leads to a smaller dipole moment of 0.78 D relative to that for 1 (2.80 D). The two stereoisomers are thus very different in their physical behaviour and properties. Of course, they are also derived from completely different plant species. Since 2 is a potent cytotoxic compound,11 one wonders whether the conformational and physical differences between 1 and 2 favour reduced or enhanced cytotoxicity in the case of 1? Only future biological testing on 1 can reveal the answer to this question.
 |
| | Fig. 13 CPK (space-fill) models of the structures of 1 and 2 viewed approximately down the C-10–C-19 bond vector. The folded down projection for ring A of 1 leads to two key short intramolecular H⋯H contacts (H-4⋯H-9 and H-2B⋯H-9). | |
IR frequency analysis.
Finally, since we carried out frequency calculations on the monomer through hexamer structures of 1 to confirm that the calculated structures were true minima, the effect of hydrogen-bonding on the hydroxy group vibrational modes in this system may be assessed. The mean O–H stretching vibration for the monomeric form of 1 had an AM1-calculated in vacuo energy of 3500(2) cm−1. This was reduced to 3459(5) cm−1 for the hydrogen-bonded hydroxy groups in the hexamer of 1, consistent with a slight reduction of the O–H bond order due to polarization as a result of hydrogen bond formation. This energy perturbation was mirrored by a change in the mean calculated O–H bond lengths for 1. In the monomer, the O–H bond distances average 0.964(0) Å, while hydrogen bonding in the hexamer slightly reduces the bond order and lengthens the mean O–H distance to 0.968(1) Å. Incidentally, the mean O–H distance from the X-ray structure of 1 measured 0.80(8) Å. Given the experimental uncertainty of locating the hydrogen atoms accurately with X-ray diffraction methods and the thermal libration of these atoms at room temperature, the AM1-calculated values are not unreasonable. As always, it is appropriate to compare the calculated stretching frequencies for key functional groups to experimental data if available. In the present system, the experimental stretching frequency of the O–H bonds was 3428 cm−1 for the solid sample in a KBr matrix. The AM1-calculated O–H stretching frequency of 3459(5) cm−1 for the supramolecular structure of 1 is thus in good agreement with experiment.
Conclusions
We have isolated a novel crystalline steroidal sapogenin (25R)-5β-spirostane-1β,3α-diol (1) from the bulbs of a southern African species of bushveld lily, namely Ornithogalum tenuifolium. The crystal and molecular structure of this compound is remarkable in that it is the first properly delineated example of a non-oligomeric supramolecular zipper based on a steroid motif or synthon. Quantum chemical simulations at the AM1 level of theory adequately reproduce the supramolecular structure of 1 and provide substantial insights into the structure and bonding of the monomeric and supramolecular structures of the system. The simulations show that self-assembly of 1 in the gas phase is characterized by cooperativity in the stepwise heats of formation. Moreover, self-assembly to form the supramolecular zipper structure is clearly electrostatically controlled in this system, as judged by the charge complementarity of the donor and acceptor hydroxy groups of the hydrogen-bonded interacting units. Finally, analysis of the stereochemistry of 1 reveals that in addition to the 1β/3α hydroxy group configuration, the 5β/10β configuration of the steroid skeleton (ring A) is an absolutely essential design requirement for the formation of the supramolecular zipper structure. The structural beauty of this naturally occurring zipper should, we hope, inspire the design of synthetic ladder-like supramolecular systems with similar or equivalent synthons.
Acknowledgements
We are grateful to Mr Martin Watson for running NMR spectra, Mr Bret Parel for acquiring mass spectra, and Mr Raj Somaru for technical assistance. Mrs Gael Whiteley of Ashburton kindly permitted collection of plant materials on her property. The staff of the Mary Gunn Library in Pretoria are thanked for facilitating access to literature. This research was funded by the National Research Foundation (NRF, Pretoria) and the University of KwaZulu-Natal.
References
- B. Stedje and I. Nordal, Nord. J. Bot., 1984, 4, 749 Search PubMed.
- B. Stedje, Plant Syst. Evol., 1984, 166, 79 Search PubMed.
-
T. S. Kellerman, J. A. W. Koetzer and T. W. Naudé, Plant Poisonings and Mycotoxicoses of Livestock in Southern Africa, Oxford University Press, Cape Town, 1988 Search PubMed.
- T. S. Pohl, N. R. Crouch and D. A. Mulholland, Curr. Org. Chem., 2000, 4, 1287 CrossRef CAS.
-
B.-E. van Wyk, F. van Heerden and B. van Oudtshoorn, Poisonous Plants of South Africa, Briza Publications, Pretoria, 2002, pp. 160–161 Search PubMed.
- T. Hirano, K. Oka, Y. Mimaki, M. Kuroda and Y. Sashida, Life Sci., 1996, 58, 789 CrossRef CAS.
- M. Kuroda, Y. Mimaki and Y. Sashida, Phytochemistry, 1999, 52, 435 CrossRef CAS.
- U. Ghannamy, B. Kopp, W. Robien and W. Kubelka, Planta Med., 1987, 53, 172 CrossRef CAS.
- R. Ferth, A. Baumann, W. Robien and B. Kopp, Z. Naturforsch., B: Chem. Sci., 1992, 47b, 1444.
- S. B. Mahato, A. N. Ganguly and N. P. Sahu, Phytochemistry, 1982, 21, 959 CrossRef CAS.
- A. D. Patil, P. W. Baures, D. S. Eggleston, L. Faucette, M. E. Hemling, J. W. Westley and R. K. Johnson, J. Nat. Prod., 1993, 56, 1451 CrossRef CAS.
- A. P. Bisson, F. J. Carver, C. A. Hunter and J. P. Waltho, J. Am. Chem. Soc., 1994, 116, 10292 CrossRef CAS.
- R. S. Hodges and C. M. Kay, Biochemistry, 1994, 33, 15501 CrossRef CAS.
- R. Kramer, J. M. Lehn and A. Marquisrigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CAS.
- M. Kubota and A. Ono, Tetrahedron Lett., 2004, 45, 5755 CrossRef CAS.
- A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856 CrossRef CAS.
- A. P. Bisson and C. A. Hunter, Chem. Commun., 1996, 15, 1723 RSC.
- J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, Chem. Commun., 2005, 1258 RSC.
- X.-M. Chen and G.-F. Liu, Chem.–Eur. J., 2002, 8, 4811 CrossRef CAS.
- M. J. Plater, S. Aiken and G. Bourhill, Tetrahedron Lett., 2001, 42, 2225 CrossRef CAS.
- J. Granifo, M. T. Garland and R. Baggio, Inorg. Chem. Commun., 2004, 7, 77 CrossRef CAS.
- M. Barboiu, E. Petit and G. Vaughan, Chem.–Eur. J., 2004, 10, 2263 CrossRef CAS.
- S. Keinan, M. A. Ratner and T. J. Marks, Chem. Phys. Lett., 2004, 392, 291 CrossRef CAS.
- V. del Amo, L. Siracusa, T. Markidis, B. Baragana, K. M. Bhattarai, M. Galobardes, G. Naredo, M. N. Perez-Payan and A. P. Davis, Org. Biomol. Chem., 2004, 2, 3320 RSC.
- A. P. Davis and J.-B. Joos, Coord. Chem. Rev., 2003, 240, 143 CAS.
- Z. Lotowski, Wiadomosci Chemiczne, 2003, 57, 1093 Search PubMed.
- C. A. La Mesa, Recent Res. Dev. Surf. Colloids, 2004, 1, 97 Search PubMed.
- K. Kato, M. Sugahara, N. Tohnai, K. Sada and M. Miyata, Cryst. Growth Des., 2004, 4, 263 CrossRef CAS.
-
G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed;
G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef.
-
R
1 = Σ||Fo| − |Fc||/Σ|Fo| and wR2 = {Σ[w(Fo2
−
Fc2)2]/Σ[wFo4]}½. R factors R1 are based on F, with F set to zero for negative F2. The criterion of F2 > 2σ(F2) was used only for calculating R1. R factors based on F2 (wR2) are statistically about twice as large as those based on F.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. P. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef.
-
Spartan ‘04, Wavefunction, Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612 USA, 2004, http://www.wavefun.com Search PubMed.
- F. Maseras and K. Morokuma, J. Comput. Chem., 1995, 16, 1170 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
-
P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, Wiley, Chichester, 2002, 2nd edn, pp. 150–232 Search PubMed.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, A39, 876 CrossRef CAS.
- K. Miyahara, F. Kumamoto and T. Kawasaki, Tetrahedron Lett., 1980, 21, 83 CrossRef CAS.
-
(a) O. Q. Munro and L. Mariah, Acta Crystallogr., Sect. B: Struct. Sci., 2004, B60, 598 CrossRef CAS;
(b) O. Q. Munro and G. Camp, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, C59, o672 CrossRef CAS.
-
(a) J. J. Novoa and M. H. Whangbo, J. Am. Chem. Soc., 1991, 113, 9017 CrossRef CAS;
(b) A. C. Testa, Spectrosc. Lett., 1999, 32, 819 CrossRef CAS;
(c) A. Kukovecz and I. Palinko, J. Mol. Struct., 1999, 482–483, 463 CrossRef CAS;
(d) F. Orsini and F. Pelizzoni, THEOCHEM, 1992, 94, 65 CrossRef CAS;
(e) J. J. Dannenberg and E. M. Evleth, Int. J. Quantum Chem., 1992, 44, 869 CAS.
- L. K. Vinson and J. J. Dannenberg, J. Am. Chem. Soc., 1989, 111, 2777 CrossRef CAS.
- R. Villar, M. J. Gil, J. I. Garcia and V. Martinez-Merino, J. Comput. Chem., 2005, 26, 1347 CrossRef CAS.
-
(a) J. E. Carpenter and F. Weinhold, J. Mol. Struct. (THEOCHEM), 1988, 169, 41 CrossRef;
(b) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS.
- M. Bures and J. Bezus, Collect. Czech. Chem. Commun., 1994, 59, 1251 CrossRef CAS.
- G. Buemi, F. Zuccarello and A. Raudino, THEOCHEM, 1988, 41, 379 CrossRef.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024 CrossRef CAS.
- M. S. Searle, M. S. Westwell and D. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1995, 141 RSC.
- I. Goldberg, Chem. Commun., 2005, 1243 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3 illustrating the supramolecular zipper structure for β,2β,3β,4β,5β,7α-hexahydroxy-spirost-25(27)-en-6-one, the DFT/AM1-calculated structure of 1, and selected H-bonding distances from the X-ray structure of 1, respectively. See DOI: 10.1039/b513734a |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.