Molecular Modelling†
Xavier Barril‡a and Robert Solivab
aSenior Scientist, Vernalis (R&D), Granta Park, Abington, Cambridge, UK CB1 6GB
bMolecular Modelling, Grup Uriach, Polígon Industrial Riera de Caldes, Av. Camí Reial 51-57, 08184, Palau-solità i Plegamans (Barcelona), Spain
First published on 19th October 2006
1 Introduction
The enormous pressure that the pharmaceutical and biotech companies are facing, has created the need to apply all available techniques to decrease attrition rates, costs and the time to market. Currently, one of the most widely applied techniques in drug discovery is computational chemistry and molecular modelling. This branch of science is centred on applying the fundamental laws of physics and chemistry to the study of molecules. In the case of drug discovery, the molecules under study are those directly or indirectly involved in human disease. The ultimate aim is to create models and simulations, which can help in the different stages of a discovery pipeline by predicting, rationalizing and estimating the properties of molecules and their interactions, thereby allowing a more rational approach to drug development.1 This whole trend is now seen both as an alternative and a complement to the more “brute-force” approach exemplified by the application of combinatorial chemistry and high-throughput screening (HTS).The fundamental factor allowing the widespread use of molecular modelling is the central paradigm of today's drug discovery, the one-disease one-target concept and its implementation. Within this paradigm, a certain human condition is associated with the role played by a particular macromolecule, whose action can be modulated with a small organic molecule in order to achieve a therapeutic effect. With this perspective, drugs are developed in a sequential way. First, a macromolecular target to treat the pathology under study must be found, a process termed as target finding. Then, the search for small molecule binders (hits) for that particular target begins, the so-called hit finding stage. Once found, these binders must be optimized in order to achieve better in vitro activity, selectivity, pharmacodynamic and pharmacokinetic properties, the stage termed as hit to lead. Then, the lead must be optimized in a series of in vivo studies, the stage of lead optimization. Only when the lead has been optimized and tested in several animal models can the project then progress to human clinical trials. Computational chemistry and molecular modelling methods have become central features of all these pre-clinical research stages of the drug-discovery process.
When applied to the study of drugs and their receptors, molecular modelling techniques are generally divided into two broad categories. Ligand-based modelling consists of a series of techniques used for creating models and predictions based solely on the structure of the small organic compounds. In contrast, structure-based drug design (SBDD)2,3 exploits the knowledge of the 3D structure of one or more biological receptors (targets, the ones sought to modulate and anti-targets, the ones sought not to interfere with) and/or their macromolecular ligands. These two broad categories are very often applied in a myriad of different combinations, so the frontier separating them is not clear-cut.
Molecular modelling as applied to SBDD has undergone a dramatic change over the last two decades. At first, the simulation of biochemical systems and their interactions was a nearly unfeasible task. The targeted macromolecules were treated in a very simplified way because of the great amounts of computation required. Often only a portion of the whole system could be dealt with, solvent effects were rarely taken into account, and the simulation of complex formation could only be carried out for a small number of molecules. This picture has changed dramatically in the last decade mainly due to two factors. First, as Moore's law stated in 1965, the number of transistors on a given chip has been doubling approximately every 2 years, with the subsequent impact on computer power. This has allowed an increase in the size of system that can be studied, the degree of accuracy of the models and the number of interactions feasible to calculate on a reasonable time scale. Second, there has been incredible progress in the experimental techniques that the different modelling tools rely on. X-ray crystallography and nuclear magnetic resonance (NMR) have been developed to a level where they are now applied routinely, which has had a tremendous impact on the number of experimentally determined molecular structures available. The number of both small molecules and macromolecules deposited in the Cambridge structural database4 and the protein data bank (PDB),5 respectively have increased dramatically. This wealth of experimental information has fuelled the refinement and application of many modelling tools, from force-field and scoring function development to homology modelling. The progress in the reliability of prediction, applicability of the different techniques and higher throughput capacity has enabled the application of structure-based molecular modelling in many phases of drug discovery, as will be reviewed below.
First, a brief outline of all the methods is presented, with references to the standard publications in each field. Then, a number of different applications are discussed and structured according to the usual progress of a drug-discovery project.
2 Methods
2.1 Quantum Chemistry Methods
Quantum chemistry is the application of quantum mechanics (QM) to problems in chemistry. QM provides the most rigorous and physically meaningful description of molecular systems, most noticeably, the electrons are explicitly considered. In its purest form, QM is used to solve the wavefunction of molecular systems without any prior knowledge of the system or need for empirically derived parameters; hence its designation as ab initio methods. Ab initio methods have to rely on a series of approximations to provide a solution to Schroëdinger's equation and to speedup calculations, but the level of theory can be chosen to provide the best trade-off between quality of results and computational cost for each specific application. As ab initio methods are computationally very demanding and become prohibitive for relatively small systems (currently a few tens of atoms), semi-empirical approximations were introduced, which make use of experimentally determined data to avoid the calculation of certain terms, particularly two-electron integrals. Density-functional theory (DFT) provides a third class of QM methods; these methods are based on the fact that, for a system in its lowest energy state, there exists a one-to-one mapping between the electron density and the wavefunction of a system. As DFT methods give direct access to electron density, they are much faster than wavefunction-based methods of similar quality, although it is also true that they rely on some adjustable parameters. Each QM method has its own benefits and weaknesses, as well as a myriad of levels of theory to choose from. Moreover, a number of methods have been described to enable the extension of QM studies to large molecular systems.6 These include further approximations that enable a better scaling of the computational cost with the number of atoms or the use of different levels of theory for different subsets of the system. It is not our intention to compare the many available options, as this has already been done in the literature.7 Our aim here is just to outline the most common applications of QM methods to SBDD; hence, the use of QM methods in ligand-based applications8 will not be discussed. First, we will introduce two types of applications that are well established, where QM provides a clear advantage over parametric methods, then we will present an emerging application that may become increasingly useful in the near future.2.2 Parametric Methods
The forces that govern the structure of molecules, both in gas and condensed phase, are generally well known and can be used to rationalize and predict the behaviour of molecules. This enables us to partition a whole system into smaller units that interact with each other following certain rules. A very successful and widely applied approach in molecular simulation consists of using a set of molecules to derive rules and parameters of more general applicability. These are known as parametric methods, which we have further divided into three major classes based on the approach used to obtain the parameters and the scope of the model. The first class is force-fields, which aim at providing a complete description of the system. The other two classes of parametric methods presented here are only concerned with the process of molecular recognition.As we will show in the next sections, parametric methods play a major role in computational simulation of biological systems, but equally important for drug discovery is the fact that chemists, in general, and molecular modellers, in particular, have a good working knowledge of the physical forces behind molecular recognition (mainly electrostatic, van der Waals, hydrogen bond, π-aromatic and hydrophobic).22 This enables them to assess automatic predictions, generate hypotheses and make timely decisions.
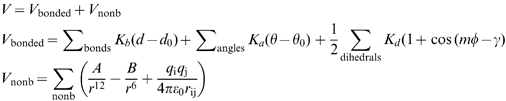 | (1) |
 | (2) |
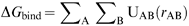 | (3) |
2.3 Solvation
Water has a strong influence in all biochemical phenomena, and specifically plays a central role in molecular recognition. It profoundly alters properties such as the dipole moment and the molecular electrostatic potential, affects the conformational and tautomeric preferences of both small molecules and their macromolecular targets, and governs the hydrophobic effect, by which non-polar molecules (or the non-polar parts) tend to aggregate to reduce the solvent exposed hydrophobic surface therefore minimizing the loss of entropy associated with the ordering of water molecules.44,45 Because ligand–receptor non-covalent association takes place in an aqueous environment, the role of water must be taken into account in order to qualitatively understand this process and also quantitatively determine its free energy.The effect of water can be introduced in different ways depending on the representation of the system (solute) under study. When the system is represented as a quantum mechanical particle, the effect of solvent can be introduced explicitly or implicitly. However, because of computational limitations, the former is seldom used and virtually always the latter is preferred. The most popular methods to account for solvent effects are the continuum methods, where these effects are introduced as a perturbational operator representing the solvent reaction field. Because the solute wavefunction and the reaction field depend on each other, they have to be solved using a self-consistent procedure. Many quantum mechanical continuum methods have been developed, varying basically on the definition of the solute/solvent boundary and the description of the reaction field and the solute charge distribution (see reviews by Cramer and Trular45 and Orozco and Luque44).
When the system is described by a classical model based on a force-field, the simplest way of taking into account the effect of water is representing it discretely. If velocities are given to such a system, as in a molecular dynamics (MD) trajectory, a solvated ensemble of the molecular system is obtained, where the atomic coordinates of both solvent and solute are produced. This provides an insight into the differential solvation of certain parts of the solute, generates radial distribution functions, and allows an assessment of whether water molecules bridge ligand and receptor, etc. When coupled to statistical mechanics, it can also be used to calculate differential free energies of solvation between different solutes with the help of thermodynamic cycles.46 However, one of the most serious drawbacks of the explicit water treatment is its computational expense.
Less computationally demanding methods have been developed to account for solvent effect on a classical system. Among the most popular are the classical continuum electrostatic methods, where the solvent is treated as a continuum environment. The solute molecule is placed in a cavity, whose permittivity usually ranges from one to eight, and surrounded by a polarizable continuum medium with a defined solvent dielectric constant. The main difference among the classical continuum electrostatic methods is in the definition of the solute/solvent boundary and how the solute–solvent electrostatic interaction energy is calculated. The most popular methods are the Poisson, or Poisson–Boltzmann (PB)47 if the effect of counterions is also taken into account, and the Generalized Born (GB) model.48
Other implicit water-treatment methods for a classical system are those derived empirically. Although less rigorous, they are still used because of their low computational cost. A first group of methods makes use of parameters for modelling screening of electrostatic interactions by water, replacing the macroscopic permittivity by a distance-dependent dielectric function. In the simplest models the latter can change linearly,49 but more complex models where it changes exponentially have also been developed.50 A second group of empirical methods is based on the solvent accessible surface area (SASA). In them, it is assumed that solvation free energy can be calculated by addition of the contribution of each atom or group of atoms. Each atom type is given a solvation parameter obtained by a fitting procedure and the contribution of each atom is based on its SASA.51
2.4 Sampling Algorithms
Once the chemical system under study has been defined and any necessary parameters have been obtained, one can proceed to run calculations on it. Usually flexible molecules have a complicated potential energy surface, with several minima and saddle points, which are a function of the nuclear coordinates. Especially interesting are the configurations that correspond to minima in the potential hypersurface as these are stable states of the system. The identification of these minima will generally consist of two steps: global exploration and local minimization. Although it is theoretically possible to systematically explore each degree of freedom, in practice, this can only be done in a reasonable time frame for very small systems; hence, stochastic methods are most commonly used in SBDD. Evolutionary computational techniques such as genetic algorithms (GA) are particularly widespread. These methods start by generating a random collection of candidate solutions whose fitness is evaluated, the best individuals are then stochastically selected and mutated or recombined to obtain a new population. This process is then repeated until a certain convergence criterion is achieved.For local optimizations, there are several minimization algorithms that search the nearest minimum in the potential surface. These can be broadly classified into two groups, those that do not use the derivatives of the potential energy with respect to the coordinates, such as the simplex method, and those that do, such as the steepest descent and conjugate gradient methods. The latter operate in an iterative procedure: (1) potential energy evaluation for a given configuration; (2) determination of the first (gradient) and second derivative of the energy with respect to the coordinates; (3) generation of a new set of coordinates in the direction of the minimum and (4) energy evaluation for the new set of coordinates. If the energy is converged the calculation stops, if not, the process is repeated again. They are often used in SBDD in several contexts, such as the initial refinement of a protein structure obtained by experimental methods or to relax the geometry and eliminate unfavourable contacts of a ligand–receptor complex.
However, the biggest limitation of a minimized molecular system is its static character. Molecules vibrate and constantly change conformation, overcoming potential energy barriers and populating an ensemble of microstates, which are globally responsible for the properties of the system. To generate such an ensemble different algorithms can be used, the most widely used of which are MD and Monte Carlo (MC).
MD is based on the application of Newton's equations of motion to describe the evolution of a classical system along time. When the system is defined with a force field, it is feasible to calculate the forces acting upon each particle (atom) by obtaining the gradient of the potential energy. Once the force on each particle is known, its acceleration can be derived, which by integration determines the velocity and position after a time increment. After generating the new set of coordinates the steps can be repeated again in an iterative fashion. The result is a set of structures that represent the evolution of the system along a time path. The only prerequisites for the calculation of a trajectory are a set of initial coordinates, as the initial velocities are randomly generated. Calculation of a trajectory develops a dynamic view of the molecular system to have an ensemble that can later on be used to calculate free energies of binding for a ligand–receptor complex52 (see below).
Restrained MD is a special case used extensively in the refinement of macromolecular structures with data derived from experimental techniques. In restrained MD, additional terms are added, which complement the original potential energy definition of the system. These terms do not have a chemical sense, but penalize those conformations that do not respect the experimental data. Thus, for instance, an NMR experiment can provide interproton distances that can be added as an extra retraint function to the original force-field in a similar way as a stretching function defines a bond between two covalently attached atoms. In this way, only the structures complying with the combined potential are allowed. This strategy is nowadays routinely used in the refinement of structures by NMR. In a similar way, distance and dihedral restraints can be derived from the alignment of two protein sequences and also added to the original force-field definition; this is a central strategy used in homology modelling, which will be presented below.
While in MD, there is a time dependency, MC also generates an ensemble of states, but in a stochastic fashion. Starting from a given conformation, a perturbation is introduced to the system by modifying a random degree of freedom by a random small quantity. Then a ratio of probabilities is computed for the trial and original configurations and, from this quantity, a decision is made to accept or reject the trial configuration. Usually, in molecular simulations, the metropolis criterion is used to decide whether the trial configuration is accepted or rejected. First, the energy change (ΔU) of the system is measured on introduction of a perturbation. If the trial configuration has lower energy than the original, it is accepted; otherwise a function of probability (ω) is calculated,
| ω = exp(−βΔU |
MD and MC as sampling algorithms are crucial in the application of statistical mechanics methods, which are considered as the most rigorous classical methods for the determination of changes in free energy of binding for a ligand–receptor complex.46 The two most frequently used methods are free energy perturbation (FEP) and thermodynamic integration (TI). These two techniques estimate free energy changes or ΔΔG between two inhibitors, thanks to the use of thermodynamic cycles.46 A perturbation is used to smoothly convert one ligand A to another ligand B with the help of a coupling parameter both in solvent and within the protein environment (see Figure 1). Because the ΔGbinding A and ΔGbinding B cannot be obtained in a simulation, the differential free energy of binding between both molecules must be obtained by solving the other two terms in the cycle, ΔG1 and ΔG2, which can be calculated in an MD or MC simulation. By subtracting both of these values, one can have an estimation of the changes in affinity between molecules A and B for the same receptor.
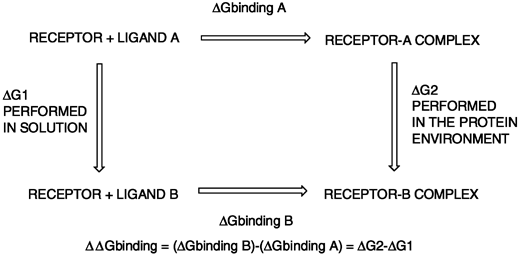 | ||
| Fig. 1 Thermodynamic cycle used to compute differences in free energy of binding between two inhibitors, A and B | ||
3 Applications
3.1 Target Evaluation
Computational chemistry typically becomes associated with a drug-discovery project at the hit-identification stage, but it is important for the modeller to understand and make a thorough assessment of the tractability of the chosen biological target, as well as the quality of the structural information. In extreme cases this may lead to the suggestion of abandoning a target or to pursue a non-structure-based strategy. The real goal of this stage is, nevertheless, to make a rational and optimal use of the available information, to identify possible pitfalls and to start a project with the greatest chance of success.Although empirical, all these limits to chemical space come from the selection pressure enforced by the functioning of the human body, and restrict the number of macromolecules that can be treated effectively with the traditional medicinal chemistry approach. Thus, although DNA could in principle be the perfect target, modulation with high potency and selectivity of a unique stretch of double stranded DNA would require a chemical agent well outside drug-like chemical space. The sheer size and number of hydrogen bond donors and acceptors that a potential drug would need to bind selectively and potently to the minor groove of a DNA duplex would exceed by far the drug-like property limits. Therefore, among the four types of macromolecules found in the human body, carbohydrates, lipids, nucleic acids and proteins, only the latter usually make druggable targets. However, not all proteins directly linked to a specific disease are druggable. Indeed, an analysis of marketed small molecule drug targets reveals that more than three quarters are enzymes and membrane receptors, proteins which usually bind small molecule endogenous ligands in a well-defined and secluded cavity,56 thus making it possible for a small chemical agent to compete with them and exert a therapeutic effect. This implies that a good portion of the human proteome might not be druggable even if closely related to human pathology.
The most relevant application of structure-based molecular modelling at the initial stage is finding the important areas of the receptors that make good binding spots for a chemical agent (see Table 1). Usually this involves detecting the active site for enzymes, membrane and nuclear receptors, where their endogenous ligands bind, as well as finding the crucial epitopes and clefts involved in many of the protein–protein interaction partners known from proteomics studies.57 Thus, for protein–protein contacts it involves finding those particular “hotspots” (in the usually huge contact surface), which contribute the most to the non-covalent association of both proteins.
| Parameter | Ideal value | Explanation |
|---|---|---|
| Shape | Deep or enclosed | The ligand–receptor interaction energy roughly correlates with the surface-contact area. Small molecules require enveloping cavities to attain sufficient binding affinity. |
| Size | Fits ligands of 300–600 Da | Small cavities may not be able to accommodate drug-like molecules. Very large cavities may not provide sufficient surface-contact area. |
| Chemical character | Mix of hydrophobic and hydrophilic. | Drug-like molecules present a balance between lipophilicity (low logP) and hydrophilicity (H-bond donor/acceptors, PSA). |
| Flexibility | Rigid | Binding to very flexible binding sites involves an entropic penalty. Flexibility of the receptor is a difficult property to model. |
For active site detection, many algorithms have been devised to highlight the possible small molecule binding points. One of the earliest attempts was developed by Goodford58 and relies on the calculation of interaction energies. Typically, the protein structure is immersed into a cubic grid, and the interaction potential between receptor and a number of different probe particles placed at each grid point is calculated (probes can be charged atoms, a water molecule, or a hydrophobic particle, etc.). The result can be visualized graphically in a series of contours of different energy values, which give a “feel” of where certain chemical groups better interact with the receptor. Because active sites are small, sterically limited and usually hydrophobic cavities, the contours help to highlight the energetically favourable sites. Alternatives to the interaction energy approach have been developed, which rely on purely geometric methods.59 An example is the Site Finder program implemented in molecular operating environment (MOE),60 which locates binding sites by first calculating empty spheres contacting four protein atoms on their boundary. The spheres that correspond to inaccessible sites of the protein, as well as those that are too exposed to solvent are eliminated, effectively leaving those that correspond to regions of tight atomic packing. The spheres are then classified as hydrophobic or hydrophilic and all those hydrophilic spheres not close to at least one hydrophobic sphere are eliminated. Finally, the spheres are clustered giving a collection of sites, which are ranked according to the number of hydrophobic contacts to the receptor. This simple technique can be efficiently used for active site detection. Parallel to this, other efforts are being directed at compiling structural information on all the active sites solved by experimental methods (e.g. the catalytic site atlas61). These databases are and will increasingly be very helpful for active site detection and even for catalytic function prediction, irrespective of the level of sequence identity of the proteins under study.
The detection of protein–protein interfaces is a great challenge for structure-based in silico techniques. In contrast to active sites, these interfaces are huge and shallow surfaces, barely distinguishable from other parts of the protein. The detection of the cluster of residues that contribute the most to binding (hot spot) within these interfaces is an even bigger challenge. A promising approach to detect the binding interface for non-obligate protein complexes has recently been reported where the interfaces are highlighted by predicting the optimal docking area (ODA) of a protein.62 The method identifies area patches with optimal docking desolvation energies using a simple accessible-surface-area (ASA) method, and it is reported to have an 80% success rate. Also recently, an extensive analysis of protein interfaces in the PDB has shown that binding interfaces can generally be detected by analysing structural (not necessarily sequential) conservation of certain aminoacids within protein families, specifically tryptophan, and less pronouncedly phenylalanine and methionine.63 These results are reinforced by an independent analysis of protein–protein pairs in which it was found that the core surface of the interface is enriched in tryptophan, tyrosine, methionine and phenylalanine residues.64 Although these methods discriminate binding interfaces from the rest of the protein, the detection of the epitopes within them that can be mimicked by a small molecule is very complicated, and will likely require a combination of in silico and experimental techniques, as has been the case in the development of VLA-4 integrin antagonists.65–67 Finally, it should be emphasized that antagonizing a protein–protein interaction is still perceived to be difficult and risky by the drug-discovery community.57 Usually the molecules showing promising in vitro activity tend to have molecular weight, hydrogen bond donor and acceptor counts on the limit of what is considered desirable, as can be seen for instance in the development of IL-2 and integrin antagonists.57,68
| No. of PDB entries | No. of unique ligands bound to proteins | |||
|---|---|---|---|---|
| All | Proteins | Protein-ligand complexes | ||
| X-ray | 26255 (85%) | 24301 (87%) | 9895 (99%) | 3362 (99%) |
| NMR | 4548 (15%) | 3776 (13%) | 132 (1%) | 73 (2%) |
| Total | 30803 | 28077 | 10027 | 3395 |
Even with the fast progress in high throughput protein crystallization,72 it will take some years before experimental structures are available for all proteins of potential pharmacological interest. However, the number of possible folds is significantly lower and structures currently available already represent most of the protein families,73 and computational methods to generate 3D models based on homology are increasingly accurate and can be used in SBDD. These will be presented next.
3.1.2.1 Considerations regarding the use of crystallographic structures. As the structure of a target underpins all computational SBDD methods, a good understanding of how the structure is obtained and its potential limitations is not only useful, but really necessary to avoid misinterpretation or loss of information. An excellent recent review has analysed in detail the use of crystallography in drug design and the limitations of the method.74 Here we will only outline the most important factors that one should consider, as listed in Table 3. The resolution of the data (expressed in Å) and the R- and R free-values provide an idea of the overall quality of the structure as a whole, whereas B factors (or temperature factors) indicate the reliability of individual atom positions. In the absence of electron density maps (which may sometimes be available75), these parameters are useful to get an idea of the experimental uncertainty associated with the coordinates. The PDB files often contain some information that can be easily overlooked, such as comments in the Remark section or multiple positions of certain atoms due to experimental double occupancies. For this reason a careful examination of the PDB files and additional information (e.g. accompanying papers) is strongly recommended. Depending on the quality of the crystal and the flexibility of the system, certain parts may not have observable electron density. If this affects the targeted site, the model will have to be completed prior to use. The number of electrons also plays an important role: while the coordinates of hydrogen atoms are almost invariably missing, the positions of heavy atoms (S, P, Cl, Br, etc.) can usually be assigned with much more confidence than first-row elements. Even when the electron density is very clear, it is not possible to distinguish between isoelectronic groups (see Table 3 for a list). For these atoms, the assignment will reflect, at best, the personal interpretation of the crystallographer based on the interactions that the group makes with its surroundings. Finally, crystallization itself is another potential source of error: as the protein is in an environment sensibly different from solution, the structure can be affected by packing or the crystallization solution; furthermore, all the proteins in the crystal are generally considered identical, meaning that heterogeneity is ignored.76
| Interpretation of PDB files | Resolution |
| R-value and R free-value | |
| B factors | |
| Partial occupancies | |
| Electron density-related issues | Isoelectronic groups: Proteins: Asn, Gln, His side chains Electronically symmetric ligands or ligand moieties Solvent/Ions: H2O = Na+ = NH4+ |
| Poor or lack of observed density: Side chains, particularly of flexible polar residues Mobile loops Domains Solvent molecules can be missing or confused with noise | |
| Hydrogen atoms not observed: Undetermined tautomeric and protonation states (e.g. His) Orientation of rotatable hydrogens (e.g. hydroxyl, water) | |
| If data is poor, the model will rely on dictionary parameters, which may be wrong for ligands.78 | |
| Crystal | Crystallization conditions: pH, salt concentrations, etc |
| Packing effects | |
| Heterogeneity neglected |
In an ideal scenario, the modeller will work closely with the structural scientists, enabling them to provide mutual feedback during the structure generation process and the modeller to receive much more than a set of coordinates. This is fundamental if high-throughput crystallography77 has to translate into high-output in drug discovery.
3.1.2.2 Homology modelling. The sequencing of entire genomes in recent years has produced many more sequences than the structural genomics initiatives can absorb. In addition, some proteins can be very difficult to crystallize, even if close homologues crystallize well, and may require substantial time and efforts to obtain a structure.79 A faster alternative is to generate a theoretical 3D model of the protein. Ab initio prediction of the structure of a protein is a formidable problem and, in spite of advances in protein folding studies,80 the most reliable method to predict the structure of a protein is by comparison with related proteins for which the structure is known, a technique known as comparative or homology modelling.81 A common first step in all homology modelling tools is to align the sequence that has to be modelled (query protein) with sequences of proteins of known structure (templates). The identity between these sequences provides a first indication of the reliability of the model; as a rule of thumb, if the level of identity is lower than 30% then the result of the alignment may be dubious and any model based on it questionable.82 The sequence alignment allows generation of an initial 3D model of the query sequence, which can be restrained to certain coordinates or dihedral values of the reference structure to produce a refined model by restrained MD.83
3.2 Hit Finding
The discovery of chemical entities with a desired biological activity is the first milestone in the quest to obtain a drug candidate. The activity of interest can only be identified with a relevant biological assay, but computational methods can be used to identify chemical structures with a greater probability of being active. This process is often referred to as VS or screening of virtual libraries. Docking is, by far, the structure-based method most commonly used in VS; de novo design provides an interesting alternative. Both methods, and particularly its application to VS, will be described here.Docking and de novo design methods are, nevertheless, seldom used as stand-alone tools for VS. This is because a wealth of experimental information from very different sources is available in a pharmaceutical research project and optimal results are obtained when several complementary methods are combined together. The integrative aspects of structure-based VS are, therefore, most important and merit particular attention. In this section, we will also consider two special cases of hit finding: template and scaffold hopping.
| Item | Options | Examples |
|---|---|---|
| Note: Please note that this is not intended as an exhaustive list and that docking packages may include several features of each class. | ||
| Scoring function | Force-field | DOCK |
| Empirical | LUDI, FlexX, AutoDock | |
| Semi-empirical | GOLD | |
| Knowledge based | — | |
| Search algorithm | Systematic | — |
| Stochastic: GA | GOLD, AutoDock | |
| Stochastic: MC | ICM, Glide | |
| Stochastic: Other | PRO_LEADS (Tabu) | |
| Deterministic (e.g. simplex minimizer) | — | |
| Ligand flexibility | None or implicit (precomputed conformers) | All |
| Incremental construction | FlexX, DOCK, Glide, Surflex | |
| Full flexibility | GOLD | |
| Receptor flexibility | None or implicit (multiple cavities) | All |
| Implicit (“soft” docking) | Most | |
| Terminal polar hydrogens | GOLD | |
| Water molecules | — | |
| Side chains | SLIDE | |
| Grid averages87,88 | DOCK, AutoDock | |
| Unified receptor description89,90 | FlexE, DOCK | |
| Full flexibility of the binding site | ICM91 | |
The early realization that the conformation of small molecules in complex with macromolecules does not generally correspond to a global minimum9,10 and that proteins undergo structural rearrangements upon binding of ligands92 highlighted the necessity to incorporate flexibility in docking algorithms. This would represent a major burden for the search algorithms because, in addition to the rotational and translational degrees of freedom of the ligand, they would have to consider the fluctuations of bond distances and angles as well as torsions. Considering the size and flexibility of macromolecular receptors and the time constraints applied to docking, this is not really feasible. At present, most docking applications consider the receptor as a rigid body and, on the ligand side, only the degrees of freedom corresponding to dihedral angles are explored, either during docking (flexible docking) or by means of pre-generated libraries of conformers (rigid docking). This consensus should enable direct comparison of different docking packages and/or protocols; nevertheless, this has traditionally been difficult and had to rely on data published by different authors, often using different test sets.93
Very recently, a profusion of studies comparing the performance of docking tools have been published.94–103 In most cases, the comparisons are made by groups not involved in the development of the evaluated software. Two main metrics have been developed to characterize docking performance. The first is predicting the position and conformation of a ligand (the binding mode) for a known protein–ligand complex structure. The second is to calculate the enrichment factors that can be obtained in VS against a particular receptor protein. Here, a large library of varied organic molecules is seeded with compounds that are known for binding to the receptor. VS orders the library of compounds on the docking score to the receptor. The enrichment factor (EF) is calculated as how many of the known compounds are found on the top 1% of this list, compared to random. Although it is still difficult to rank the docking tools according to their performance, a number of trends have emerged.
- Most docking programs will correctly predict the binding mode for 70–80% of the protein–ligand pairs within an RMSD of 2 Å
- The programs providing best results in binding mode prediction are also best in VS experiments.
- Programs with empirical scoring functions that have benefited from large and diverse validation sets currently available tend to provide the best results (e.g. Glide, GOLD and Surflex).
- The results are largely receptor dependent. Some programs are more consistent than others (e.g. Glide).
- Runtimes can vary widely; some programs (e.g. FRED) have specifically been developed for massive screening.
Overall, many docking tools have proved their usefulness in controlled VS experiments and their performance is expected to improve because this is a very competitive area, which is under very active development and receives considerable attention both from companies and academic groups (see Table 5). The published comparative studies have also played a fundamental role in identifying and making large and diverse test sets publicly available, which will facilitate further comparisons and encourage the developers to thoroughly validate their docking software.
| Program | Developer | Citations (out of 10) |
|---|---|---|
| DOCK | UCSF | 8 |
| GOLD | CCDC | 8 |
| FlexX | Tripos | 7 |
| GLIDE | Schrödinger | 6 |
| ICM | MolSoft LLC | 3 |
| Ligand fit | Accelrys | 3 |
| FRED | OpenEye | 2 |
| QXP (FLO+) | Thistlesoft | 2 |
| AutoDock | Scripps | 1 |
| DOCKVISION | DockVision Inc | 1 |
| SLIDE | Michgan State U. | 1 |
| SURFLEX | Discovery Partners Int. | 1 |
The two main areas that are expected to receive most attention in the near future are: (i) improvement of the docking scoring functions and (ii) introducing the flexibility of the receptor in docking applications. The first includes further optimization of empirical and knowledge-based scoring functions, driven by the incessant increase of available experimental data,104,105 as well as more rigorous physical-based scoring functions, which should consider polarization and/or solvation effects.21 The excellent results obtained with consensus scoring106 suggest that most docking packages will implement several scoring schemes to facilitate its application. Regarding the flexibility of the receptor, a number of recent studies have shown that VS results can improve when it is accounted for either implicitly107 or explicitly.89,108 Nevertheless, it has been recently shown that flexible receptor docking is very prone to generating false positives and it should be cautiously applied to VS.109
Docking has been widely applied to hit discovery with remarkable success,93,110,111 but the benefits of VS can only be properly assessed when compared with random screening. Unfortunately, head-to-head comparisons of HTS and docking-based VS are very rare. Merck researchers screened two different subsets of the corporate collection against dihydropicolinate reductase; the first subset was screened whole, while the second was pre-filtered with the docking program FLOG;112 the corresponding hit rates were ≤0.2% and 6% resulting in a ∼30-fold improvement.113 A second comparison was provided by a project to discover novel inhibitors of protein tyrosine phosphatase-1B.114 In this case HTS and VS cannot be directly compared because: (1) different libraries were screened, the HTS one was not very drug like; (2) the assay conditions used for VS hits were more permissive than the ones used in HTS and (3) the VS hits were screened using a medium-throughput assay probably more accurate and sensitive than the HTS. As a result, the authors reported a value of 1700 as an upper estimate of the EF. Finally, a docking-based VS performed at Vernalis to identify Hsp90 inhibitors resulted in a hit rate of 1%, approximately 500-fold greater than HTS.93 It is interesting to note that in the first case, the EF is in agreement with those reported with pure docking VS experiments using seeded libraries (usually in the 10–50 range93), while the other two studies report EFs that are far greater than can be reasonably expected from docking. This can be explained because in the latter cases docking was complemented with a variety of other methods, ranging from drug-like filters to visual inspection of the predicted binding modes and also because they benefited from low- or medium-throughput screening assays, generally more accurate than HTS. This highlights the importance of integrating docking into a wider VS strategy, which will be described below.
The chemical structures proposed by de novo methods are novel in most cases. Paradoxically, this is a major limitation because synthetic tractability is generally ignored by the ligand construction algorithms and because the predicted binding affinity is very poor. These deficiencies often lead to proposed chemical structures of difficult synthesis and low probability of being active and, ultimately, to waste of valuable synthetic resources. Recent developments in the field have been directed to tackle this issue using two complementary approaches:118 (a) use de novo design program to explore the substitution pattern of known binders and (b) to prioritize the output of the programs by its chemical accessibility. Chemical accessibility has been addressed from different angles, including the use of substructural searches to identify commercially available compounds similar to the de novo designed molecules,119 explicit use of synthetic routes120 or by limiting the fragment libraries to substructures of drug-like molecules.121 These recent studies resulted in successful discovery of new chemical series. It is worth noting that de novo design methods are highly complementary to fragment screening methods.122,123 The latter identifies small weak binders that need to grow to become proper leads, the former can greatly benefit from constraining the search to chemical scaffolds experimentally known to bind the active site, because both the limitations of the scoring function and issues around chemical accessibility can be partially overcome.121 In the light of recent successes and the increasing importance of fragment screening, a revived interest in de novo methods is granted.
| Period | Concept | Library composition/Filters |
|---|---|---|
| <1990s | Random screening | Historic collections |
| Natural products or extracts | ||
| Early 1990s | Combinatorial chemistry | Combinatorial libraries127 |
| Early 1990s | Enhanced information content | Molecular diversity128,129 |
| Late 1990s | Drug likeness | Rule of 554 |
| Solubility and permeability130 | ||
| Late 1990s | Frequent hitters | Reactivity131,132 |
| Self aggregation133,134 | ||
| Early 2000s | Lead likeness | Lead like filters135 |
| Ligand efficiency136 | ||
| Early 2000s | Focused libraries | Privileged scaffolds137,138 |
| Target families and chemogenomics139,140 |
Chemoinformatics also provides tools to analyse and extract information from large volumes of data. Particularly, it is extensively used to generate predictive models, which can then be used to profile compound collections or virtual libraries. Some examples are provided in Table 7.
| Class | Property |
|---|---|
| Note: For specific examples see recent reviews by Davis and Riley143 and Oprea and Matter.144 | |
| Solubility | Aqueous solubility |
| Organic solubility | |
| Permeability and distribution | Partition coefficient (LogP) |
| Passive absorption (caco-2) | |
| Blood-brain barrier | |
| Volume of distribution | |
| Plasma protein binding | |
| P-glycoprotein (PGP) substrate | |
| Metabolism | Cytochrome P450 (CYP) inhibition: CYP2D6 CYP3A4 |
| Toxicity | HERG Binding |
| Compound quality or relevance | Drug-likeness |
| Target specificity141 | |
| Hit probability142 | |
- Computational methods. An inspection of recent accounts of successful docking-based VSs reveal that docking is almost invariably supplemented with empirical information,93,111 this includes the so-called guided docking approaches145,146 as well as substructural searches,147 drug-like filters or in silico Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) profiling.144
- Experimental screening methods. HTS and fragment screening methods are highly complementary with VS and should be interconnected rather than perceived as competing technologies.148Table 8 provides several examples of how information generated with experimental methods can be used to refine the VS protocol. Ultimately, iterative cycles of virtual and real screenings can be envisaged to identify and evolve those chemical series with greatest potential in a minimum amount of time and cost.
| Information | Common sources | Possible uses |
|---|---|---|
| Receptor structure | X-ray | Direct use (docking, de novo) |
| Homology modelling | Extraction of interaction patterns149 | |
| Comparison with other binding pockets150 | ||
| Property filters: Size Polarity Charge “Warhead” moiety | ||
| Ligands | Published information | Use of ligand-based methods to pre-filter the library: |
| HTS | Descriptors | |
| Fragment screening | Similarity | |
| Pharmacophore | ||
| VS test experiments to optimize docking or de novo protocols: Choice of Scoring Function Definition of receptor (e.g. water molecules) | ||
| Learning tools to identify binders | ||
| Binding modes | X-ray of protein–ligand complexes | Focused docking (tethered scaffold) |
| Focused de novo design (around known scaffolds) | ||
| Similarity-guided docking151 | ||
| Pharmacophore-constrained docking | ||
| Learning tools to detect incorrect binding modes152 |
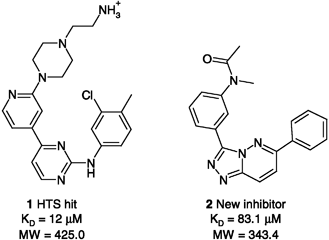 | ||
| Fig. 2 Reference compound (1) and example of hit (2) identified in a scaffold-hopping exercise (Rush et al. 2005)155 | ||
Chemogenomics aspires at describing the interaction of all possible drugs with all possible targets.158 To a certain extent, this can be done experimentally,159 but considering the vast size of both the proteome and the chemical collections, a complete interaction matrix is simply out of reach by any experimental means. A more focused approach is therefore used, where drug-discovery techniques are applied in parallel to several members of a given protein family. Here the aim is to exploit synergies gained from targeting closely related binding sites. Central to this approach is the premise that there is a balance between cross reactivity and selectivity, and the confidence that this balance can be tilted at will in one or the other direction. For example, hit identification is facilitated by searching libraries of compounds designed to be active against a certain protein family (i.e., non-specific compounds). Specificity can be built in at later stages, often by exploiting certain areas of the binding site known to be diverse within the family. A chemogenomics approach does not require structural information but when this is available, as is the case of the large family of protein kinases, a whole new array of methods can be applied.139,140,160 Just to mention a recent example, Vertex scientists have used the experimental binding mode of frequent kinase hitters to derive a pharmacophore model, which has the ablity to recognize molecules that bind to protein kinases in a non-specific manner.161
3.3 Hit to Lead
Once a hit is found, whether it is by experimental or computational means, the next big step in a discovery pipeline is to turn it into a lead. The hit must be optimized to improve both potency and ADMET properties, with the help of in vitro and in vivo screens. Knowledge of the way the hit interacts with the target is crucial to guide potency improvements and gaining insight on where to modify its scaffold and/or chains in order to modulate several ADMET properties such as solubility, metabolic stability and toxicity. This makes the application of structure-based modelling techniques at this stage of development as important as in hit identification. We will first consider the problem faced in elucidating the binding mode of the hit, Second, once this is known, the strategies and modelling techniques used to increase its biological activity, and finally, review some cases where details of the ligand–receptor complex have allowed the rational modification of the hit in order to improve ADMET properties, making special emphasis on selectivity issues potentially linked to toxicity.If the hit has been found with a modelling method such as high-throughput docking, a pharmacophore built from a crystal structure, or other approaches, there may be straightforward clues as to how it is interacting with its receptor. There are numerous instances where active hits selected from high-throughput docking have later been confirmed to bind the way they were predicted by the docking program.93,110 However, it must be borne in mind that, although in the ideal cases the docking pose can be very close to the actual location, there might be many cases in which the rigidity of the receptor prevents finding some of the right ligand–receptor interactions. In those cases where induced fit is of relevance, refinement of the docked pose by more computer intensive techniques such as MD or MC, can alleviate some of the distorted interactions and give a more realistic model. One of such cases has recently been published on the binding mode of hydantoin-based antagonists of lymphocyte-associated antigen-1 (LFA-1).163 First the antagonists were docked to the I-domain of LFA-1 with restraints derived from a combination of experimental techniques such as antibody mapping and photoaffinity labelling. The proposed binding mode was then refined by a series of explicit solvent MD runs followed by minimization. The final modelled structure was afterwards seen to have only 0.64 Å RMSD from the experimentally derived one.163
Irrespective of the source and technique used in hit finding, sometimes multiple reasonable binding modes are possible. In those cases where a SAR around the hit is available, the changes in activity can give clues on what is the correct binding mode among those detected by docking. Nevertheless, there is frequently no SAR available at the beginning of the hit to lead phase. In such cases, it is reasonable to use some of the in silico techniques routinely used for improving the potency of a hit such as linear response (LR) or MM-PBSA, (see a full description below). Because these techniques more reliably predict the ΔG of binding than the prototypical scoring functions used for docking, they can more successfully identify the correct binding mode of a compound as opposed to incorrect (“decoy”) poses generated by a docking program. An illustration of this strategy was reported for the elucidation of the binding mode of efavirenz to HIV-1 RT.164 Efavirenz was first docked to the crystal structure of HIV-1 RT with the Dock program, and five possible binding modes were found. Next, MM-PBSA calculations were carried out on a 500 ps MD trajectory to the five possible binding modes. One of them was clearly pointed out by these calculations as the more stable, having a binding free energy of 7 kcal/mol more favourable than the second pose. Subsequent cocrystallization of the HIV-1 RT-efavirenz complex confirmed the correctness of the modelled complex.
Once the 3D structure of the receptor and the binding mode of a hit are known, one of the simplest techniques that can be used as a guide to improve the potency is a molecular interaction energy analysis of the target's active site (described above for active site detection). Typically, the hit is removed, only the active site is immersed into a cubic grid, and the interaction potentials between receptor and the probes are calculated. This can be subsequently translated into new chemical modifications to the hit being optimized, which are consistent with the interaction energy analysis. Although this technique is still in use, the major drawback of this type of calculations is their qualitative nature, which does not allow to use them as a prioritizing tool.
Among the low-throughput techniques available for hit optimization and quantitative rank ordering of small (ca. 10–100 compounds) sets, one of the less computationally demanding ones is the COMparative BINding Energy (COMBINE) method.166 This method is similar to the classical QSAR methodology. QSAR techniques correlate binding affinities with a set of physicochemical descriptors of the ligands. Similarly, in COMBINE the free energy of binding is expressed as a linear combination of different weighted terms. However, the terms are individual residue-based ligand–receptor interaction energies (electrostatic and van der Waals) computed by MM minimizations of the set of molecules under study in the active site of the receptor. The interaction energies are first subjected to a chemometrical analysis devised to separate the mechanistically important ones from the background noise. Because only MM minimizations are required, the sampling achieved is very crude, but translates into a higher throughput capacity of the method. COMBINE was first tested on a set of 26 human synovial fluid phospholipase A2 inhibitors,166 with an encouraging predictive ability. The method has later been refined and adapted to incorporate desolvation effects, which were shown to increase the predictive ability of the linear regression model in a set of HIV-1 protease inhibitors.167 A further application was reported on the use of COMBINE for the derivation of a predictive regression model for influenza neuraminidase type A inhibitors168 in which the role played by a particular crystallographic water molecule was found to improve the model. Very recently, the use of this technique has been coupled to a docking engine and found to improve the recognition of actives vs. inactives as compared to only docking energies for a series of factor Xa inhibitors.169 While COMBINE represents a step beyond docking in terms of sampling and accuracy of rank-order prediction, this method still has several limitations. Radically different binding modes by different scaffolds may involve different residue interaction patterns, which could lead to inaccurate predictions. Because the entropic term is not accounted for, only series of molecules with very similar flexibility can be properly addressed. Also, the minimization procedure applied for relaxing the different complexes might not be sufficient to find the correct interactions. Finally, because it is a parametric method, a set of known experimental binding activities must be available for a training set of molecules before the model can be built, which limits its applicability.
The linear interaction energy (LIE) or LR first introduced by Aqvist et. al.170 represents a step further in the modelling of ligand–receptor complexes. Although it is also a regression method where the absolute free energy of association is calculated by a sum of different weighted contributions, it is more thorough than COMBINE because it includes solvent explicitly and performs ensemble averaging derived from extensive sampling of configurational space. The method requires two simulations per inhibitor, one freely in solution and the second on the solvated inhibitor–receptor complex. This allows calculation of the van der Waals and electrostatic interaction energy of the ligand with its environment in both states, bound and unbound. The free energy of binding is then expressed as a linear combination of these two energy differences. The two differences must first be calibrated with a training set in order to derive the weighting terms and build a predictive model. The extensive sampling can be achieved by both MC or MD simulations. Although originally the method only accounted for differences in van der Waals and electrostatic interactions, it was later expanded to account for other terms such as changes in SASA. Additional descriptors such as changes in the number of hydrogen bonds, internal energy of the ligand or the number of rotatable bonds were added to the original LR approximation in subsequent versions of the method,171–173 effectively making it an ensemble averaged empirical scoring function analogous to the ones used in docking. Thus, in a study of 20 thrombin inhibitors, regression models were built based on 3–5 descriptors that reproduced experimental binding affinities with an r2 of 0.7–0.8 and rms errors of 1–1.3 kcal/mol, an accuracy suitable for hit optimization.171 The terms found most important for the prediction of binding affinities were the internal energy change and loss of hydrogen bonds for the ligand upon binding, enhancement of van der Waals contacts with the protein and number of rotatable bonds. However, in another study conducted on a set of 40 HIV reverse transcriptase inhibitors, the most important terms were not identical, being of special importance the removal of exposed hydrophobic surface area upon binding, the so-called hydrophobic effect.172 This illustrates the difficulty in finding a universal LR expression that can be used to predict the affinity of any organic molecule for any biological receptor.
Changes in the microenvironment of the targets and the actual nature of the ligand–receptor interactions translate into sensible variations of the different terms. In this respect, a very recent communication analysed the feasibility of obtaining a universal LR equation for the prediction of affinities for the kinase family.174 Three different kinase systems, lck, p38 and cdk2 were chosen. As the first step, regression models were built for each one of them separately by using three training sets of inhibitors, one for each target, with reasonably good correlation coefficients in the 0.7–0.8 range. The terms found to be most significant were changes in van der Waals and Coulombic ligand–receptor interactions and changes in hydrogen bonds upon binding for lck and cdk2 (see expression 1 and 2).
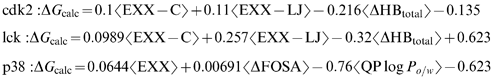
As a second step, combinations of two kinase datasets were used to derive an LR equation that was later used to predict the activities of the third kinase. The correlation coefficients in these three cases between predicted and experimental ΔGs were 0.53, 0.7 and 0.7. Although the three models shared the same types of descriptors, the coefficients multiplying them varied. As a final step, the three kinase datasets were combined (totalling 148 compounds) to obtain a single “universal” LR equation for the kinases (see expression (4)).
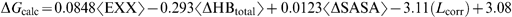
The biggest alternative to the LR methodology is the MM-PB/GB(SA) approximation developed by the Kollman group.52 In this new method one predicts the free energy based on an MD simulation in explicit solvent and counterions. The dehydrated trajectory is later postprocessed, and the free energy is calculated as a sum of the molecular mechanical term (which accounts for bond, angle, torsion, van der Waals and electrostatic terms), a solvation free energy term calculated with the PB or the GB method plus a surface area term, and a final term that accounts for entropy changes estimated by quasiharmonic or normal-mode analysis. The biggest advantage of the method with respect to LR is that it is non-parametric, and thus one does not need to utilize a set of structures and experimental activities to train the model. Furthermore, the method can be applied for estimating the free energy of ligand–receptor association based solely on a single MD run, that of the complex, instead of 2 as in the LR method.52 One of its first applications was the study of a set of avidin inhibitors. In this study, a remarkable correlation r2=0.92 was achieved. Furthermore, it was stated that the method could yield better results than LR, since the latter gave an r2=0.55 for the same set of molecules.175 In a recent application of the method, calculated and experimental binding free energies were compared for a set of cathepsin D inhibitors generated by a combinatorial library approach.176 Although the free energies of association calculated with a standard docking tool such as Dock were found not to correlate with the experimental values, the MM-PB(SA) estimated affinities gave impressive results, with an r2=0.98 and an average error of approximately 1 kcal/mol, enough to be used as a prioritizing tool.
A very recent application of the MM-PB(SA) approach has been disclosed in which this technique is used in combination with experimental techniques for the development of high affinity phosphodiesterase inhibitors.177 First, a fragment-based high-throughput X-ray crystallography screening was performed in order to find low molecular weight inhibitors with only marginal in vitro activities. At this stage of scaffold discovery, from a library of 20,000 compounds, 316 were found to give more than 30% inhibition at 200 μM. From this pool, 107 were cocrystalized with the target PDE4. One of the interesting scaffolds was found to be a pyrazolo derivative (PCEE) with an IC50 of 82 μM and only 168 Da molecular weight. In a second step, this newly found scaffold was tested to see whether its binding mode was consistent with chemical optimization. In order to make sure that the binding mode of the core would be maintained, a small series of derivatives were synthesized and cocrystallized in a second round based on the information derived from the first round of crystallography. From this pool of new derivatives, a phenyl-substituted derivative (PhPCEE, 244 Da. molecular weight) was chosen because of its improved activity, with an IC50of 270 nM. In an effort to further increase its activity, from PhPCEE and the available reagents, more than 100 compounds were designed in silico. These molecules were docked to the crystal structure of PDE4 bound to PhPCEE and their ΔG of binding was predicted with the MM-PB(SA) approach. Out of this group, 10 compounds were synthesized and their IC50 values were obtained. The correlation found between predicted and actual values was 0.92, confirming the good reliability of the technique. The most active compound tested showed an IC50 of 21 nM, that is, a 4000-fold increase with respect to the starting structure.177
The only structure-based modelling techniques considered more reliable than the ones mentioned above are those based on statistical mechanics.1,52 FEP and TI represent the most rigorous approach to simulating free energy changes, and should therefore be also applied in the drug-discovery process. However, these methods have two severe limitations, great computational expense and problems with convergence, which limits its application to small structural changes between the two inhibitors to be compared. For these reasons, they are considered impractical and very seldom used in the drug-discovery context. To illustrate the reliability of this type of calculations a study was published in which the changes in potency of a congeneric series of p38 inhibitors were calculated with scoring functions and also with the more rigorous TI calculations.165 The results of this publication clearly pointed to the complete unreliability of the high-throughput techniques for the prediction of small changes in free energy of binding, and how the latter can be effectively used in a hit-optimization context. Because of its precision, this technique has recently been applied in a considerable number of other situations, such as the study of the effect of protein mutations on complex formation (for instance, see the study of HIV-RT mutations and how they affect the binding of several drugs178), the change in orientation of lateral chains in an active site depending on the actual electronic structure of the inhibitor (e.g. see the study of COX2 complexed with rofecoxib and celecoxib and other diaryl-heterocyclic inhibitors179), for the validation of a proposed binding mode (see e.g. the binding of tacrine-huprine hybrids to acetylcholinestarase180), and many others.
Finally, there exist other less thoroughly tested methods to be used in the hit to lead process. One of such methods has recently been proposed as an alternative to LR and MM-PBSA.181 In this method, the free energy of binding is calculated from descriptors obtained from a MD trajectory performed in vacuo. The binding free energy is expressed as a sum of weighted terms, just as in LR. The terms are an electrostatic interaction energy, a term accounting for the buried surface upon complexation, and a solvation term which is calculated by postprocessing the MD trajectory and solving the PB equation at selected snapshots. The advantage of the method with respect to MM-PBSA is that the MD simulation is performed in vacuo, in contrast to the former, which is carried out in explicit solvent. Also, it does not need an MD trajectory of the ligand alone, as in LR. This method has only been tested on a training series of HIV protease inhibitors, and a remarkable r2 of 0.91 was obtained. However, on being used for the prediction of a 25-member test set the correlation coefficient dropped to 0.64, although it must be borne in mind that experimental free energies of binding only spanned 3 orders of magnitude. The novelty of this method together with the lack of additional applications precludes its comparison with other methods to be used in the hit to lead stage.
Although structure-based modelling techniques can give clues that directly impact ADME optimization, toxicity in particular deserves a special attention. Many of today's interesting targets with known 3D structure belong to large protein families such as kinases, nuclear receptors and proteases. These families have from a few dozens to several hundred members in the human genome (see for example an analysis of the human kinome184). The degree of structural homology within these families is high. Usually the active sites of many family members are nearly identical in terms of electrostatics and shape, making the development of potent and selective inhibitors a challenging task. Usually, a lack of selectivity for these targets translates into toxicity problems. This has fuelled the rise of chemogenomics approaches to drug discovery,158 whereby biological activities of series of compounds are studied on a family wide-basis instead of on a single target. Very interesting in silico applications have recently been published for the kinase family. One of them150 attempted to classify a group of kinases using 3D molecular interaction potentials derived from their structure. A second one classified this family of enzymes on the basis of small molecule selectivity.160 A third analysis shows how chemogenomics can be used for finding or avoiding dual inhibition of a pair of related enzymes.140 Undoubtedly progress in the structural determination of many of the target family members will spur the development of more complete and reliable in silico chemogenomics studies, which will have a positive impact on toxicity liabilities due to the rational fine tuning of selectivity within protein families.
4 Conclusion
There are continuous methodological developments in computational chemistry which, coupled with an ever increasing availability of computational power, are rapidly advancing the capacity of the field to make an impact not only in the drug discovery arena but also in the broader field of molecular biology. Our tools remain, nevertheless, imperfect and inaccurate. This is particularly true for binding and conformational free energies when a macromolecule is involved. But the good news is that molecular modelling techniques have reached a stage where they are definitely useful. An inspection of high-impact medicinal chemistry journals shows that molecular modelling is an integral part of the drug-discovery process in most research organizations and, most importantly, the combination of computational chemistry with detailed knowledge of the structure of pharmacological targets has been particularly successful, and a number of drugs reaching the market have, in fact, resulted from this fertile interaction.185 As the knowledge about biological systems continues growing at gigantic pace, molecular modelling is destined to assume an ever more central role in the integration of information of genetic, structural, biological pathways or chemical origin and, with it, increasing responsibility to deliver effective medicines and to make possible the anxiously awaited increase of productivity in the pharmaceutical industry.References
- W.L. Jorgensen, The many roles of computation in drug discovery, Science, 2004, 303, 1813–1818 CrossRef CAS.
- Ajay and M.A. Murcko, Computational methods to predict binding free energy in ligand–receptor complexes, J. Med. Chem., 1995, 38, 4953–4967 CrossRef.
- H.J. Bohm, Computational tools for structure-based ligand design, Prog. Biophys. Mol. Biol., 1996, 66, 197–210 CrossRef CAS.
- F.H. Allen and R. Taylor, Research applications of the Cambridge structural database (CSD), Chem. Soc. Rev., 2004, 33, 463–475 RSC.
- H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov and P.E. Bourne, The protein data bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- K. Morokuma, New challenges in quantum chemistry: quests for accurate calculations for large molecular systems, Philos. Transact. A Math. Phys. Eng Sci., 2002, 360, 1149–1164 Search PubMed.
- R.A. Friesner, Chemical theory and computation special feature: ab initio quantum chemistry: methodology and applications, Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 6648–6653 CrossRef CAS.
- E. Besalu, X. Girones, L. Amat and R. Carbo-Dorca, Molecular quantum similarity and the fundamentals of QSAR, Acc. Chem. Res., 2002, 35, 289–295 CrossRef CAS.
- M.C. Nicklaus, S. Wang, J.S. Driscoll and G.W. Milne, Conformational changes of small molecules binding to proteins, Bioorg. Med. Chem., 1995, 3, 411–428 CrossRef CAS.
- E. Perola and P.S. Charifson, Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding, J. Med. Chem., 2004, 47, 2499–2510 CrossRef CAS.
- B. Hernandez, F.J. Luque and M. Orozco, Tautomerism of xanthine oxidase substrates hypoxanthine and allopurinol, J. Org. Chem., 1996, 61, 5964–5971 CrossRef CAS.
- C. de Graaf, N.P. Vermeulen and K.A. Feenstra, Cytochrome p450 in silico: an integrative modeling approach, J. Med. Chem., 2005, 48, 2725–2755 CrossRef CAS.
- A. Warshel, Computer simulations of enzyme catalysis: methods, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 425–443 CrossRef CAS.
- R.A. Friesner and V. Guallar, Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis, Annu. Rev. Phys. Chem., 2005, 56, 389–427 CrossRef CAS.
- J.C. Schoneboom, S. Cohen, H. Lin, S. Shaik and W. Thiel, Quantum mechanical/molecular mechanical investigation of the mechanism of C–H hydroxylation of camphor by cytochrome P450cam: theory supports a two-state rebound mechanism, J. Am. Chem. Soc., 2004, 126, 4017–4034 CrossRef.
- L. Ridder and A.J. Mulholland, Modeling biotransformation reactions by combined quantum mechanical/molecular mechanical approaches: from structure to activity, Curr. Top. Med. Chem., 2003, 3, 1241–1256 CrossRef CAS.
- C. Hensen, J.C. Hermann, K. Nam, S. Ma, J. Gao and H.D. Holtje, A combined QM/MM approach to protein–ligand interactions: polarization effects of the HIV-1 protease on selected high affinity inhibitors, J. Med. Chem., 2004, 47, 6673–6680 CrossRef CAS.
- K. Raha and K.M. Merz Jr., A quantum mechanics-based scoring function: study of zinc ion-mediated ligand binding, J. Am. Chem. Soc., 2004, 126, 1020–1021 CrossRef CAS.
- E. Nikitina, V. Sulimov, V. Zayets and N. Zaitseva, Semiempirical calculations of binding enthalpy for protein–ligand complexes, Int. J. Quant. Chem., 2004, 97, 747–763 CrossRef CAS.
- V. Vasilyev and A. Bliznyuk, Application of semiempirical quantum chemical methods as a scoring function in docking, Theor. Chem. Acc., 2004, 112, 313–317 CAS.
- K. Raha and K.M. Merz Jr., Large-scale validation of a quantum mechanics based scoring function: predicting the binding affinity and the binding mode of a diverse set of protein–ligand complexes, J. Med. Chem., 2005, 48, 4558–4575 CrossRef CAS.
- H.J. Bohm and G. Schneider (eds), Protein–Ligand Interactions: From Molecular Recognition to Drug Design, Weinheim, Wiley-VCH, 2003. Search PubMed.
- W.D. Cornell, P. Cieplak, C.I. Baylay, I.R. Gould, K.M. Merz Jr., D.M. Ferguson, D.C. Spellmeyer, T. Fox, J.W. Caldwell and P.A. Kollman, A second generation force field for the simulation of proteins and nucleic acids, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- N. Foloppe and A.D. MacKerell Jr., All-atom empirical force field for nucleic acids: I. parameter optimization based on small molecule and condensed phase macromolecular target data, J. Comput. Chem., 2000, 21, 86–104 CrossRef CAS.
- C. Oostenbrink, A. Villa, A.E. Mark and W.F. van Gunsteren, A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS.
- W.L. Jorgensen, D.S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- T.A. Halgren, Merck molecular force field. I. Basis, form, scope, parameterization and performance of MMFF94, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- K. Lindorff-Larsen, R.B. Best, M.A. DePristo, C.M. Dobson and M. Vendruscolo, Simultaneous determination of protein structure and dynamics, Nature, 2005, 433, 128–132 CrossRef CAS.
- J. Gasteiger and M. Marsili, Iterative partial equalization of orbital electronegativity – a rapid access to atomic charges, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- M. Rarey, S. Wefing and T. Lengauer, Placement of medium-sized molecular fragments into active sites of proteins, J. Comput. Aided Mol. Des., 1996, 10, 41–54 CrossRef CAS.
- M.D. Eldridge, C.W. Murray, T.R. Auton, G.V. Paolini and R.P. Mee, Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes, J. Comput. Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS.
- H.J. Bohm, The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure, J. Comput. Aided Mol. Des., 1994, 8, 243–256 CAS.
- H.J. Bohm, Prediction of binding constants of protein ligands: a fast method for the prioritization of hits obtained from de novo design or 3D database search programs, J. Comput. Aided Mol. Des., 1998, 12, 309–323 CrossRef CAS.
- H.J. Bohm, The computer program LUDI: a new method for the de novo design of enzyme inhibitors, J. Comput. Aided Mol. Des., 1992, 6, 61–78 CAS.
- G. Jones, P. Willett and R.C. Glen, Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation, J. Mol. Biol., 1995, 245, 43–53 CrossRef CAS.
- C.M. Venkatachalam, X. Jiang, T. Oldfield and M. Waldman, LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites, J. Mol. Graph. Model., 2003, 21, 289–307 CrossRef CAS.
- J.B.O. Mitchell, R.A. Laskowski, A. Alex and J.M. Thornton, BLEEP – potential of mean force describing protein–ligand interactions: I. Generating potential, J. Comput. Chem., 1999, 20, 1165–1176 CrossRef CAS.
- J.B.O. Mitchell, R.A. Laskowski, A. Alex, M.J. Forster and J.M. Thornton, BLEEP – potential of mean force describing protein–ligand interactions: II. calculation of binding energies and comparison with experimental data, J. Comput. Chem., 1999, 20, 1177–1185 CrossRef CAS.
- D.K. Gehlhaar, G.M. Verkhivker, P.A. Rejto, C.J. Sherman, D.B. Fogel, L.J. Fogel and S.T. Freer, Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming, Chem. Biol., 1995, 2, 317–324 CrossRef CAS.
- I. Muegge and Y.C. Martin, A general and fast scoring function for protein–ligand interactions: a simplified potential approach, J. Med. Chem., 1999, 42, 791–804 CrossRef CAS.
- H. Gohlke, M. Hendlich and G. Klebe, Knowledge-based scoring function to predict protein–ligand interactions, J. Mol. Biol., 2000, 295, 337–356 CrossRef CAS.
- H. Lu, L. Lu and J. Skolnick, Development of unified statistical potentials describing protein-protein interactions, Biophys. J., 2003, 84, 1895–1901 CrossRef CAS.
- H. Lu and J. Skolnick, Application of statistical potentials to protein structure refinement from low resolution ab initio models, Biopolymers, 2003, 70, 575–584 CrossRef CAS.
- M. Orozco and F.J. Luque, Theoretical methods for the description of the solvent effect in biomolecular systems, Chem. Rev., 2000, 100, 4187–4226 CrossRef CAS.
- C.J. Cramer and D.G. Trular, Implicit solvation models: equilibria, structure, spectra, and dynamics, Chem. Rev., 1999, 99, 2161–2200 CrossRef CAS.
- P.A. Kollman, Free energy calculations: applications to chemical and biochemical phenomena, Chem. Rev., 1993, 93, 2395–2417 CrossRef CAS.
- W.H. Orttung, Direct solution on the Poisson equation for biomolecules of arbitrary shape,polarizability density and charge distribution, Ann. N. Y. Acad. Sci., 1977, 303, 22–37 CrossRef CAS.
- W.C. Still, A. Tempczyk, R.C. Hawley and T. Hendrickson, Semianalytical treatment of solvation for molecular mechanics and dynamics, J. Am. Chem. Soc., 1990, 112, 6127–6129 CrossRef CAS.
- J.M. Blaney, P.K. Weiner, A. Dearing, P.A. Kollman, E.C. Jorgensen, S.J. Oatley, J.M. Burridge and J.F. Blake, Molecular mechanics simulation of protein–ligand interactions: binding of thyroid hormone analogs to prealbumin, J. Am. Chem. Soc., 1982, 104, 6424–6434 CrossRef CAS.
- E.L. Mehler and T. Solmajer, Electrostatic effects in proteins: comparison of dielectric and charge models, Protein Eng., 1991, 4, 903–910 CrossRef CAS.
- D. Eisenberg and A.D. McLachlan, Solvation energy in protein folding and binding, Nature, 1986, 319, 199–203 CrossRef CAS.
- P.A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D.A. Case and T.E. Cheatham, III Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS.
- I. Muegge, Selection criteria for drug-like compounds, Med. Res. Rev., 2003, 23, 302–321 CrossRef CAS.
- C.A. Lipinski, F. Lombardo, B.W. Dominy and P.J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev., 1997, 23, 3–25 CrossRef.
- D.F. Veber, S.R. Johnson, H.Y. Cheng, B.R. Smith, K.W. Ward and K.D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- A.L. Hopkins and C.R. Groom, The druggable genome, Nat. Rev. Drug Discov., 2002, 1, 727–730 CrossRef CAS.
- M.R. Arkin and J.A. Wells, Small-molecule inhibitors of protein–protein interactions: progressing towards the dream, Nat. Rev. Drug Discov., 2004, 3, 301–317 CrossRef CAS.
- P.J. Goodford, A computational procedure for determining energetically favorable binding sites on biologically important macromolecules, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS.
- M. Hendlich, F. Rippmann and G. Barnickel, LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins, J. Mol. Graph. Model., 1997, 15, 359–363–389.
- Molecular Operating Environment (MOE), version 2003.02; Chemical Computing Group Inc., Montreal, Canada, 2003..
- C.T. Porter, G.J. Bartlett and J.M. Thornton, The catalytic site atlas: a resource of catalytic sites and residues identified in enzymes using structural data, Nucleic Acids Res., 2004, 32, D129–D133 CrossRef CAS.
- J. Fernandez-Recio, M. Totrov, C. Skorodumov and R. Abagyan, Optimal docking area: a new method for predicting protein–protein interaction sites, Proteins, 2005, 58, 134–143 Search PubMed.
- B. Ma, T. Elkayam, H. Wolfson and R. Nussinov, Protein–protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 5772–5777 CrossRef CAS.
- P. Chakrabarti and J. Janin, Dissecting protein–protein recognition sites, Proteins, 2002, 47, 34–343.
- D.Y. Jackson, Alpha 4 integrin antagonists, Curr. Pharm. Des., 2002, 8, 1229–1253 Search PubMed.
- T.R. Gadek, D.J. Burdick, R.S. McDowell, M.S. Stanley, J.C. Marsters Jr., K.J. Paris, D.A. Oare, M.E. Reynolds, C. Ladner, K.A. Zioncheck, W.P. Lee, P. Gribling, M.S. Dennis, N.J. Skelton, D.B. Tumas, K.R. Clark, S.M. Keating, M.H. Tilley, J.W. Beresini, L.G. Presta and S.C. Bodary, Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule, Science, 2002, 295, 1086–1089 CrossRef CAS.
- J.W. Tilley, L. Chen, A. Sidduri and N. Fotouhi, The discovery of VLA-4 antagonists, Curr. Top. Med. Chem., 2004, 4, 1509–1523 CrossRef CAS.
- G.X. Yang and W.K. Hagmann, VLA-4 antagonists: potent inhibitors of lymphocyte migration, Med. Res. Rev., 2003, 23, 369–392 CrossRef CAS.
- K. Lundstrom, Structural genomics of GPCRs, Trends Biotechnol., 2005, 23, 103–108 CrossRef CAS.
- M.L. Pusey, Z.J. Liu, W. Tempel, J. Praissman, D. Lin, B.C. Wang, J.A. Gavira and J.D. Ng, Life in the fast lane for protein crystallization and X-ray crystallography, Prog. Biophys. Mol. Biol., 2005, 88, 359–386 CrossRef CAS.
- A.S. Altieri and R.A. Byrd, Automation of NMR structure determination of proteins, Curr. Opin. Struct. Biol., 2004, 14, 547–553 CrossRef CAS.
- R. Hui and A. Edwards, High-throughput protein crystallization, J. Struct. Biol., 2003, 142, 154–161 CrossRef CAS.
- C.A. Orengo and J.M. Thornton, Protein families and their evolution – a structural perspective, Annu. Rev. Biochem., 2005, 74, 867–900 CrossRef CAS.
- A.M. Davis, S.J. Teague and G.J. Kleywegt, Application and limitations of X-ray crystallographic data in structure-based ligand and drug design, Angew. Chem. Int. Ed Engl., 2003, 42, 2718–2736 CrossRef CAS.
- G.J. Kleywegt, M.R. Harris, J.Y. Zou, T.C. Taylor, A. Wahlby and T.A. Jones, The uppsala electron-density server, Acta Crystallogr. D. Biol. Crystallogr., 2004, 60, 2240–2249 CrossRef.
- M.A. DePristo, P.I. de Bakker and T.L. Blundell, Heterogeneity and inaccuracy in protein structures solved by X-ray crystallography, Structure. (Cambridge), 2004, 12, 831–838 Search PubMed.
- T.L. Blundell and S. Patel, High-throughput X-ray crystallography for drug discovery, Curr. Opin. Pharmacol., 2004, 4, 490–496 CrossRef CAS.
- G.J. Kleywegt, K. Henrick, E.J. Dodson and D.M. van Aalten, Pound-wise but penny-foolish: how well do micromolecules fare in macromolecular refinement?, Structure. (Cambridge), 2003, 11, 1051–1059 Search PubMed.
- G.E. Dale, C. Oefner and A. D'Arcy, The protein as a variable in protein crystallization, J. Struct. Biol., 2003, 142, 88–97 CrossRef CAS.
- R. Day and V. Daggett, All-atom simulations of protein folding and unfolding, Adv. Protein Chem., 2003, 66, 373–403 CAS.
- B. Contreras-Moreira, P.W. Fitzjohn and P.A. Bates, Comparative modelling: an essential methodology for protein structure prediction in the post-genomic era, Appl. Bioinformatics, 2002, 1, 177–190 Search PubMed.
- P. Koehl and M. Levitt, Sequence variations within protein families are linearly related to structural variations, J. Mol. Biol., 2002, 323, 551–562 CrossRef CAS.
- A. Sali, L. Potterton, F. Yuan, H. van Vlijmen and M. Karplus, Evaluation of comparative protein modeling by MODELLER, Proteins, 1995, 23, 318–326 CrossRef CAS.
- I.D. Kuntz, J.M. Blaney, S.J. Oatley, R. Langridge and T.E. Ferrin, A geometric approach to macromolecule–ligand interactions, J. Mol. Biol., 1982, 161, 269–288 CAS.
- N. Brooijmans and I.D. Kuntz, Molecular recognition and docking algorithms, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 335–373 CrossRef CAS.
- R.D. Taylor, P.J. Jewsbury and J.W. Essex, A review of protein-small molecule docking methods, J. Comput. Aided Mol. Des., 2002, 16, 151–166 CrossRef CAS.
- R.M. Knegtel, I.D. Kuntz and C.M. Oshiro, Molecular docking to ensembles of protein structures, J. Mol. Biol., 1997, 266, 424–440 CrossRef CAS.
- F. Osterberg, G.M. Morris, M.F. Sanner, A.J. Olson and D.S. Goodsell, Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock, Proteins, 2002, 46, 34–40 CrossRef CAS.
- B.Q. Wei, L.H. Weaver, A.M. Ferrari, B.W. Matthews and B.K. Shoichet, Testing a flexible-receptor docking algorithm in a model binding site, J. Mol. Biol., 2004, 337, 1161–1182 CrossRef CAS.
- H. Claussen, C. Buning, M. Rarey and T. Lengauer, FlexE: efficient molecular docking considering protein structure variations, J. Mol. Biol., 2001, 308, 377–395 CrossRef CAS.
- M. Totrov and R. Abagyan, Flexible protein–ligand docking by global energy optimization in internal coordinates, Proteins, 1997,((Suppl. 1)), 215–220 CrossRef CAS.
- S.J. Teague, Implications of protein flexibility for drug discovery, Nat. Rev. Drug Discov., 2003, 2, 527–541 CrossRef CAS.
- X. Barril, R.E. Hubbard and S.D. Morley, Virtual screening in structure-based drug design, Mini Rev. Med. Chem., 2004, 4, 779–791 Search PubMed.
- M.D. Cummings, R.L. DesJarlais, A.C. Gibbs, V. Mohan and E.P. Jaeger, Comparison of automated docking programs as virtual screening tools, J. Med. Chem., 2005, 48((4)), 962–976 CrossRef CAS.
- M. Kontoyianni, L.M. McClellan and G.S. Sokol, Evaluation of docking performance: comparative data on docking algorithms, J. Med. Chem., 2004, 47((3)), 558–565 CrossRef CAS.
- M. Kontoyianni, G.S. Sokol and L.M. McClellan, Evaluation of library ranking efficacy in virtual screening, J. Comput. Chem., 2005, 26((1)), 11–22 CrossRef CAS.
- E. Kellenberger, J. Rodrigo, P. Muller and D. Rognan, Comparative evaluation of eight docking tools for docking and virtual screening accuracy, Proteins, 2004, 57((2)), 225–242 Search PubMed.
- E. Perola, W.P. Walters and P.S. Charifson, A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance, Proteins, 2004, 56((2)), 235–249 Search PubMed.
- R.T. Kroemer, A. Vulpetti, J.J. McDonald, D.C. Rohrer, J.Y. Trosset, F. Giordanetto, S. Cotesta, C. McMartin, M. Kihlen and P.F. Stouten, Assessment of docking poses: interactions-based accuracy classification (IBAC) versus crystal structure deviations, J. Chem. Inf. Comput. Sci., 2004, 44, 871–881 CrossRef CAS.
- J.A. Erickson, M. Jalaie, D.H. Robertson, R.A. Lewis and M. Vieth, Lessons in molecular recognition: the effects of ligand and protein flexibility on molecular docking accuracy, J. Med. Chem., 2004, 47, 45–55 CrossRef CAS.
- B.D. Bursulaya, M. Totrov, R. Abagyan and C.L. Brooks, III Comparative study of several algorithms for flexible ligand docking, J. Comput. Aided Mol. Des., 2003, 17((11)), 755–763 CrossRef CAS.
- T. Schulz-Gasch and M. Stahl, Binding site characteristics in structure-based virtual screening: evaluation of current docking tools, J. Mol. Model.(Online), 2003, 9((1)), 47–57 Search PubMed.
- C. Bissantz, G. Folkers and D. Rognan, Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations, J. Med. Chem., 2000, 43((25)), 4759–4767 CrossRef CAS.
- R. Wang, X. Fang, Y. Lu, C.Y. Yang and S. Wang, The PDBbind database: methodologies and updates, J. Med. Chem., 2005, 48, 4111–4119 CrossRef CAS.
- L. Hu, M.L. Benson, R.D. Smith, M.G. Lerner and H.A. Carlson, Binding MOAD (Mother Of All Databases), Proteins, 2005, 60, 333–340 Search PubMed.
- P.S. Charifson, J.J. Corkery, M.A. Murcko and W.P. Walters, Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins, J. Med. Chem., 1999, 42, 5100–5109 CrossRef CAS.
- A.M. Ferrari, B.Q. Wei, L. Costantino and B.K. Shoichet, Soft docking and multiple receptor conformations in virtual screening, J. Med. Chem., 2004, 47, 5076–5084 CrossRef CAS.
- C.N. Cavasotto and R.A. Abagyan, Protein flexibility in ligand docking and virtual screening to protein kinases, J. Mol. Biol., 2004, 337, 209–225 CrossRef CAS.
- X. Barril and S.D. Morley, Unveiling the full potential of flexible receptor docking using multiple crystallographic structures, J. Med. Chem., 2005, 48, 4432–4443 CrossRef CAS.
- B.K. Shoichet, Virtual screening of chemical libraries, Nature, 2004, 432, 862–865 CrossRef CAS.
- J.C. Alvarez, High-throughput docking as a source of novel drug leads, Curr. Opin. Chem. Biol., 2004, 8, 365–370 CrossRef CAS.
- M.D. Miller, S.K. Kearsley, D.J. Underwood and R.P. Sheridan, FLOG: a system to select ‘quasi-flexible’ ligands complementary to a receptor of known three-dimensional structure, J. Comput. Aided Mol. Des., 1994, 8, 565–582 CrossRef.
- A.M. Paiva, D.E. Vanderwall, J.S. Blanchard, J.W. Kozarich, J.M. Williamson and T.M. Kelly, Inhibitors of dihydrodipicolinate reductase, a key enzyme of the diaminopimelate pathway of myobacterium tuberculosis, Biochim. Biophys. Acta, 2001, 1545, 67–77 CAS.
- T.N. Doman, S.L. McGovern, B.J. Witherbee, T.P. Kasten, R. Kurumbail, W.C. Stallings, D.T. Connolly and B.K. Shoichet, Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B, J. Med. Chem., 2002, 45, 2213–2221 CrossRef CAS.
- R.S. Bohacek, C. McMartin and W.C. Guida, The art and practice of structure-based drug design: a molecular modeling perspective, Med. Res. Rev., 1996, 16, 3–50 CrossRef CAS.
- H.O. Villar and R.T. Koehler, Comments on the design of chemical libraries for screening, Mol. Divers., 2000, 5, 13–24 CrossRef CAS.
- M.A. Murcko, An introduction to de novo ligand design, in Practical Applications of Computer-Aided Drug Design, P.S. Charifson (ed), Marcel Dekker, New York, 1997, 304–354. Search PubMed.
- T. Honma, Recent advances in de novo design strategy for practical lead identification, Med. Res. Rev., 2003, 23, 606–632 CrossRef CAS.
- T. Honma, K. Hayashi, T. Aoyama, N. Hashimoto, T. Machida, K. Fukasawa, T. Iwama, C. Ikeura, M. Ikuta, I. Suzuki-Takahashi, Y. Iwasawa, T. Hayama, S. Nishimura and H. Morishima, Structure-based generation of a new class of potent Cdk4 inhibitors: new de novo design strategy and library design, J. Med. Chem., 2001, 44, 4615–4627 CrossRef CAS.
- H.M. Vinkers, M.R. de Jonge, F.F. Daeyaert, J. Heeres, L.M. Koymans, J.H. van Lenthe, P.J. Lewi, H. Timmerman, K. Van Aken and P.A. Janssen, SYNOPSIS: SYNthesize and OPtimize System in Silico, J. Med. Chem., 2003, 46, 2765–2773 CrossRef CAS.
- D. Douguet, H. Munier-Lehmann, G. Labesse and S. Pochet, LEA3D: a computer-aided ligand design for structure-based drug design, J. Med. Chem., 2005, 48, 2457–2468 CrossRef CAS.
- D.A. Erlanson, R.S. McDowell and T. O'Brien, Fragment-based drug discovery, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS.
- D.C. Rees, M. Congreve, C.W. Murray and R. Carr, Fragment-based lead discovery, Nat. Rev. Drug Discov., 2004, 3, 660–672 CrossRef CAS.
- J.R. Archer, History,evolution and trends in compound management for high throughput screening, Assay. Drug Dev. Technol., 2004, 2, 675–681 Search PubMed.
- A.R. Leach and M.M. Hann, The in silico world of virtual libraries, Drug Discov. Today, 2000, 5, 326–336 CrossRef CAS.
- A. Schuffenhauer, M. Popov, U. Schopfer, P. Acklin, J. Stanek and E. Jacoby, Molecular diversity management strategies for building and enhancement of diverse and focused lead discovery compound screening collections, Comb. Chem. High Throughput Screen., 2004, 7, 771–781 CrossRef CAS.
- E.M. Gordon, R.W. Barrett, W.J. Dower, S.P. Fodor and M.A. Gallop, Applications of combinatorial technologies to drug discovery,2. Combinatorial organic synthesis,library screening strategies, and future directions, J. Med. Chem., 1994, 37, 1385–1401 CrossRef CAS.
- Y.C. Martin, R.D. Brown, M.G. Bures, Quantifying diversity, in Combinatorial Chemistry and Molecular Diversity in Drug Discovery, E.M. Gordon and J.F. Kerwin Jr., (eds) Wiley-Liss, New York, 1998, 369–385. Search PubMed.
- W.H. Moss, D.H. Green and M.R. Pavia, Molecular diversity, Annu. Rep. Med. Chem., 1993, 28, 315–324 Search PubMed.
- C.A. Lipinski, Drug-like properties and the causes of poor solubility and poor permeability, J. Pharmacol. Toxicol. Methods, 2000, 44, 235–249 CrossRef CAS.
- M. Hann, B. Hudson, X. Lewell, R. Lifely, L. Miller and N. Ramsden, Strategic pooling of compounds for high-throughput screening, J. Chem. Inf. Comput. Sci., 1999, 39, 897–902 CrossRef CAS.
- G.M. Rishton, Reactive compounds and in vitro false positives in HTS, Drug Discov. Today, 1997, 2, 382–384 CrossRef CAS.
- S.L. McGovern, B.T. Helfand, B. Feng and B.K. Shoichet, A specific mechanism of nonspecific inhibition, J. Med. Chem., 2003, 46, 4265–4272 CrossRef CAS.
- J. Seidler, S.L. McGovern, T.N. Doman and B.K. Shoichet, Identification and prediction of promiscuous aggregating inhibitors among known drugs, J. Med. Chem., 2003, 46, 4477–4486 CrossRef CAS.
- T.I. Oprea, A.M. Davis, S.J. Teague and P.D. Leeson, Is there a difference between leads and drugs? A historical perspective, J. Chem. Inf. Comput. Sci., 2001, 41, 1308–1315 CrossRef CAS.
- A.L. Hopkins, C.R. Groom and A. Alex, Ligand efficiency: a useful metric for lead selection, Drug Discov. Today, 2004, 9, 430–431 CrossRef.
- G.W. Bemis and M.A. Murcko, Properties of known drugs, 2. Side chains, J. Med. Chem., 1999, 42, 5095–5099 CrossRef CAS.
- G.W. Bemis and M.A. Murcko, The properties of known drugs. 1. Molecular frameworks, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS.
- M. Vieth, J.J. Sutherland, D.H. Robertson and R.M. Campbell, Kinomics: characterizing the therapeutically validated kinase space, Drug Discov. Today, 2005, 10, 839–846 CrossRef CAS.
- E. ter Haar, W.P. Walters, S. Pazhanisamy, P. Taslimi, A.C. Pierce, G.W. Bemis, F.G. Salituro and S.L. Harbeson, Kinase chemogenomics: targeting the human kinome for target validation and drug discovery, Mini Rev. Med. Chem., 2004, 4, 235–253 Search PubMed.
- H. Briem and J. Gunther, Classifying “kinase inhibitor-likeness” by using machine-learning methods, Chembiochem., 2005, 6, 558–566 CrossRef CAS.
- A. Teckentrup, H. Briem and J. Gasteiger, Mining high-throughput screening data of combinatorial libraries: development of a filter to distinguish hits from nonhits, J. Chem. Inf. Comput. Sci., 2004, 44, 626–634 CrossRef CAS.
- A.M. Davis and R.J. Riley, Predictive ADMET studies, the challenges and the opportunities, Curr. Opin. Chem. Biol., 2004, 8, 378–386 CrossRef CAS.
- T.I. Oprea and H. Matter, Integrating virtual screening in lead discovery, Curr. Opin. Chem. Biol., 2004, 8, 349–358 CrossRef CAS.
- X. Fradera and J. Mestres, Guided docking approaches to structure-based design and screening, Curr. Top. Med. Chem., 2004, 4, 687–700 CrossRef CAS.
- J.M. Jansen and E.J. Martin, Target-biased scoring approaches and expert systems in structure-based virtual screening, Curr. Opin. Chem. Biol., 2004, 8, 359–364 CrossRef CAS.
- C. Merlot, D. Domine, C. Cleva and D.J. Church, Chemical substructures in drug discovery, Drug Discov. Today, 2003, 8, 594–602 CrossRef CAS.
- J. Bajorath, Integration of virtual and high-throughput screening, Nat. Rev. Drug Discov., 2002, 1, 882–894 CrossRef CAS.
- N. Majeux, M. Scarsi, J. Apostolakis, C. Ehrhardt and A. Caflisch, Exhaustive docking of molecular fragments with electrostatic solvation, Proteins, 1999, 37, 88–105 CrossRef CAS.
- T. Naumann and H. Matter, Structural classification of protein kinases using 3D molecular interaction field analysis of their ligand binding sites: target family landscapes, J. Med. Chem., 2002, 45, 2366–2378 CrossRef CAS.
- X. Fradera, R.M. Knegtel and J. Mestres, Similarity-driven flexible ligand docking, Proteins, 2000, 40, 623–636 CrossRef CAS.
- A.B. Garmendia-Doval, S.D. Morley and S. Juhos, Docking filtering using cartesian genetic programming, in Lecture Notes in Computer Science, P. Liardet, P. Collet and C. Onlupt (eds), Springer-Verlag GmbH, Berlin, 2004, 189–200. Search PubMed.
- S. Renner, V. Ludwig, O. Boden, U. Scheffer, M. Gobel and G. Schneider, New inhibitors of the Tat-TAR RNA interaction found with a “fuzzy” pharmacophore model, Chembiochem., 2005, 6, 1119–1125 CrossRef CAS.
- G. Schneider, W. Neidhart, T. Giller and G. Schmid, “Scaffold-hopping” by topological pharmacophore search: a contribution to virtual screening, Angew. Chem. Int. Edit., 1999, 38, 2894–2896 CrossRef CAS.
- T.S. Rush III, J.A. Grant, L. Mosyak and A. Nicholls, A shape-based 3-D scaffold hopping method and its application to a bacterial protein–protein interaction, J. Med. Chem., 2005, 48, 1489–1495 CrossRef.
- Y.Z. Chen and D.G. Zhi, Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule, Proteins, 2001, 43, 217–226 CrossRef CAS.
- N. Paul, E. Kellenberger, G. Bret, P. Muller and D. Rognan, Recovering the true targets of specific ligands by virtual screening of the protein data bank, Proteins, 2004, 54, 671–680 Search PubMed.
- J. Mestres, Computational chemogenomics approaches to systematic knowledge-based drug discovery, Curr. Opin. Drug Discov. Devel., 2004, 7, 304–313 Search PubMed.
- A.F. Fliri, W.T. Loging, P.F. Thadeio and R.A. Volkmann, Biological spectra analysis: linking biological activity profiles to molecular structure, Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 261–266 CrossRef CAS.
- M. Vieth, R.E. Higgs, D.H. Robertson, M. Shapiro, E.A. Gragg and H. Hemmerle, Kinomics-structural biology and chemogenomics of kinase inhibitors and targets, Biochim. Biophys. Acta, 2004, 1697, 243–257 CAS.
- A.M. Aronov and M.A. Murcko, Toward a pharmacophore for kinase frequent hitters, J. Med. Chem., 2004, 47, 5616–5619 CrossRef CAS.
- A. Baxter, S. Brough, A. Cooper, E. Floettmann, S. Foster, C. Harding, J. Kettle, T. McInally, C. Martin, M. Mobbs, M. Needham, P. Newham, S. Paine, S. St Gallay, S. Salter, J. Unitt and Y. Xue, Hit-to-lead studies: the discovery of potent, orally active, thiophenecarboxamide IKK-2 inhibitors, Bioorg. Med. Chem. Lett., 2004, 14, 2817–2822 CrossRef CAS.
- K. Last-Barney, W. Davidson, M. Cardozo, L.L. Frye, C.A. Grygon, J.L. Hopkins, D.D. Jeanfavre, S. Pav, C. Qian, J.M. Stevenson, L. Tong, R. Zindell and T.A. Kelly, Binding site elucidation of hydantoin-based antagonists of LFA-1 using multidisciplinary technologies: evidence for the allosteric inhibition of a protein–protein interaction, J. Am. Chem. Soc., 2001, 123, 5643–5650 CrossRef CAS.
- J. Wang, P. Morin, W. Wang and P.A. Kollman, Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA, J. Am. Chem. Soc., 2001, 123, 5221–5230 CrossRef CAS.
- D.A. Pearlman and P.S. Charifson, Are free energy calculations useful in practice? A comparison with rapid scoring functions for the p38 MAP kinase protein system, J. Med. Chem., 2001, 44, 3417–3423 CrossRef CAS.
- A.R. Ortiz, M.T. Pisabarro, F. Gago and R.C. Wade, Prediction of drug binding affinities by comparative binding energy analysis, J. Med. Chem., 1995, 38, 2681–2691 CrossRef CAS.
- C. Perez, M. Pastor, A.R. Ortiz and F. Gago, Comparative binding energy analysis of HIV-1 protease inhibitors: incorporation of solvent effects and validation as a powerful tool in receptor-based drug design, J. Med. Chem., 1998, 41, 836–852 CrossRef CAS.
- T. Wang and R.C. Wade, Comparative binding energy (COMBINE) analysis of influenza neuraminidase-inhibitor complexes, J. Med. Chem., 2001, 44, 961–971 CrossRef CAS.
- M. Murcia and A.R. Ortiz, Virtual screening with flexible docking and COMBINE-based models. Application to a series of factor Xa inhibitors, J. Med. Chem., 2004, 47, 805–820 CrossRef CAS.
- J. Aqvist, C. Medina and J.E. Samuelsson, A new method for predicting binding affinity in computer-aided drug design, Protein Eng., 1994, 7, 385–391 CrossRef CAS.
- A.C. Pierce and W.L. Jorgensen, Estimation of binding affinities for selective thrombin inhibitors via Monte Carlo simulations, J. Med. Chem., 2001, 44, 1043–1050 CrossRef CAS.
- R.C. Rizzo, J. Tirado-Rives and W.L. Jorgensen, Estimation of binding affinities for HEPT and nevirapine analogues with HIV-1 reverse transcriptase via Monte Carlo simulations, J. Med. Chem., 2001, 44, 145–154 CrossRef CAS.
- R.C. Rizzo, M. Udier-Blagovic, D.P. Wang, E.K. Watkins, M.B. Kroeger Smith, R.H. Smith Jr., J. Tirado-Rives and W.L. Jorgensen, Prediction of activity for nonnucleoside inhibitors with HIV-1 reverse transcriptase based on Monte Carlo simulations, J. Med. Chem., 2002, 45, 2970–2987 CrossRef CAS.
- Y. Tominaga and W.L. Jorgensen, General model for estimation of the inhibition of protein kinases using Monte Carlo simulations, J. Med. Chem., 2004, 47, 2534–2549 CrossRef CAS.
- B. Kuhn and P.A. Kollman, Binding of a diverse set of ligands to avidin and streptavidin: an accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models, J. Med. Chem., 2000, 43, 3786–3791 CrossRef CAS.
- S. Huo, J. Wang, P. Cieplak, P.A. Kollman and I.D. Kuntz, Molecular dynamics and free energy analyses of cathepsin D-inhibitor interactions: insight into structure-based ligand design, J. Med. Chem., 2002, 45, 1412–1419 CrossRef CAS.
- G.L. Card, L. Blasdel, B.P. England, C. Zhang, Y. Suzuki, S. Gillette, D. Fong, P.N. Ibrahim, D.R. Artis, G. Bollag, M.V. Milburn, S.H. Kim, J. Schlessinger and K.Y. Zhang, A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design, Nat. Biotechnol., 2005, 23, 201–207 CrossRef CAS.
- M. Udier-Blagovic, J. Tirado-Rives and W.L. Jorgensen, Validation of a model for the complex of HIV-1 reverse transcriptase with nonnucleoside inhibitor TMC125, J. Am. Chem. Soc., 2003, 125, 6016–6017 CrossRef CAS.
- R. Soliva, C. Almansa, S.G. Kalko, F.J. Luque and M. Orozco, Theoretical studies on the inhibition mechanism of cyclooxygenase-2. Is there a unique recognition site?, J. Med. Chem., 2003, 46, 1372–1382 CrossRef CAS.
- X. Barril, M. Orozco and F.J. Luque, Predicting relative binding free energies of tacrine-huperzine A hybrids as inhibitors of acetylcholinesterase, J. Med. Chem., 1999, 42, 5110–5119 CrossRef CAS.
- V. Zoete, O. Michielin and M. Karplus, Protein-ligand binding free energy estimation using molecular mechanics and continuum electrostatics. Application to HIV-1 protease inhibitors, J. Comput. Aided Mol. Des., 2003, 17, 861–880 CrossRef CAS.
- P.F. Cirillo, C. Pargellis and J. Regan, The non-diaryl heterocycle classes of p38 MAP kinase inhibitors, Curr. Top. Med. Chem., 2002, 2, 1021–1035 CrossRef CAS.
- P.F. Jackson and J.L. Bullington, Pyridinylimidazole based p38 MAP kinase inhibitors, Curr. Top. Med. Chem., 2002, 2, 1011–1020 CrossRef CAS.
- G. Manning, D.B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, The protein kinase complement of the human genome, Science, 2002, 298, 1912–1934 CrossRef CAS.
- L.W. Hardy and A. Malikayil, The impact of structure-guided drug design on clinical agents, Curr. Drug Disc., 2003, 3, 15–20 Search PubMed.
Footnotes |
| † This is Chapter 3 taken from the book Structure Based Drug Discovery (Edited by Roderick E. Hubbard) which is part of the RSC Biomolecular Sciences series. |
| ‡ Current address: ICREA and Department de Fisicoquimica, Facultat de Farmacia, Universitat de Barcelona, Av. Joan XXIII, Barcelona s/n 08028, Spain |
| This journal is © The Royal Society of Chemistry 2006 |