Fragment screening: an introduction†‡
Andrew R.
Leach
a,
Michael M.
Hann
a,
Jeremy N.
Burrows
b and
Ed J.
Griffen
c
aGlaxoSmithKline Research and Development, Gunnels Wood Road, Stevenage, Herts, UK SG1 2NY
bAstraZeneca R&D, Södertälje, S-151 85, Södertälje, Sweden
cAstraZeneca R&D, Alderley Park, Macclesfield, UK, SK10 4TG
First published on 2nd August 2006
1 Introduction
The development of a new therapeutic drug creates many challenges as a drug has to possess many attributes for it to be an effective medicine. To discover within one molecule all the properties needed to ensure target specificity and potency, bioavailability, appropriate duration of action and lack of toxicity is a very tough challenge and unfortunately often ends in failure even after many years of work. In addition, higher standards of drug efficacy are always needed and drugs for new diseases and targets that test our current understanding of science are increasingly our aims. Undoubtedly, starting close to the desired end point would be a good strategy for drug discovery. But, the sheer diversity of molecular species1 that might be considered as drug molecules suggests that a process of evolution by selection will invariably be the way in which new drugs are discovered. The concepts of fragment screening have arisen from the realisation that finding a sensible starting point for the evolution of a new molecule is a key factor in modern drug discovery. The fragment screening approach involves finding molecules which are substantially smaller in relative terms to the probable size of a final drug-like molecule, yet which still have some of the activity at the required target protein and so may act as good starting points for a lead optimisation drug discovery programme.2 The concept of drug-likeness
Modern technologies for the synthesis and screening of large numbers of compounds have provided some unique challenges and opportunities in drug discovery. It became clear early in the application of methods, such as libraries, chemistry and high throughput screening (HTS), that success would not result simply from an increase in throughput (both in terms of the numbers of molecules synthesised and the numbers of molecules screened)2 rather, design and selection would also be very important. A key contribution in this realisation was that of Lipinski et al.,3,4 who examined a series of clinically tested drug molecules to try to determine whether they possessed any distinguishing properties. This led Lipinski to propose a “rule of fives”, which constitutes a set of simple rules designed to suggest whether or not a molecule is likely to have absorption problems due to poor solubility and/or poor permeability. The rule of fives states that poor oral absorption and/or distribution are more likely when- 1. The molecular weight (MW) is greater than 500 Da.
- 2. log P > 5.
- 3. There are more than five hydrogen bond donors (defined as the sum of OH and NH groups).
- 4. There are more than 10 hydrogen bond acceptors (defined as the number of N and O atoms).
The rule of fives is usually implemented by flagging compounds that exceed two or more of the above parameters; Lipinski and colleagues found that fewer than 10% of the data set of clinical drug candidates had any combinations of two parameters outside the desired range.
Following from Lipinski's publication several other groups reported analyses of collections of non-drugs and drugs with the aim of identifying the most probable properties that distinguish “drug-like” molecules. Such analyses were usually done by selecting and comparing sets of drug and non-drug molecules published in the literature. For example, Veber et al.5 proposed that the number of rotatable bonds (≤10) and the polar surface area (<140 Å2) were two important properties to obtain oral bioavailability in the rat. The polar surface area is defined as the part of the molecular surface due to oxygen or nitrogen atoms or from hydrogen atoms attached to nitrogen or oxygen atoms, and is a useful descriptor for QSAR analysis.6 Many of these properties are of course highly correlated.5,7 In another study,8 most “drug-like” compounds were found to have between 0 and 2 hydrogen bond donors, between 2 and 9 hydrogen bond acceptors, between 2 and 8 rotatable bonds and between 1 and 4 rings.
The Lipinski Rule of 5 is essentially an oral drug-likeness filter that can be applied both to “real” compounds (e.g. from those in a company's screening collection or being offered for purchase) and to “virtual” compounds (i.e. molecules that have not yet been made). The simplicity of Lipinski's “rule of 5” and ease of calculation were important reasons for its widespread adoption. Other types of in silico filters have been similarly proposed and used.9,10 Some of these filters are used to spot compounds that contain reactive or otherwise undesirable functionality, such as Michael acceptors, alkyl halides or aldehydes. Another type of filter uses a mathematical model, often based on simple calculated properties, to score or classify molecules according to their degree of “drug-likeness”. Several types of mathematical model have been used; these are often based on a series of molecular descriptors and other properties. Multiple linear regression, neural networks and genetic algorithms are often used to construct such models, analogous to those long-used in the field of QSAR.11
3 The evolution of lead-likeness and fragment screening
As the various concepts of drug-likeness gained credence they were incorporated into many areas of drug discovery. However, a further refinement of the concept was introduced in 1999 by Teague, Davis and Oprea from AstraZeneca.12 They analysed a series of compounds from the literature to identify the original leads that the medicinal chemists had used to develop the drug from thus coming up with pairs of leads and drugs. A variety of properties were then calculated for these pairs of molecules so as to ascertain how the properties of leads might differ (if at all) from those of the final evolved drugs. They found that many properties showed a statistically significant change in their value and this gave rise to the concept of lead-likeness as something different to drug-likeness. Several properties increased in the sense that optimised drugs are more complex than their initial leads. For example, the molecular weight and logP increase, as do the numbers of hydrogen bond donors and acceptors in going from a lead to a drug. In a further publication,8 this group expanded their data set and reported the differences shown in Table 1 between the median values of the various properties.| Property | Increase |
|---|---|
| Data from Oprea8 | |
| Molecular weight | 69 Da |
| Hydrogen bond acceptors | 1 |
| Rotatable bonds | 2 |
| Number of rings | 1 |
| C log P | 0.43 |
| Hydrogen bond donors | 0 |
Shortly after publication of the first AstraZeneca paper, Hann, Harper and Leach13 from GlaxoSmithKline (GSK) published their analysis of a much larger data set, comprising a different set of lead/drug pairs, this time derived from the extensive compendium previously published by Sneader14 (see Table 2).
Although covering different data sets, both studies demonstrated that when drug discovery programmes are analysed the initial hits have statistically different properties to those of the final drugs. There are a number of possible explanations for these observations. Initial hits from HTS are often less potent than the ultimate drug needs to be, and improving potency is often most easily achieved by adding additional functionality. This in turn increases the molecular weight together with properties such as the numbers of donors and acceptors. Thus medicinal chemists tend to add mass to a compound in pursuit of potency. Log P is another property that often increases during lead optimisation. It is not always clear whether this reflects the addition of specific hydrophobic interactions with the target (giving increased potency), or more non-specific hydrophobic interactions that yield increased potency due to increase in the apparent concentration of a lipophilic drug in the lipophilic environment of a membrane-bound target. Another reason for the differences between leads and drugs, at least in the case of the Sneader data set, is that many of the leads were small hormones such as biogenic amines. These starting points are of such low complexity (i.e. low molecular weight) that adding mass is almost the only thing that can be done in evolving the drug!
More recently other compilations and analyses of drug sets have been described which give further insights into physicochemical characteristics of drugs (ref. 56).
As a complement to these analyses of historical data, a number of groups have treated the problem of lead-likeness and its impact on drug discovery from a more theoretical perspective. One such analysis was included in the paper of Hann and colleagues13 discussed above. They presented a simple model to predict how the probability of finding a hit varies with the complexity of the molecule. In this model the ligand and its binding site are represented as simple bitstrings of interaction points. The number of interaction points in the ligand and receptor are considered as measures of the complexity of the system. The bitstrings represent molecular properties of the ligand that might influence binding, such as shape, electrostatics and other properties such as lipophilicity. In the model, a ligand has to exactly match all of the bitstrings of the binding site for the interaction to be counted as contributing to the probability of a successful interaction. Thus, each positive element in the ligand must match a negative element in the binding site and vice versa. Figure 1 illustrates a number of examples of successful and unsuccessful matches in the case of a ligand with three features and a receptor with nine. It is then possible to calculate the probability that a ligand of size L will match a binding site of size B. A typical result is shown in Figure 2 for a binding site of size 12 and varying size ligand. Thus, the probability that a ligand of size 2, 3, 4,…can match the binding site in 1, 2, 3,…ways is calculated. Also shown is the total probability that a ligand can match in any way, which is the sum of these individual matches. As shown in the figure, the chance that a ligand can match at all shows a smooth and rapid decay to zero as the complexity increases. From the point of view of drug discovery, the probability that the ligand has just one (i.e. unique) match is an important situation, as this helps the development of an unambiguous binding mode. In the example shown, this probability passes through a maximum at a ligand complexity of 3.
 | ||
| Fig. 1 For the simple Hann model an exact match between ligand and receptor has to take place for a successful interaction to be recorded. Here a ligand of complexity of three points of interaction (+ + −) is being matched in various positions against a receptor whose complexity is nine features and has the pattern (− − + + − − + − +). | ||
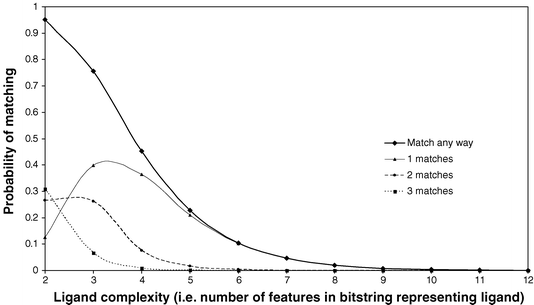 | ||
| Fig. 2 The probability of finding 1, 2 or matches (and total matches by summation) for varying ligand complexity using a receptor with 12 interaction sites in the Hann model. | ||
In the second part of the Hann model, the probability of being able to actually measure the binding of a ligand as the complexity of the interaction increases is considered. The complexity is used as a crude indicator of the likely strength of the interaction if it is able to occur following the criterion of the first part of the model. As the number of pairwise matches increases, the probability of measuring experimentally the interaction also increases. This is indicated in Figure 3 as the hyperbolic curve, indicating that if the number of interactions is below a certain number then it is not possible to measure the binding because it will be very weak. There is then a rapid increase in the probability, consistent with the notion that once the potency exceeds some threshold it will then be possible to measure the interaction and so the probability equals 1. Finally the probability of a “useful event” is defined as the product of the two probability distributions. This probability then reflects the true balance of the probability of having matching interactions and also being able to measure it.
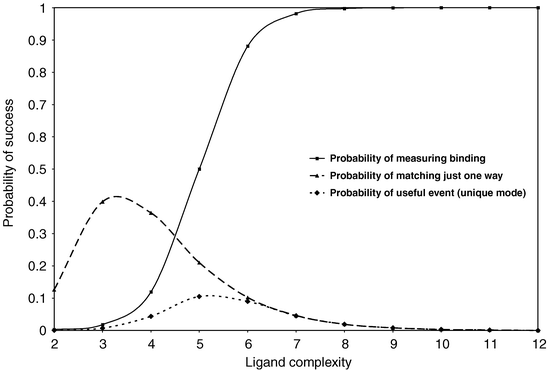 | ||
| Fig. 3 The product of multiplying the probability of a single match by the probability of being able to experimentally detect the binding event. The resulting probability of a “useful event” passes through a maximum due to the nature of the two contributing and underlying probabilities. | ||
The two probabilities can be seen to have competing distributions in that the probability of finding a match falls while the probability of measuring the interaction increases as the number of interactions contributing to the complexity of the successful interaction increases. These distributions will clearly vary for different model criteria and for real systems. However, the combined effect is that the probability of a “useful event” will always have a bell shape because of the competing component distributions. At low complexity, the probability of a useful event is zero because even though there is a high probability that the properties match there is not enough of them to contribute to an observable binding in a real assay. At high complexity, the probability of getting a complete match is vanishingly small, although if such a match does occur it will be easily measured. In the intermediate region there is the highest probability of a useful event being found. Here there is an acceptable probability of both having a match and being able to measure it.
More recently, the terms fragments and fragment screening have become synonymous with the concept of screening less complex molecules to increase the probability of finding hits. Essentially, a fragment reflects the medicinal chemist's realisation that the compounds being used are probably not of the size that will reflect the complete needs of the final drug but will however provide an attainable starting point.
Another aspect of the problem of molecular complexity and the value in using less complex ligands or fragment approaches concerns the effectiveness with which chemical space can be sampled. A number of groups have estimated how many potential “drug-like” molecules exist. This refers to molecules that might be considered to generally fall within what is considered “drug-like” space in terms of the element types, the ways in which the elements are bonded together and certain properties, such as molecular weight and the ratio of heteroatoms to carbon atoms. These estimates1,15 vary quite widely, but all agree that drug like chemical space is very large—many orders of magnitude greater than the number of compounds that have been made to date and indeed probably large enough to use up more material than that is available on Earth or probably the known universe! The challenge in drug discovery is to effectively explore the vastness of potential compounds so as to identify those that not only possess the necessary activity at the target(s) but will also have appropriate ADMET (absorption, distribution, metabolism, excretion and toxicity) properties to enable development into an effective drug.
Again these concepts are best explored with model systems and Figure 4 shows a representation of a target protein with two binding sites. If we have a set of five simple binding elements (molecular fragments) that could possibly bind at either site, then to exhaustively identify all the possible molecules that could bind to both binding sites would require the synthesis of the full 25-member combinatorial library. By contrast, if we could identify the best constituent fragments independently then we need to only synthesise one full molecule containing two active fragments. This assumes that the properties of the fragments are additive, which is not always a safe assumption in medicinal chemistry but is the default method by which drug discovery initially operates. It also requires that we have a spatial insight as to how to join them correctly. If we are able to identify molecular fragments that bind at the individual sites and then combine them, rather than trying to identify the complete molecule in one step by making the full combinatorial library, then this enables the search of chemical space to be explored iteratively and in an additive, rather than multiplicative, manner. For example, for a target containing S subsites that are combined using L linkers then the complete combinatorial library would be of the order MSLS−1 in size, where M is the number of members (or monomers) to be included in the combinatorial library. However, the fragment library would only be of the order of M members and if only these are tested initially then they can act as a surrogate for the much larger full combinatorial library providing some very dramatic savings in both synthesis and assays.
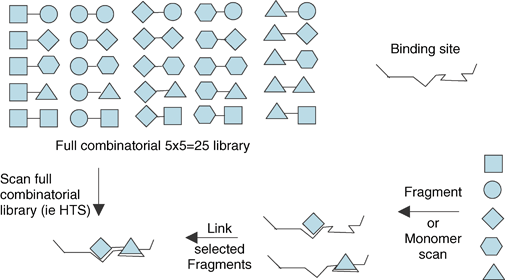 | ||
| Fig. 4 For a set of five fragments there are 25 different pairwise combinations. By contrast, if the fragments are screened individually then the desired combination can be obtained directly. | ||
Thus a 1000-member fragment library with 20 linkers would combinatorially give a 20 million-member library for a two-site target, but could be surveyed initially with 1000 assay points if the initial screening was done with the monomers.
Another way to approach these issues is to consider the amount of sampling that can be done at any given level of complexity. Provided biological properties do follow an additive (or quasi-additive) behaviour then fragment screening approaches provide better sampling of the corresponding chemical space. This can be illustrated as follows. Figure 5 shows the number of carboxylic acids (of all types) registered in the GSK registry system and has been plotted as a function of binned molecular weight (top curve). As can be seen, the number of carboxylic acids in a particular molecular weight band initially increases rapidly following an approximately exponential curve. At about 150 Da this exponential behaviour stops. Although the most populated bin is at around a molecular weight of 400 (max in top curve) the growth in numbers per bin relative to the previous one is maximal at about 150 Da (max in bottom curve). Therefore the GSK acid set is significantly under-sampling, the virtual space of carboxylic acids in a way that gets progressively worse as the molecular weight increases. Thus, when operating in a lower molecular weight region (e.g. <350 Da, typical of many fragment sets) the set of available acids provides a more effective sampling than at a higher molecular weight region (say 450 Da). This is schematically illustrated in Figure 6, which includes an extrapolation of the initial rate of increase as if all carboxylic acids were available for consideration. Such an exponential increase has been shown in an exhaustive theoretical enumeration of all possible molecules (not just acids) containing up to only 12 non-hydrogen atoms by Fink et al.,16 who estimated that there was nearly 145 million such compounds (for molecules containing up to 25 atoms, or MW ca. 350 Da, it was estimated that there was ca. 1025 possible structures!). It can readily be seen that sampling is more effective in the lower molecular weight region because there is a smaller divergence between the numbers of available compounds and the number theoretically possible.
 | ||
| Fig. 5 The number of carboxylic acids registered at GSK with a given molecular weight (top) together with the change in 25 Da increments (bottom) relative to previous bin. | ||
 | ||
| Fig. 6 Graph to illustrate that the GSK available carboxylic acids reasonably represents the total number possible for low molecular weight at higher molecular weight values the divergence of the two curves means that there is very poor representation. | ||
One of the consequences of using a fragment-based lead discovery strategy is that the activities of the molecules initially identified will often be lower than for larger, more drug-like molecules. This could result in an interesting fragment of relatively weak potency being overlooked in favour of more complex molecules that may have higher initial potency but which are ultimately less developable. As a result of this, several groups have proposed the use of binding affinities normalised by molecular weight so as to have ways to compare molecules that takes into account their size. A particularly useful concept in this context is the maximal affinity of a ligand. This is equal to the maximum free energy of interaction that a compound might be expected to express in an interaction with a biological macromolecule. This was first introduced by Andrews and colleagues17 by studying a set of ligands with known binding affinities from the literature. The functional groups present in each ligand were identified and counted and a multiple linear regression analysis was performed in order to determine the contributions of each functional group to the observed binding affinity (together with an entropy term related to the freezing of translational and rotatable degrees of freedom). These are given in Table 3 although it should be borne in mind that there are large standard deviations likely on each figure due to the method used. Summing the corresponding contributions for any novel ligand thus gives a maximal binding energy that might be expected if all functional groups make their optimal contribution.
| Functional group | Energy (kcal−1 per group) |
|---|---|
| sp2carbon | 0.7 |
| sp3 carbon | 0.8 |
| N+ | 11.5 |
| N | 1.2 |
| CO−2 | 8.2 |
| OH | 2.5 |
| C=O | 3.4 |
| O, S | 1.1 |
| Halogens | 1.3 |
| PO2−4 | 10.0 |
Use of this in HTS analysis throws up a key issue. The difference in coefficients for a protonated and non-protonated amine is very high—∼10kcal, meaning that the choice of whether an amine is protonated or not in the binding site can alter the estimate by 8 log units of potency—essentially making the prediction very unreliable in its absolute sense. Nevertheless as an intellectual tool, this remains valuable.
More recently, Kuntz and colleagues18 analysed a data set of 160 ligands. Figure 7 shows the ratio of binding affinity to heavy atom plotted as a function of the number of heavy atoms in the ligand. The initial slope of this graph has a value of approximately 1.5 kcal mol−1 and this was therefore proposed as the maximal free energy contribution per heavy atom averaged across all functional group types represented in the 160 ligands.
 | ||
| Fig. 7 Free energy of binding plotted against number of heavy atoms. The initial line has a slope of 1.5 kcal/mol-atom18 | ||
When a molecule contains more than ca. 15 heavy atoms the free energy tends to increase little with molecular mass. The reasons for this are not fundamental thermodynamic ones, but due to the properties of very tight binding ligands (such as very long dissociation times). Moreover, many assays are configured so that affinities greater than nanomolar cannot be effectively measured. This results in an artificial ceiling to the plot.
Although the initial line has a slope of 1.5kcal/mol-atom, all molecules with less than six heavy atoms form interactions that would not be considered representative of typical drug molecule interactions (e.g. heavy metals and carbon monoxide). If the data are reanalysed by excluding such compounds and focussing on the compounds with less than 25 heavy atoms (i.e. <∼330 Da) then a different conclusion can be drawn (see Figure 8).
 | ||
| Fig. 8 Plot of free energy of binding per heavy atom vs. number of heavy atoms for more restricted set of fragment like compounds | ||
Now the initial high binding per atom drops quite quickly and almost asymptotically to around 0.3kcal/heavy atom, and this fits better with the results published by other researchers at Pfizer. They coined the term “ligand efficiency” for the experimental binding affinity per heavy atom and have proposed that it is a useful parameter to use when prioritising the output from HTS or other screening strategies.19 They suggest a lower limit on the ligand efficiency, which can be estimated by assuming that the goal is to achieve a binding constant of 10nM in a molecule with molecular weight of 500 Da (as needed to be consistent with Lipinski's rules). An analysis of the Pfizer screening collection revealed that the mean molecular mass for a heavy atom in their “drug-like” compounds is 13.3 and so a molecule with a molecular weight of 500 Da and a binding constant of 10 nM would have 38 heavy atoms and therefore a ligand efficiency of 0.29 kcal mol−1 per heavy atom. This is significantly less than the maximal value of 1.5 kcal mol−1 per atom (Figure 6), but fits better with the more drug-like molecules found in the Kuntz data set (Figure 8). The Pfizer proposal was that the hits with the highest ligand efficiencies are the best ones to consider for optimisation, provided that all other factors such as synthetic accessibility are equal.
An extension of these ideas enables other properties to be taken into account. Thus, to achieve compounds with a not too high log P while still retaining potency, the difference between the log potency and the log D can be utilised. Burrows and colleagues,20 at AstraZenecca, have proposed that when this term is greater than 2 log units then it is likely that the compound will be a good lead compound. Further analysis and comparison of a number of potential ligand efficiency metrics from a survey of drug hunting projects that delivered clinical candidates Griffen et al.,55 suggested two other metrics with statistical validity: potency (pIC50)/non-hydrogen atom >0.2 and potency minus serum protein binding affinity constant (logKapp) > 1. Simultaneous expansion of these ideas was provided by Abad-Zapatero and Metz,21 who in addition to normalising the binding affinity by molecular weight (i.e. ligand efficiency), also defined a surface-binding efficiency index in which the polar surface area was used as the normalising factor.
When a molecule with low nanomolar potency is split into two fragments the individual potencies of the derived fragments will be considerably less than half of the potent molecule. The converse, of course, is that if one can successfully link two weakly potent fragments then this may afford a low nanomolar compound. Why the expected affinity of the joined molecule should be larger than the sum of the affinities of the two individual fragments has been extensively discussed in the literature.22,23 The reason is that a fragment loses significant rigid-body translational and rotational entropy when it forms the intermolecular complex. This unfavourable entropic term is only weakly dependent on molecular weight. Thus, whereas two unfavourable terms have to be overcome when two fragments bind, this is replaced by just one unfavourable term for the combined molecule. This however does ignore the fact that combining the two separate binding entities into one may also not be completely compatible with the binding configuration and conformation of the individual entities as first identified.
Another theoretical insight into drug discovery using small molecular fragments was provided by Rejto and Verkhiver,24 who hypothesised that the primary molecular recognition event between a protein binding site is due to a core fragment, or “molecular anchor” which can then be grown into a complete ligand. Their initial analysis was on the interactions between the FK506 binding protein (FKBP-12) and fragments of its inhibitor FK506 (Figure 9). FK506 contains a pipecolinyl moiety that is the key anchor into the binding site. As the pipecolinyl moiety has only a weak binding affinity per se, they argued that the binding affinity might not be the only factor in determining when a fragment is a good molecular anchor.
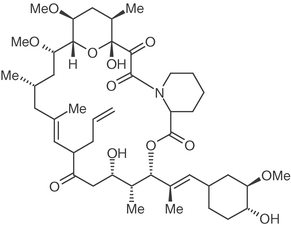 | ||
| Fig. 9 Structure of FK506. | ||
When several different fragments derived from the FK506 structure were docked into the binding site on FKBP-12 it was seen that the pipecolinyl fragment settled into a single binding mode, while the other fragments gave multiple binding modes of comparable (predicted) binding affinity. Therefore, the binding energy landscape for a successful molecular anchor needs to have one binding mode much lower in energy than the next lowest level; by contrast unsuccessful fragments (of a similar size) would have an energy spectrum in which many different binding modes would have approximately the same energy (Figure 10). This stability gap was the unique feature of successful fragments compared with random ones.
 | ||
| Fig. 10 The preferred binding energy distribution for a molecular anchor must have a significant gap between the lowest and the next lowest energy mode (left) rather than many binding modes of similar energy (right). In this way a unique binding mode is adopted. | ||
4 Finding fragments by screening
Advances in HTS technologies has allowed increasingly large compound collections to be effectively screened for the desired activity. Typically HTS is performed with ligand concentrations around 10 μM and is regularly successful in hit generation; however a number of screens do still fail to deliver attractive hit series vs. targets. Some of these targets are a priori not believed to be chemically interactable (i.e. the molecular properties necessary to bind to the target protein and illicit a biological response are compatible with those properties necessary for oral dosing).25 This can be explained in various ways including (i) assays or assay technology configured inappropriately, (ii) problems with screening collections (solubility and stability)26 or (iii) lack of appropriate compounds in the screening library, i.e. no potent actives in the library ‘waiting’ to be found. This is unfortunate given the time, effort and expense in building, running and analysing such libraries and screens. One way to avoid these two latter issues is to ensure that screening collections are continually updated with new compounds which are derived from novel and ‘lead-like’ libraries of compounds (through synthesis or acquisition) and which cover the chemical space that is missing (as discussed above).12 In the absence of any knowledge of where to start it is often difficult to design novel enriching libraries other than to ensure chemical diversity compared to that which is already available and that the compounds are chemically tractable for further analogue constructions and have favourable drug metabolism and pharmacokinetic (DMPK) properties.One alternative approach to this whole problem is to screen libraries of ‘ultra-lead like’ fragments (which are much smaller than compounds typically screened in HTS) at much higher concentrations allowing for the detection of probably only weakly binding compounds but which are small and novel as starting points from which new areas of chemistry can be developed.54 Not only is this attractive on the already discussed theoretical arguments but it can also be extremely valuable in identifying truly novel cores, scaffolds, and warheads which can then give competitive advantage in terms of patentability. Analogues or libraries prepared from these fragment hits can then significantly enhance the diversity of the original library and hopefully give a way in dealing with related targets that may have been previously intractable.
There are essentially two ways in which fragment screening can be carried out. First biochemical screening (often referred to as High Concentration Screening (HCS)) and second biophysical and direct structure based screening (using, for instance, NMR or X-ray methods).27–29
4.1 High concentration screening using a biochemical assay
This involves the use of typical biochemical assays but performing them in such a way that they are robust to higher concentrations of ligands that are to be tested. This concentration is typically in the 1 mM range. The major advantages of this approach are that the assays are fast, quantitative in principle, and use widely available technologies for detection. Also, only small amounts of protein are necessary and consequently assays involving GPCR or ion channel targets may be considered. However, there are also many potential problems with this approach. Not every assay is suitable for this approach; for instance, the concentrations of added ligand may interfere with the assay through undesirable mechanisms or may be toxic to a cell, if the assay is cell-based. In addition, there can be problems with the identification of false positives as a result of compound aggregation at the concentration of ligands used,30 interference with assay end point (e.g. optical interference fluorescence, quenching, toxicity, etc.)9,31 or disruption of the protein by unfolding or precipitation. Additionally, false negatives can occur due to lack of effective solubility of compounds.4.2 Biophysical and direct structure determination screening
An alternative approach is to screen at high concentration using a more direct biophysical assay or structure determination.In addition, for 2D methods, labelled protein is required which results in further expense, time and effort. It is much slower than a biochemical assay but can be faster than X-ray crystallography if appropriate protein is available.
In general, these biophysical and structure-based methods are more robust than biochemical screening though not always technically feasible. Alternative direct biophysical approaches include affinity detection by mass spectrometry34 and surface plasmon resonance (BIAcore).27
Another method that has been pioneered at Sunesis involves the introduction of tags (usually individual cysteine residue) into the environment of the binding site. These are then used to capture (by disulfide formation) probes that also contain a free thiol moiety. These probes are in a library of fragments, which can be screened against the protein, and binding is detected by a mass spectrometric procedure. The disposition of bound fragments is then found by protein crystallography. The Sunesis group have shown that fragments can often adopt novel insertion modes into the protein surface and that these fragments can then be grown (directed by structure-based design) to give larger molecules with more interactions. Eventually, the disulfide tag is dispensed so as to leave a non-covalent compound with specific and novel interactions. This method has most recently been exploited to explore the design of novel GPCR inhibitors.35
In practice, screening for fragments is often performed using a variety of the above approaches, e.g. biochemical screening followed by X-ray crystallography on the hits. The real value of the NMR and/or X-ray methods is that they give structural insights which aid in decisions about what to make next in the search for increased potency and specificity.
5 The design of fragment screening sets
Three key issues need to be considered when designing and implementing a fragment-screening library: (1) how many molecules are included in the set? (2) which molecules to include in the set? and (3) which method(s) is going to form the basis for the detection of binding? As these problems are closely related a compromise often has to be reached. All experimental assay techniques impose some kind of constraint on the properties of the molecules involved. Thus the size of a library that is needed will be critically dependent on the expected testing concentration, as this will affect the number of compounds that can be followed up. For instance, an HTS library (screened at 10 μM) might include 106 compounds, a high concentration library (screened at 100 μM) might include 104 compounds and a small fragment library (to be screened by NMR or X-ray at 1 mM) might include 103 compounds.There are many approaches that can be used for the actual design of the content of screening sets. Generally, the objective is to cover appropriate chemical space with a testable number of compounds and to use existing knowledge to weigh compound selection. One simple, general approach to the problem considers a process whereby a core compound set is established based on knowledge. To this is added an outer layer of compounds based on neighbours, diversity, calculated properties, and other information (Figure 11).
 | ||
| Fig. 11 Concept of growing a screening collection. | ||
A distinction is often made between the large, “diverse” sets such as those used in HTS and the smaller, “focussed” sets that are selected with a specific target or group of related targets in mind. The methods that can be used to construct diverse and focussed screening sets have been discussed and reviewed extensively in the literature and so will not be considered in detail, except where there are specific issues arising from the use of fragments. One factor that is worth considering here is the balance between diversity and focus. The knowledge plot shown in Figure 12 is a particularly useful way of representing this balance. This relates the different levels of knowledge about targets to the level of diversity required in the screening set. This diagram suggests that the need for diversity is inversely proportional to the knowledge that is available on the biological target.
 | ||
| Fig. 12 The knowledge plot illustrates that the degree of diversity needed has an inverse relationship to the amount of knowledge that is available to be used. | ||
The key factors to be considered when constructing a fragment library include:
- 1. Availability. Screening compounds at 1 mM will require a 100 mM DMSO concentration if a final DMSO concentration of 1% is acceptable. Depending on the volume required this can easily consume 25 mg of the compound.
- 2. Availability of close analogues. It is a great advantage to have available close analogues of any hits in the screening library to help confirm actives, to build up clusters and define SAR. Alternatively, ready access to analogues by array synthesis is useful.
- 3. Solubility. Although related to lipophilicity, other factors contributing to solubility are difficult to predict and model and therefore this property can only be safely obtained through measurement.
- 4.
- Purity
- . Given compound concentrations and the sensitivities of many assays, levels of contaminants or impurities need to be kept to a minimum to avoid identification of false positives, particularly those impurities that could lead to irreversible inhibition. This is usually ascertained through
- LCMS
- and NMR on each sample in the library.
- 5. Molecular size/ weight. Lipophilicity and other parameters need to be constrained (see below).36
- 6. Absence of reactive functionality. Compounds containing such moieties can be removed using computationally derived filters.37
- 7.
- Opportunity for synthetic elaboration
- . Clearly, it is important that this ‘scope’ is properly defined in discussion with medicinal chemists. A carboxylic acid may provide a handle for further elaboration but if a fragment hit possessing an acid is identified it is likely that the acid provides a key interaction
- per se
- that would be destroyed by further chemistry (
- e.g
- . amide formation). Usually, the inclusion of synthetic handles on library compounds is a decision based on an organisational rationale (
- i.e
- . hits with functional groups that can be readily derivatised are more likely to be followed up for operational reasons). Although this is seductive, it makes more sense to first understand the binding mode of a fragment and then evolve or join fragments using linking groups that will give the correct geometry for further improvements in binding, rather than letting synthetic ease drive towards inactive compounds. Screening prototypical reaction products,
- e.g.
- methylated oxime fragments,
- circumvents the incompatibility of the nature of the fragment “hit” and its subsequent chemical transformation.
- 8. Reduced chemical complexity. As has been stated a complex molecule is less likely to be able to use all its features for binding and the number of compounds required to cover chemical space increases with molecular size and complexity.53
This latter point can be illustrated in practical terms by considering the interaction of a benzoate with two different guanidine containing active sites (Figure 13).53 The less complex benzoate can interact with both sites, whereas the two more complex examples described have greater specificity. Naturally it may be that the less complex benzoate is a weaker binder—which demonstrates the balance required between the number of compounds in a set and their complexity and the concentration at which the set is screened.
 | ||
| Fig. 13 Increased complexity gives increased specificity but only if it is compatible with the binding site. | ||
Several computational techniques have been used in the selection of fragments to include in lead-like screening sets. A key parameter is the physicochemical properties of the ligands and a more restrictive set of parameters, than typical Lipinski criteria, is often used. Within the lead-likeness arena an analogous rule has also been adopted at Astex.36 These workers analysed the hits obtained by screening their own collections of fragments using X-ray crystallography at a variety of targets (e.g. kinases and proteases). From this analysis evolved the idea that a “rule of threes” might be appropriate to help in selecting good fragments to try. This rule requires that the molecular weight be <300; that the number of hydrogen bond donors and hydrogen bond acceptors should be ≤3, and that the calculated octanol/water partition coefficient (using C log P) should be ≤3. Three or fewer rotatable bonds and a polar surface area of 60 Å2 or less were also proposed as useful criteria.
As some of the screening techniques used in fragment-based discovery are limited in capacity (compared to HTS), it is usually necessary to further refine the initial set of compounds that meet such simple filters; some form of further selection is required. One useful way to do this is to identify fragments related to those that commonly occur in drug-like molecules. Certain fragments (often referred to as privileged structures) are those that appear frequently in drug molecules. While some of the most common of these privileged structures have been defined “by hand”, a number of computational methods have also been developed to systematically identify appropriate fragments from collections of drug-like molecules. The fragments may be sorted by frequency and after removal of trivial examples, such as simple alkyl groups, the highest scoring fragments are selected.
Bemis and Murcko39,40 defined an hierarchial approach in which a molecule is converted into its graphical representation and then broken down into sub-ring systems, linker atoms and side chains. The ring systems and linkers together define a framework, as illustrated in Figure 14. The top scoring frameworks were identified by applying this algorithm to the Comprehensive Medicinal Chemistry database and are shown in Figure 15. It was found that just 32 frameworks (after removing atom types) accounted for 50% of the 5120 drug molecules in the entire set. An alternative approach that is widely used is the Retrosynthetic Combinatorial Analysis Procedure (RECAP).41 In RECAP the fragmentation is performed by successively cleaving bonds that can be easily formed in a reaction sequence, such as amides and ethers. These methods can be further used to help in identifying more appropriate synthetic fragments that may be directly used in further array chemistries.
 | ||
| Fig. 14 The creation of rings, linkers, side chains and frameworks from the molecular graph. | ||
 | ||
| Fig. 15 Top-scoring frameworks in drug molecules as identified by Bemis and Murcko. | ||
Several groups have published on how they have derived fragment-inspired screening collections.
- At GSK the term reduced complexity screening (RCS) is used to cover the fragment screening activities. For this purpose a screening set was developed by taking a large set of available in-house and external compounds and applying a series of 2D substructure and property to identify potential candidates for inclusion in the set (heavy atoms < 22, rotatable bonds < 6, donors < 3, acceptors < 8, C log P < 2.2). The selection criteria also required there to be a synthetic handle present in order to facilitate the rapid synthesis of further analogues. Note the use of heavy atoms rather than molecular weight as a criterion as this avoids deselecting molecules, which may contain, for instance bromine atoms, which may prove to be useful synthetic handles at a later stage. The GaP diversity measure based on 3D pharmacophore keys was then used to select a subset of compounds from the initial filtered selection.42,43
- Scientists at AstraZeneca have used a broadly similar approach to select a set of 2000 compounds for what they term HCS. This set was designed to have a roughly equal proportion of acidic, basic and neutral compounds (with a small number of zwitterionic molecules) and with a pre-defined physicochemical property distribution.20
- Astex scientists have described the construction of screening sets for use in X-ray crystallographic fragment screening.
- Again, sets directed against a specific target or groups of related targets have been constructed together with a more general-purpose set. The starting point for the latter was a fragmentation analysis of
- molecules, which identified a small set of commonly found, simple organic ring systems. These ring systems were then combined with a set of desirable side chains. Three sources of side chains were used: those observed frequently in
- drug
- molecules, lipophilic/secondary side chains (intended to pick up hydrophobic interactions in a protein binding site) and a set of nitrogen substituents. Each of the relevant side chains was combined with the ring systems to give a virtual library (of size 4513); the structures in this virtual library were then compared against databases of compounds available from external sources giving a final set of 327 compounds.
- At Vernalis, scientists have described four generations of a low molecular weight fragment library for use in NMR-based screening.
- 45
- As in other examples,
- in silico
- property calculations were developed to automate the selection process. Both “general-purpose” sets together with those directed towards particular protein targets (kinases) were constructed. 3D-based descriptors were also used (analogous to those employed by the GSK group) as a measure of diversity and complexity.
6 Turning fragment hits into leads
As it is frequently the case that the activity of initial hits will be weak (sometimes very weak), then a key requirement is to increase the potency. This is done by synthetic manipulations to the structure to give a sustainable lead series, which will be more akin to a series that might be found from traditional HTS. Other properties besides potency will also need to be taken into account (e.g. selectivity against other targets and ADMET properties). However, these non-efficacy parameters need to be balanced vs. the potency/complexity of the compound. For instance, oral bioavailability data on a fragment can be wholly inappropriate, given the potential for altered absorption mechanisms (paracellular vs. transcellular) and even the data from many in vitro screens (e.g. P450 inhibition) are unlikely to be very useful as the structure is likely to evolve substantially from the early hit.A number of scenarios for getting potent drug-like molecules from fragment lead-like hits have been identified:29 fragment evolution, fragment linking, fragment self-assembly and fragment optimisation.
6.1 Fragment evolution
Fragment evolution is most like standard lead optimisation, requiring the addition of functionality that binds to additional parts of the target protein. The fact that the starting point is a small molecule means that there should be plenty of opportunity for this approach before hitting the “Lipinski rules”. Where structural information is available on the binding mode of the initial fragment hit, e.g. from X-ray crystallography29 or NMR spectroscopy,46 then structure-based design approaches can give rapid direction and progress.53 When structural information is not available then the screening of appropriate analogues of the original hit would be performed in order to try and establish a traditional structure–activity relationship.Scientists at AstraZeneca20 have summarised the profiles of fragment “hits” vs. a range of different target classes which, through active drug discovery programmes, have delivered “hits” from these fragments that are comparable to those delivered via alternative hit generation strategies (Figure 16). In each of these cases fragment evolution has been the adopted strategy and in many cases, X-ray crystallography and NMR have supported.
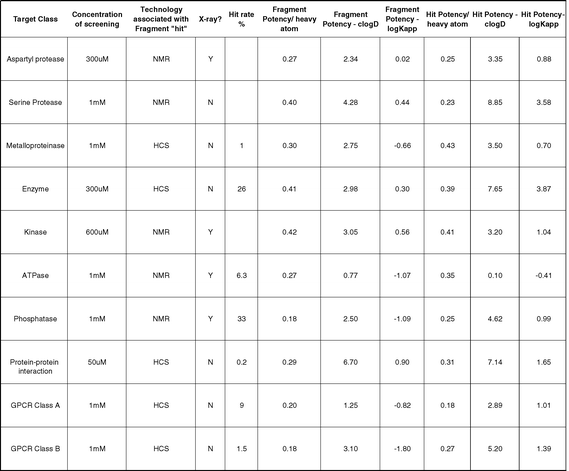 | ||
| Fig. 16 “Fragment” refers to initial weak fragment hit and “hit” refers to compound profile typical of series post-HTS at the start of Hit-to-Lead work. Ligand efficiencies for fragment and hit are given in units of pIC50/non-H atom. In addition, potency relative to C log D and log Kapp (binding constant to albumin) are given for information. HCS = high concentration screening. | ||
Two examples from AstraZeneca for two GPCR targets have demonstrated the rapidity with which progress can be made even without synthesis.47 First, screening a set of 600 compounds vs. a Class A GPCR identified 72 actives at 1 mM of which 60 actives repeated from solid retest. One of these actives (pIC50 3.2) had a near-neighbour search performed against it and in the next screening round a more complex potent neighbour (pIC50 5.4) was identified. Interestingly, one of the original fragment hits turned out to have a potency approaching 10 μM (pIC50 4.9)—a small highly attractive start point for chemistry; so fragment screening can unearth potent hits with high ligand efficiency. Second, the same set of 600 compounds was screened vs. a class B GPCR and this led to 29 actives, of which nine repeated from solid retest. Near-neighbour screening of a weak hit (pIC50 3.2) then promptly identified a more complex neighbour to be a submicromolar antagonist (pIC50>6).
Scientists at AstraZeneca were interested in developing novel inhibitors of the phosphatase PTP1b.48 Following some excellent medicinal chemistry design, a sulfahydantoin motif (compound 1) was synthesised as a phosphate mimetic and screened vs. the target. This fragment demonstrated weak activity and was first confirmed as a binder by NMR (∼1–3 mM). Subsequently, the X-ray crystal structure of this fragment bound to the target was solved showing it to be adopting a specific high-energy conformation as well as clearly identifying other potential binding sites (Figure 17).
 | ||
| Fig. 17 X-ray crystal structure of sulfur hydantoin (compound 1) bound in PTPIB. | ||
Consequently, an ortho-substituent was inserted to stabilise the twisted aryl-sulfahydanto in conformation (compound 2, 150 μM) and then an additional aryl ring was added that gave a further rise in potency (compound 3). Thus, following the X-ray information, in the process of a few well-designed compounds' potency was improved from 3 mM to 3 μM as shown in Figure 18.
 | ||
| Fig. 18 Evolution of PTP1b inhibitors from initial 3 mM hit compound. | ||
Another example of the fragment evolution approach is in the design and synthesis of DNA gyrase inhibitors.49 In this example, a set of potential low molecular weight inhibitors (termed “needles” at Roche) was docked into the active site or was identified via 3D pharmacophore searching. From these calculations a set of compounds was identified for testing; from a total of ca. 3000 compounds tested 150 real hits were obtained. Various hit validation techniques were employed to confirm which of these bound to the desired active site. The subsequent optimisation then made extensive use of X-ray structural information; during the optimisation the activity of the compounds increased by approximately four orders of magnitude (Figure 19).
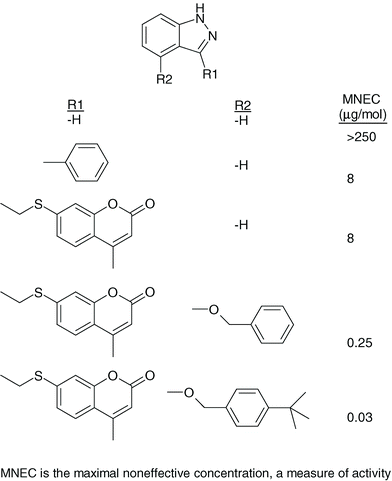 | ||
| Fig. 19 Evolution of indazole inhibitors of DNA gyrase. | ||
6.2 Fragment linking
This is illustrated in Figure 20; this involves joining two fragments that have been identified to bind at adjacent sites. Even in those cases where it is possible to find fragments binding to more than one site, the linking step can also be difficult to achieve. Having access to structural information is almost a perquisite: otherwise there is a large combinatorial and random search to be done in order to find the effective linking scheme. | ||
| Fig. 20 Fragment linking schematic representation. | ||
An example of the fragment-based linking approach was the identification of a potent inhibitor of the FK506-binding protein (FKBP) using the SAR-by-NMR method developed at Abbott.50 First, compounds that bound weakly to FKBP were identified. These included a trimethyoxyphenyl pipecolinic acid derivative (Kd=2.0 μM). A second round of screening was then performed using the same library but in the presence of saturating amounts of this pipecolinic acid fragment. This led to the identification of a benzanilide derivative that bound with an affinity of 0.8 mM. Screening of close analogues enabled the SAR to be expanded, and thus a model for the binding of these fragments to be developed. Four compounds that linked the two sites were then synthesised and found to have nanomolar activities (see Figure 21).
 | ||
| Fig. 21 FK506 fragment linking example.50 | ||
6.3 Fragment self-assembly
This involves the use of reactive fragments that link together to form an active inhibitor in the presence of the protein target. The essence of the approach is that the protein serves as a template and therefore selects those combinations of reagents that act as inhibitors. One of the first examples of this method was the reaction between four amines and three aldehydes to give imines (subsequently reduced to amines).51 Although 12 possible amines could arise from this reaction, when performed in the presence of carbonic anhydrase, the proportion of one specific amine was increased at the end of the reaction and this was presumed to correspond to the most active inhibitor (Figure 22). | ||
| Fig. 22 Fragment self-assembly brings together components on the active site, which are then able to react together with each other. Example shown is for inhibitors of carbonic anhydrase. | ||
6.4 Fragment optimisation
This involves the optimisation or modification of only a part of the molecule, often to enhance properties other than the inherent potency of the original molecule or to deal with some problem. An example of this approach is the incorporation of alternative S1-binding fragments into a series of trans-lactam thrombin inhibitors.13 The complexity of the synthesis of the trans-lactam system made it desirable to have a mechanism to prioritise potential S1 substituents in advance of committing chemistry resource (Figure 23). A novel proflavin displacement assay was developed to identify candidate fragments that bound at S1. This was possible because proflavin had been shown by X-ray crystallography to bind into the S1 pocket of thrombin and this provided the basis for a simple absorbance-based assay to assay at high concentration fragments which might bind just in S1. One fragment so discovered was 2-aminoimidazole whose binding mode in this region of the enzyme was then confirmed using X-ray crystallographic analysis.52 It was subsequently incorporated into the trans-lactam series of inhibitors. | ||
| Fig. 23 2-Amino imidazole was identified as a novel thrombin S1 binding group. This moiety was subsequently incorporated into the trans-lactam series as shown; an illustration of fragment optimisation. | ||
7 Summary
There are clearly many different philosophies associated with adapting fragment screening into mainstream Drug Discovery Lead Generation strategies. Scientists at Astex, for instance, focus entirely on strategies involving use of X-ray crystallography and NMR. However, AstraZeneca uses a number of different fragment screening strategies. One approach is to screen a 2000 compound fragment set (with close to “lead-like” complexity) at 100 μM in parallel with every HTS such that the data are obtained on the entire screening collection at 10 μM plus the extra samples at 100 μM; this provides valuable compound potency data in a concentration range that is usually unexplored. The fragments are then screen-specific “privileged structures” that can be searched for in the rest of the HTS output and other databases as well as having synthesis follow-up. A typical workflow for a fragment screen within AstraZeneca is shown below (Figure 24) and highlights the desirability (particularly when screening >100 μM) for NMR and X-ray information to validate weak hits and give information on how to optimise them.20 | ||
| Fig. 24 Workflow used at AZ for making use of HCS methods. | ||
In this chapter, we have provided an introduction to the theoretical and practical issues associated with the use of fragment methods and lead-likeness. Fragment-based approaches are still in an early stage of development and are just one of many interrelated techniques that are now used to identify novel lead compounds for drug development. Fragment based screening has some advantages, but like every other drug hunting strategy will not be universally applicable. There are in particular some practical challenges associated with fragment screening that relate to the generally lower level of potency that such compounds initially possess. Considerable synthetic effort has to be applied for post-fragment screening to build the sort of potency that would be expected to be found from a traditional HTS. However, if there are no low-hanging fruit in a screening collection to be found by HTS then the use of fragment screening can help find novelty that may lead to a target not being discarded as intractable. As such, the approach offers some significant advantages by providing less complex molecules, which may have better potential for novel drug optimisation and by enabling new chemical space to be more effectively explored. Many literature examples that cover examples of fragment screening approaches are still at the “proof of concept” stage and although delivering inhibitors or ligands, may still prove to be unsuitable when further ADMET and toxicity profiling is done. The next few years should see a maturing of the area, and as our understanding of how the concepts can be best applied, there are likely to be many more examples of attractive, small molecule hits, leads and candidate drugs derived from the approaches described.54
References
- P. Ertl, Cheminformatics analysis of organic substituents: Identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups, J. Chem. Inf. Comput. Sci., 2003, 43, 374–380 CrossRef CAS.
- D. F. Horrobin, Innovation in the pharmaceutical industry, J. Roy. Soc. Med., 2000, 93, 341–345 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef.
- C. A. Lipinski, Drug-like properties and the causes of poor solubility and poor permeability, J. Pharmacol. Toxicol., 2001, 44, 235–249 Search PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- D. E. Clark and S. D. Pickett, Computational methods for the prediction of ‘drug-likeness’, Drug Discovery Today, 2000, 5, 49–58 CrossRef CAS.
- H. van de Waterbeemd, D. A. Smith and B. C. Jones, Lipophilicity in PK design: Methyl, ethyl, futile, J. Comput. Aided Mol. Des., 2001, 15, 273–286 CrossRef CAS.
- T. I. Oprea, A. M. Davis, S. J. Teague and P. D. Leeson, Is there a difference between leads and drugs? A historical perspective, J. Chem. Inf. Comput. Sci., 2001, 41, 1308–1315 CrossRef CAS.
- G. M. Rishton, Nonleadlikeness and leadlikeness in biochemical screening, Drug Discovery Today, 2003, 8, 86–96 CrossRef CAS.
- W. P. Walters and M. A. Murcko, Library filtering systems and prediction of drug-like properties, in Methods and principles in medicinal chemistry, Vol 10 (Virtual Screening for Bioactive Molecules), ed. H. J. Bohm and G. Schneider, 2000, pp. 15–32 Search PubMed.
- A. R. Leach and V. J. Gillet, An Introduction to Cheminformatics, Kluwer, Dordrecht, 2004 Search PubMed.
- S. J. Teague, A. M. Davis, P. D. Leeson and T. Oprea, The design of leadlike combinatorial libraries, Angew. Chem., Int. Ed., 1999, 38, 3743–3748 CrossRef CAS.
- M. M. Hann, A. R. Leach and G. Harper, Molecular complexity and its impact on the probability of finding leads for drug discovery, J. Chem. Inf. Comput. Sci., 2001, 41, 856–864 CrossRef CAS.
- W. Sneader, Drug Prototypes and their Exploitation, Wiley, New York, 1996 Search PubMed.
- T. I. Oprea, Property distribution of drug-related chemical databases, J. Comput. Aided Mol. Des., 2000, 14, 251–264 CrossRef CAS.
- T. Fink, H. Bruggesser and J.-L. Reymond, Virtual exploration of the small-molecule chemical universe below 160 Daltons, Angew. Chem., Int. Ed., 2005, 44, 1504–1508 CrossRef CAS.
- P. R. Andrews, D. J. Craik and J. L. Martin, Functional group contributions to drug-receptor interactions, J. Med. Chem., 1984, 27, 1648–1657 CrossRef CAS.
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, The maximal affinity of ligands, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9997–10002 CrossRef CAS.
- A. L. Hopkins, C. R. Groom and A. Alex, Ligand efficiency: A useful metric for lead selection, Drug Discovery Today, 2004, 9, 430–431 CrossRef.
- J. N. Burrows, High concentration screening: Integrated lead generation. Oral presentation, Soc. Med. Res.: Trends Drug Discovery, 2004 Search PubMed http://webcasts. prous.com/SMR_DEC_2004/program.asp.
- C. Abad-Zapatero and J. T. Metz, Ligand efficiency indices as guideposts for drug discovery, Drug Discovery Today, 2005, 10, 464–469 CrossRef.
- M. I. Page and W. P. Jencks, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1678–1683 CAS.
- C. W. Murray and M. L. Verdonk, The consequences of translational and rotational entropy lost by small molecules on binding to proteins, J. Comput. Aided Mol. Des., 2002, 16, 741–753 CrossRef CAS.
- P. A. Rejto and G. M. Verkhivker, Unraveling principles of lead discovery: From unfrustrated energy landscapes to novel molecular anchors, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8945–8950 CrossRef CAS.
- R. Deprez-Poulain and B. Deprez, Facts, figures and trends in lead generation, Curr. T. Med. Chem., 2004, 4, 569–580 Search PubMed.
- C. A. Lipinski, Chemistry quality and the medicinal chemistry-biology interface. Oral presentation, 6th Winter Conference on Medicinal and Bioorganic Chemistry, 2004 Search PubMed.
- D. A. Erlanson, R. S. McDowell and T. O'Brien, Fragment-based drug discovery, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS.
- R. Carr and H. Jhoti, Structure-based screening of low-affinity compounds, Drug Discovery Today, 2002, 7, 522–527 CrossRef CAS.
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Fragment-based lead discovery, Nat. Rev. Drug Discovery, 2004, 3, 660–672 CrossRef CAS.
- S. L. McGovern, B. T. Helfand, B. Feng and B. K. Shoichet, A specific mechanism of nonspecific inhibition, J. Med. Chem., 2003, 46, 4265–4272 CrossRef CAS.
- G. M. Rishton, Reactive compounds and in vitro false positives in HTS, Drug Discovery Today, 1997, 2, 382–384 CrossRef CAS.
- D. Lesuisse, G. Lange, P. Deprez, D. Bénard, B. Schoot, G. Delettre, J.-P. Marquette, P. Broto, V. Jean-Baptiste, P. Bichet, E. Sarubbi and E. Mandine, SAR and X-ray: A new approach combining fragment-based screening and rational drug design: Application to the discovery of nanomolar inhibitors of Src SH2, J. Med. Chem., 2002, 45, 2379–2387 CrossRef CAS.
- P. J. Hajduk, G. Sheppard, D. G. Nettesheim, E. T. Olejniczak, S. B. Shuker, R. P. Meadows, D. H. Steinman, G. M. Carrera, P. A. Marcotte, J. Severin, K. Walter, H. Smith, E. Gubbins, R. Simmer, T. F. Holzman, D. W. Morgan, S. K. Davidsen, J.B. Summers and S.W. Fesik, Discovery of potent nonpeptide inhibitors of stromelysin using SAR by NMR, J. Am. Chem. Soc., 1997, 119, 5818–5827 CrossRef CAS.
- F. J. Moy, K. Haraki, D. Mobilio, G. Walker, R. Powers, K. Tabei, H. Tong and M. M. Siegel, MS/NMR: A structure-based approach for discovering protein ligands and for drug design by coupling size exclusion chromatography, mass spectrometry, and nuclear magnetic resonance spectroscopy, Anal. Chem., 2001, 73, 571–581 CrossRef CAS.
- E. Buck and J. A. Wells, Disulfide trapping to localize small-molecule agonists and antagonists for a G protein-coupled receptor, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2719–2724 CrossRef CAS.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, A ‘rule of three’ for fragment-based lead discovery?, Drug Discovery Today, 2003, 8, 876–877 CrossRef.
- P. Roche, O. Schneider, J. Zuegge, W. Guba, M. Kansy, A. Alanine, K. Bleicher, F. Danel, E.-M. Gutknecht, M. Rogers-Evans, W. Neidhart, H. Stalder, M. Dillon, E. Sjögren, N. Fotouhi, P. Gillespie, R. Goodnow, W. Harris, P. Jones, M. Taniguchi, S. Tsujii, W. von der Saal, G. Zimmermann and G. Schneider, Development of a virtual screening method for identification of ”Frequent Hitters” in compound libraries, J. Med. Chem., 2002, 45, 137–142 CrossRef.
- D. J. Maly, I. C. Choong and J. A. Ellman, Combinatorial target-guided ligand assembly: Identification of potent subtype-selective c-Src inhibitors, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2419–2424 CrossRef CAS.
- G. W. Bemis and M. A. Murcko, The properties of known drugs. 1. Molecular frameworks, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS.
- G. W. Bemis and M. A. Murcko, The properties of known drugs. 2. Side chains, J. Med. Chem., 42, 5095–5099 Search PubMed.
- X-Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hann, RECAP-retrosynthetic combinatorial analysis procedure: A powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry, J. Chem. Inf. Comput. Sci., 1998, 38, 511–522 CrossRef CAS.
- A. R. Leach, D. V. S. Green, M. M. Hann, D. B. Judd and A. C. Good, Where are the GaPs? A rational approach to monomer acquisition and selection, J. Chem. Inf. Comput. Sci., 2000, 40, 1262–1269 CrossRef CAS.
- M. M. Hann, A. R. Leach and D. V. S. Green, Computational chemistry, molecular complexity and screening set design, in Chemoinformatics in Drug Discovery, ed. T. Oprea, Wiley-VCH, New York, 2005 Search PubMed.
- M. J. Hartshorn, C. W. Murray, A. Cleasby, M. Frederickson, I. J. Tickle and H. Jhoti, Fragment-based lead discovery using X-ray crystallography, J. Med. Chem., 2004, 48, 403–413.
- N. Baurin, F. Aboul-Ela, X. Barril, B. Davis, M. Drysdale, B. Dymock, H. Finch, C. Fromont, C. Richardson and H. Simmonite, Design and characterization of libraries of molecular fragments for use in NMR screening against protein targets, J. Chem. Inf. Comput. Sci., 2004, 44, 2157–2166 CrossRef CAS.
- M. Schade and H. Oschinat, NMR fragment screening: Tackling protein-protein interaction targets, Curr. Opin. Drug Discov. Dev., 2005, 8, 365–373 Search PubMed.
- G. Wilkinson and A. Brown, High concentration screening of GPCRs. AstraZeneca, personal communication, 2004.
- E. Black, J. Breed, A. L. Breeze, K. Embrey, R. Garcia, T. W. Gero, L. Godfrey, P. W. Kenny, A. D. Morley, C. A. Minshull, A. D. Pannifer, J. Read, A. Rees, D. J. Russell, D. Toaderb and J. Tucker, Structure-based design of protein tyrosine phosphatase-1B inhibitors, Bioorg. Med. Chem. Lett., 2005, 15, 2503–2507 CrossRef CAS.
- H.-J. Boehm, M. Boehringer, D. Bur, H. Gmuended, W. Huber, W. Klaus, D. Kostrewa, H. Kuehne, T. Luebbers, N. Muenier-Keller and F. Mueller, Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods, and 3D guided optimisation. A promising alternative to random screening, J. Med. Chem., 2000, 43, 2664–2674 CrossRef CAS.
- S. B. Shuker, P. J. Hajduk, R. P. Meadows and S. W. Fesik, Discoverying high-affinity ligands for proteins: SAR by NMR, Science, 1996, 274, 1531–1534 CrossRef CAS.
- I. Huc and J.-M. Lehn, Virtual combinatorial libraries: Dynamic generation of molecular and supramolecular diversity by self-assembly, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS.
- E. Conti, C. Rivetti, A. Wonacott and P. Brick, X-ray and spectrophotometric studies of the binding of proflavin to the S1 specificity pocket of human α-thrombin, FEBS Lett., 1998, 425, 229–233 CrossRef CAS.
- R. S. Bohacek, C. Martin and W. C. Guida, The art and practice of structure-based drug design: A molecular modelling approach, Med. Res. Rev., 1996, 16, 3–50 CrossRef CAS.
- D. Fattori, Molecular recognition: The fragment approach in lead generation, Drug Discovery Today, 2004, 9, 229–238 CrossRef CAS.
- E. J. Griffen and J. N. Burrows, manuscript, in preparation.
- M. Vieth, M. G. Siegel, R. E. Higgs, I. A. Watson, D. H. Robertson, K. A. Savin, G. L. Durst and P. A. Hipskind, Characteristic physical properties and structural fragments of marketed oral drugs, J. Med. Chem., 2004, 47, 224–232 CrossRef CAS.
Footnotes |
| † This is Chapter 5 taken from the book Structure-based Drug Discovery (Edited by Roderick E. Hubbard) which is part of the RSC Biomolecular Sciences series. |
| ‡ The HTML version of this article has been enhanced with colour images. |
| This journal is © The Royal Society of Chemistry 2006 |