Hot off the Press
In the Hot off the Press section of Molecular BioSystems members of the Editorial Board and their research groups highlight recent literature for the benefit of the community. This month the highlighted topics include steps towards personalized medicine, viral encapsulation of gold nanoparticles, regulation of heat shock protein 70, new insights into the molecular pathogenesis of Alzheimer’s disease, inhibition of Bcr-abl kinase in chronic myelogenous leukemia, detection of DNA using a self-fuelled DNA machine and a traditional solution to a new problem.
Viral encapsulation of gold nanoparticles
Viruses are the media’s favourite “bad guys” and the main source of concern when incurable diseases are in question. However, these parasitic species on the border of the living and the dead have many interesting properties, some of which we can use to our advantage. For example plant virion capsids have been used in host–guest chemistry and supramolecular assembly, therefore being a potent tool for nanotechnology. The ability of self assembly of viral protein shells was also a main focus of Stefan Franzen and his co workers’ research published recently.In their paper, the use of red clover necrotic mosaic virus (RCNMV) coat proteins (CP) to encapsulate gold nanoparticles was described. Normally, assembly of RSNM virus begins with recognition of a specific virion RNA sequence, called the origin of assembly or OAS, by coat protein. The binding of proteins to the OAS RNA initiates the assembly process and formation of a capsid. Knowing that, the researchers were able to prepare gold nanoparticles containing 20 base DNA hairpins with the loop complementary to RNA-1 of the RCNM virus. When RNA-1 is added to DNA modified gold, it hybridizes to the loop therefore forming an artificial origin of assembly (Fig. 1). Subsequent addition of RCNM coat protein and overnight incubation results in formation of virus-like particles, which contain encapsulated gold. After purification, TEM showed that the average size of the particles was 33.5 nm and only gold nanoparticles of sizes that correspond or are smaller than the size of the inner capsid hole (17 nm) can be encapsulated. Several control experiments also showed that the encapsulation is not successful if all requirements (presence of stem-loop DNA, RNA-1 and viral protein) are not met.
Taking into account the growing significance of nanoparticles for cell targeting applications, this could be a successful way of overcoming some limits of intracellular targeting. As the authors point out, viral protein capsids represent a system that can be exploited as a transport container, which could also be modified with specific protein or peptides to ease the cell penetration.
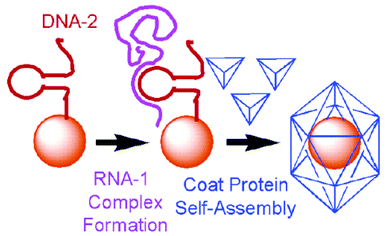 | ||
| Fig. 1 Encapsulation of gold nanoparticles. The procedure mimics the encapsulation of the genome using an oligonucleotide (DNA-2) to capture the larger polycistronic RNA-1. The loop–RNA complex forms the origin of assembly that binds to the coat proteins. Reprinted with permission from J. Am. Chem. Soc., April 12, 2006, volume 128, issue 14, pages 4502–4503. Copyright 2006 American Chemical Society. | ||
L. Loo, R. H. Guenther, V. R. Basnayake, S. A. Lommel, S. Franzen, Controlled encapsidation of gold nanoparticles by a viral protein shell, J. Am. Chem. Soc., 2006, 128, 4502–4503.
Reviewed by: Ljiljana Fruk, Universität, Dortmund, GermanyFull circle: the life of Hsp70
Heat shock protein 70 (Hsp70) is a molecular chaperone that is induced by cellular stress through regulation by heat shock transcription factor 1 (HSF1). Upon induction, Hsp70 levels are elevated and only recede back to basal amounts when the cell returns to an unstressed state. Qian et al. have previously shown that the co-chaperone/E3 ligase carboxy terminus of Hsp70-binding protein (CHIP) stimulates the transcriptional activation of HSF1 and subsequently Hsp70 expression while also triggering the degradation of chaperone-bound substrates by the addition of ubiquitin. In this paper, the group investigate the regulation of HSF1 and chaperone levels by CHIP and uncover a novel form of chaperone regulation during stress recovery of a cell.This paper reveals a new function associated with CHIP, regulation of Hsp70 independent of HSF1. The experiments show that CHIP targets Hsp70 for destruction when the chaperone-bound substrates in the cell are depleted, providing an elegant mechanism for maintaining the balance between the level of this chaperone and its substrates. From these data we can begin to understand how Hsp70 and potentially other chaperones are regulated. CHIP can indirectly stimulate Hsp70 expression, degrade the chaperone-bound substrates and then target Hsp70 for destruction during stress recovery by the cell, essentially controlling Hsp70 expression from beginning to end. This paper is the first to demonstrate how a chaperone can be regulated during stress recovery of a cell. Even more intriguing is that these results present a novel form of biphasic regulation of Hsp70 levels by one protein, CHIP. It will be interesting to evaluate the scope of this type of mechanism for the regulation of chaperone levels.
Shu-Bing Qian, Holly McDonough, Frank Boellmann, Douglas M. Cyr and Cam Patterson, CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70, Nature, 2006, 440, 551–555.
Reviewed by: Melissa O'Neal, University of Texas Southwestern Medical Center, Dallas, USANew insights into the molecular pathogenesis of Alzheimer's disease
Although the neuropathological hallmarks of Alzheimer's disease (AD) contain both extracellular amyloid-β (Aβ) peptide plaques and intracellular neurofibrillary tangles consisting of microtubule-associated protein tau, the relationship between these two phenotypes remains undefined and the predominant hypothesis for the cause of AD is the accumulation of the former plaque that is toxic to neurons. Lu and his colleagues have presented data to suggest that the prolyl isomerase Pin1 might be the molecular connector of the two neuropathological hallmarks associated with AD.Previously, it had been demonstrated that phosphorylation of transmembrane amyloid precursor protein (APP) on the Thr668–Pro motif is elevated in the brains of AD patients and Pin1 could restore the function of AD-associated phosphorylated tau protein and protect neurodegeneration. The authors hypothesized that Pin1 might also act on the pThr668–Pro motif to regulate APP processing and Aβ production. They found that Pin1 specifically interacts with phosphorylated APP and the interaction depends on both the pThr–Pro motif in APP and the WW domain of Pin1, which is a domain known to interact with the pSer/Thr–Pro motif. To further confirm this binding interaction and the consequence of this binding, NMR spectroscopy was employed to show that Pin1 can indeed bind to a 21-residue phosphopeptide corresponding to G659–Q679 of APP and more importantly, the binding accelerates the p-Thr668–Pro peptide bond to undergo cis to trans isomerization by several orders of magnitude compared to uncatalyzed isomerization.
To characterize the biological consequence of Pin1 binding to phosphorylated APP, the effect of Pin1 overexpression on Aβ secretion in CHO–APP cells, which is stably expressing the wild-type human APP751 isoform was investigated. The authors found that the total Aβ secretion was decreased by ∼40% in mitotic cells that have elevated levels of phosphorylated APP. Furthermore, Pin1 knockout in cells drastically increased the total Aβ secretion. Consistent with the cellular data, Pin1−/− mice also presented increased levels of insoluble Aβ42, the major toxic species, in the brain at 15 months old. On the other hand, the other APP processing products including soluble Aβ40, Aβ42 and insoluble Aβ40 did not show significant change compared to the Pin1+/+ mice.
Taken together, these data support the model shown in Fig. 2. In the presence of Pin1, the cis isomer of phosphorylated APP is converted into the trans isomer, which undergoes non-amyloidogenic processing resulting in decreased levels of toxic Aβ. In AD patients, however, the Pin1 is downregulated/inhibited by oxidation. Therefore, the cis-APP cannot be isomerized to the trans form, resulting in increased amyloidogenic APP processing to present elevated levels of toxic Aβ. In conjunction with previous data, deregulation of Pin1 in AD patients might account for the formation of both amyloid-β (Aβ) peptide plaques and neurofibrillary tangles.
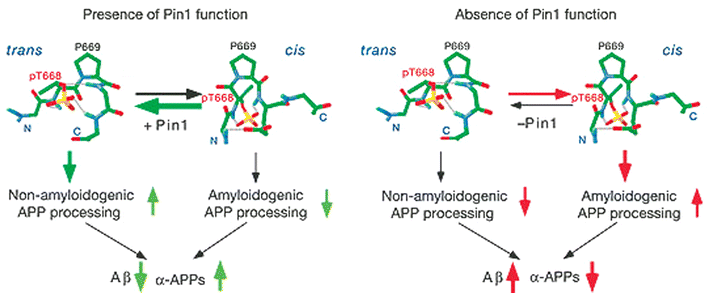 | ||
| Fig. 2 Pin1 affects the balance of amyloidogenic and non-amyloidogenic processing of phosphorylated APP by catalyzing the cis→trans isomerization of pThr–Pro motif. Adapted by permission from Macmillan Publishers Ltd: Nature, 2006, 440, 528–534 (http://www.nature.com), copyright 2006. | ||
Lucia Pastorino, Anyang Sun, Pei-Jung Lu, Xiao Zhen Zhou, Martin Balastik, Greg Finn, Gerburg Wulf, Jormay Lim, Shi-Hua Li, Xiaojiang Li, Weiming Xia, Linda K. Nicholson and Kun Ping Lu, The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production, Nature, 2006, 440, 528–534.
Reviewed by Xiangshu Xiao, University of Texas Southwestern Medical Center, Dallas, USA“Pharmaco-metabonomics”: a step towards personalized medicine
Designed drugs adapted to special needs of the patients are the ultimate goal of modern medicine and the pharmaceutical industry. However, the first difficulties in achieving this goal arise from the inability to predict the individual response to a particular drug. It has been known for a while that the genetic information alone is not enough to make a proper prediction of the effect the drugs could have on a particular organism. Now a group of authors have suggested a novel, co called pharmaco-metabonomic approach towards personalized drug treatment. This approach does not take into account the specific genetic data but the information on the pre-drug dose metabolite content which can then be related to the post-drug effects using a mathematical model.Knowing that metabolite profiles of biofluids reflect a range of different factors like age, sex, diet and disease, T. A. Clayton et al. decided to test the hypothesis (Fig. 3) that the drug efficiency or toxicity can be predicted without any genetic information, but solely on the basis of a pre-drug treatment metabolite profile. An experiment on 65 rats using paracetamol as a test drug was conducted. Urine samples of rats before and after paracetamol treatment were collected, analysed by 1H NMR and correlated to the extent of liver damage observed after treatment. The results showed a statistically significant relationship between the variation in the pre-treatment data and paracetamol-induced effects on liver damage. In the example, a high level of a chemical called taurine in pre-treatment rats led to less post-treatment liver damage. On the other hand, high levels of trimethylamine-N-oxide and betain caused higher incidence of liver damage. This was a clear demonstration that the concept of pharmaco-metabonomics works and that the drug induced response could be predicted from the pre-treatment metabolite profile. Thus, in principle, drug types and particular doses could be individually tailored and the adverse drug reactions avoided. It is important to point out that the ability to do so could lead to a real pharmaceutics revolution not only in relation to improved individual healthcare, but also in a broader scientific sense concerning the synthesis of drugs as well as understanding of their metabolitic pathways.
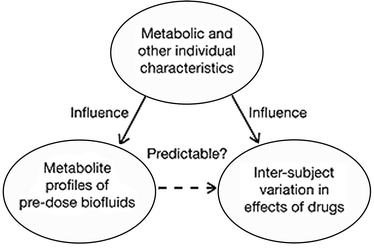 | ||
| Fig. 3 The pharmaco-metabonomic hypothesis. Adapted by permission from Macmillan Publishers Ltd: Nature, 2006, 440, 1073–1077 (http://www.nature.com), copyright 2006. | ||
T. A. Clayton, J. C. Lindon, O. Cloarec, H. Antti, C. Charuel, G. Hanton, J.-P. Provost, J.-L. Le Net, D. Baker, R. J. Walley, J. R. Everett, J. K. Nicholson, Pharmaco-metabonomic phenotyping and personalized drug treatment, Nature, 2006, 440, 1073–1077.
Reviewed by: Ljiljana Fruk, Universität, Dortmund, GermanyIn recent years, there has been growing recognition of the need for an “individualized” approach to medicine. Administration of therapeutics based upon the specific characteristics of an individual would guide both drug selection and dosage and may dramatically increase the efficacy of a treatment while significantly reducing the likelihood and severity of adverse effects. Pharmacogenetic strategies, which utilize the genetic profile of an individual as a predictive tool, provide an incomplete picture as they do not account for environmental factors that influence how a patient will metabolize a given drug. It is well known that an individual’s metabolic phenotype, which is influenced by factors such as age, disease, and the co- or pre-administration of other drugs, can dramatically affect their reaction to a drug. Thus, in a recent article, Jeremy Nicholson and coworkers have proposed that a “pharmaco-metabonomic” approach, where the pre-dose metabolic profile of an individual is used as a predictive tool (Fig. 3), could dramatically increase our ability to anticipate the effects of a therapeutic agent on an individual patient.
The authors present proof-of-principle experiments in which they examined urine samples obtained from rats prior to paracetamol (acetaminophen) treatment to determine if there was a correlation between the pre-dose metabolic profile and both the post-dose drug metabolite profile and adverse effects on the liver. Using 1H nuclear magnetic resonance spectroscopy, they analyzed both pre- and post-dose urine samples and identified a significant association between the pre-dose profile and the quantities and ratios of a number of paracetamol metabolites (Fig. 4). In particular, a strong correlation between the pre-dose data and the ratio of paracetamol glucuronide to paracetamol was observed. Nicholson and coworkers also detected a statistically significant relationship between the pre-dose urinary profile (specifically, levels of taurine, trimethylamine-N-oxide and betaine) and the post-dose histological outcome. Together, these data provide evidence that pharmaco-metabonomic studies may serve as a powerful predictive tool and suggest that pre-dose metabolic profiling could dramatically decrease the occurrence and severity of adverse drug effects. Thus, they have presented an approach that will likely be important in the development of a “personalized” therapeutic strategy.
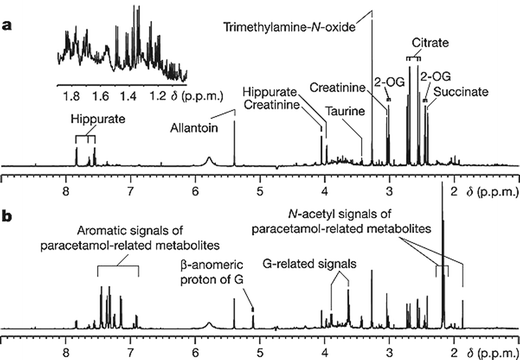 | ||
| Fig. 4 1H NMR spectra of pre-dose (a, −48 h to −24 h) and post-dose (b, 0 to +24 h) urine samples from a rat dosed with paracetamol (600 mg kg−1). The inset in a is an expansion of the δ1.9–1.0 region, indicating the complexity of the endogenous profile and the richness of the embedded information. 2-OG, 2-oxoglutarate; G, paracetamol glucuronide. Reprinted by permission from Macmillan Publishers Ltd: Nature, 2006, 440, 1073–1077 (http://www.nature.com), copyright 2006. | ||
T. A. Clayton, J. C. Lindon, O. Cloarec, H. Antti, C. Charuel, G. Hanton, J.-P. Provost, J.-L. Le Net, D. Baker, R. J. Walley, J. R. Everett, J. K. Nicholson, Pharmaco-metabonomic phenotyping and personalized drug treatment, Nature, 2006, 440, 1073–1077.
Reviewed by: Erin E. Carlson, The Scripps Research Institute, California, USAInhibition of Bcr-abl kinase in chronic myelogenous leukemia
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder characterized by Bcr-abl, a fusion protein with deregulated tyrosine kinase activity resulting from a translocation of chromosome 9 and 22. Imatinib (Gleevec) is a small molecule Bcr-abl inhibitor that binds to the ATP binding site and an adjacent allosteric site in the kinase domain and stabilizes the protein in its inactive conformation. Although it has been remarkably successful for the treatment of CML, a significant proportion of patients chronically treated with imatinib develop resistance because of the incidence of mutations in the bcr-abl kinase domain. Therefore, many efforts are underway to identify new pharmacological agents that could target this kinase by a distinct mechanism and thus have potent activity against imatinib-resistant Bcr-abl mutants. In this paper the authors described the identification of a new class of compounds that inhibit Bcr-abl kinase activity through an allosteric non-ATP competitive mechanism. To identify compounds having selective cytotoxicity towards Bcr-abl-dependent cell lines, they used a differential cytotoxicity screen. Generally screening to find a kinase inhibitor is performed with recombinant catalytic kinase domain at low ATP concentration that might be ATP-competitive in the cell. However this cell-based screen could provide an unbiased method to identify ATP-noncompetitive compounds that target any protein essential to Bcr-abl transformation. Through screening of a combinatorial library containing ~50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds and subsequent medicinal chemistry efforts, GNF-2 was identified as a highly selective inhibitor. GNF-2 was shown to selectively inhibit Bcr-abl-dependent cell growth as well as proliferation of several imatinib-resistant mutant cells. It also showed inhibition of autophosphorylation and phosphorylation of downstream substrates. Interestingly GNF-2 is inactive in vitro against c-abl kinase as well as any of 63 kinases tested in biochemical assays. In spite of a lack of in vitro activity, GNF-2 was demonstrated to specifically bind to abl by means of immobilization and affinity chromatography. Therefore the authors hypothesized that GNF-2 inhibited cellular Bcr-abl activity through an allosteric non-ATP competitive mechanism of action through an alternative cellular target. Molecular docking studies and mutations in the myristoyl cleft lead them to propose that GNF-2 may stabilize the inactive conformation of Bcr-abl upon binding to the myristoyl binding pocket. They found that the combination of imatinib and GNF-2 was effective in inhibiting Bcr-abl transformed cell growth than was treated with imatinib and GNF-2 alone. This work showed the beauty of cell-based screening to identify novel inhibitors of Bcr-abl and GNF-2 type compounds may be potentially useful for the treatment of Gleevec-resistant CML.
000 compounds and subsequent medicinal chemistry efforts, GNF-2 was identified as a highly selective inhibitor. GNF-2 was shown to selectively inhibit Bcr-abl-dependent cell growth as well as proliferation of several imatinib-resistant mutant cells. It also showed inhibition of autophosphorylation and phosphorylation of downstream substrates. Interestingly GNF-2 is inactive in vitro against c-abl kinase as well as any of 63 kinases tested in biochemical assays. In spite of a lack of in vitro activity, GNF-2 was demonstrated to specifically bind to abl by means of immobilization and affinity chromatography. Therefore the authors hypothesized that GNF-2 inhibited cellular Bcr-abl activity through an allosteric non-ATP competitive mechanism of action through an alternative cellular target. Molecular docking studies and mutations in the myristoyl cleft lead them to propose that GNF-2 may stabilize the inactive conformation of Bcr-abl upon binding to the myristoyl binding pocket. They found that the combination of imatinib and GNF-2 was effective in inhibiting Bcr-abl transformed cell growth than was treated with imatinib and GNF-2 alone. This work showed the beauty of cell-based screening to identify novel inhibitors of Bcr-abl and GNF-2 type compounds may be potentially useful for the treatment of Gleevec-resistant CML.
F. J. Adrián, Q. Ding, T. Sim, A. Velentza, C. Sloan, Y. Liu, G. Zhang, W. Hur, S. Ding, P. Manley, J. Mestan, D. Fabbro and N. S. Gray, Allosteric inhibition of Bcr-abl-dependent cell proliferation, Nat. Chem. Biol., 2006, 2, 95–102.
Reviewed by: Hyun-Suk Lim, University of Texas Southwestern Medical Center, Dallas, USADetection of DNA using a self-fuelled DNA machine
Significant research efforts have been undertaken to investigate the possible application of various self-replicating systems and to design efficient nanomachines. Itamar Willner and colleagues have described the design of an enzyme/DNA scission machine for analysis of Tay Sachs genetic disorder mutants. The machine is based on a DNA hairpin structure where the loop is complementary to the nucleic acid of interest. When the complementary strand is hybridized to the hairpin, the loop opens up. Since it contains a specific pre-designed base sequence, it can be cleaved by Fok1 enzyme yielding shorter byproducts. One of these products consists of a Fok1–DNA template which acts as a catalytic cutting machine. When this machine interacts with the fuel, which is FAM/TAMRA doubly labelled nucleic acid, a new complex is formed. This complex can be cleaved again by Fok1 and a new scission process leads to several residues (FAM labelled fluorescent nucleic acid, TAMRA containing oligonucleotide and hairpin byproduct) and the regeneration of the starting Fok1–DNA template which can be reused again in the process. The machine operation could be followed by fluorescence measurements at different stages and it allowed the detection of target mutant DNA at 10−10 M. This value was additionally improved by designing slightly modified self-replicating systems based on the same principle with a sensitivity limit of 1 × 10−14 M. This novel use of the self-replication principle for DNA sensing could in the future, according to the authors, substitute PCR amplification and further design changes could enable detection of a single DNA copy.
Y. Weizmann, Z. Cheglakov, V. Pavlov, I. Willner, Autonomous fueled mechanical replication of nucleic acid templates for the amplified optical detection of DNA, Angew.Chem., Int. Ed., 2006, 45, 2238–2242.
Reviewed by: Ljiljana Fruk, Universität Dortmund, GermanyNew problem…traditional solution
Advances in combinatorial chemistry and high-throughput drug screening are accelerating the flow of bioactive molecules into the drug discovery pipeline. Flow through the pipeline however slows at the point of bioactive target identification. Peterson and Kirschner have demonstrated an effective solution to this problem under certain conditions. Using Xenopus cytoplasmic extracts the investigators screened for small molecules that could inhibit liposomal PIP2-induced actin polymerization. A tetracyclic indole coined pirl1 was isolated in the screen for its ability to inhibit actin polymerization at low micro-molar concentrations. At this point the researchers faced the difficult yet increasingly common problem of having to identify the functional target of a bioactive molecule with a presumable modest target affinity. More often than not groups attempt to solve this problem with some form of affinity chromatography combined with mass spectrometry. This often leads to the isolation of individual proteins that are highly abundant and may or may not be the bioactive target. A more traditional and perhaps more effective route towards target identification was presented within this work. In a biochemical approach analogous to a traditional genetic high-copy suppressor screen, non-treated Xenopus extracts were fractioned and assayed for the ability to suppress pirl1 activity. With this assay two distinct target complexes, Arp 2/3 and Cdc 42/RhoGDI, were found to suppress pirl1 activity. Subsequent analysis demonstrated Arp 2/3 to be an indirect and downstream target of pirl1 activity, while modulation of the nucleotide exchange rate of CDC42 via binding to RhoGDI was the mechanism by which pirl1 directly inhibited actin polymerization. Given the fact that most proteins simultaneously function within several complexes inside of the cell, this approach has the advantage of identifying a drug target within the context of the salient multi-protein complex. Although this approach is suited for in vitro drug screens, the combination of traditional biochemistry, mass spectrometry, and chemical genetics should prove to be an effective approach when tackling a target identification problem.
J. R. Peterson, A. M. Lebensohn, H. E. Pelish, M. W. Kirschner, Biochemical suppression of small-molecule inhibitors: a strategy to identify inhibitor targets and signaling pathway components, Chem. Biol., 2006, 13(4), 443–452.
Reviewed by: Thomas Kodadek, University of Texas Southwestern Medical Center, Dallas, USA| This journal is © The Royal Society of Chemistry 2006 |